
Abstract
Dihydrofolate reductase (DHFR) is a crucial enzyme that catalyzes the conversion of folic acid. Its reserved properties and significance in both human (h-DHFR) and mycobacterium (mt-DHFR) make it a challenging target for developing drugs against cancer and bacterial infections. Although methotrexate (MTX) is commonly used for cancer therapy and bacterial infections, it has a toxic profile. In this study, we aimed to identify selective and non-toxic inhibitors against h-DHFR and mt-DHFR using an in silico approach. From a data set of 8 412 inhibitors, 11 compounds passed the toxicity and drug-likeness tests, and their interaction with h-DHFR and mt-DHFR was studied by performing molecular docking. To evaluate the inhibitory activity of the compounds against mt-DHFR, five known reference ligands and the natural ligand (dihydrofolate) were used to generate a pharmacophoric map. Two potential selective inhibitors for mt-DHFR and h-DHFR were selected for further investigation using molecular dynamics for 100 ns. As a result, BDBM18226 was identified as the best compound selective for mt-DHFR, non-toxic, with five features listed in the map, with a binding energy of –9.6 kcal/mol. BDBM50145798 was identified as a non-toxic selective compound with a better affinity than MTX for h-DHFR. Molecular dynamics of the two best ligands suggest that they provide more stable, compact, and hydrogen bond interactions with the protein. Our findings could significantly expand the chemical space for new mt-DHFR inhibitors and provide a non-toxic alternative toward h-DHFR for the respective treatment of tuberculosis and cancer therapy.
Keywords
Introduction
Dihydrofolate reductase (DHFR) enzyme is a key player in the folate metabolic pathway that is necessary for biosynthesis of DNA, RNA, and proteins, it catalyzes the nicotinamide adenine dinucleotide phosphate (NADPH)-dependent reduction of dihydrofolate to tetrahydrofolate and is critical for the synthesis of purines, thymidine, and several amino acids. Inhibition of this enzyme leads to the arrest of DNA synthesis and cell destruction. 1 The enzyme has been widely investigated as a drug target for medical chemistry. 1 DHFR inhibition has been proven as an effective agent for treating bacterial infections 2 and cancer therapy. 3
Tuberculosis (TB), is one of the ten causes of death worldwide and the primary infection source from a single infectious agent. 4 With the increase in TB treatment, a series of drug-resistant strains have emerged, diagnosis and therapy of multidrug-resistant-TB (MDR-TB) continues to be a major hurdle and is far from being fully solved. 5 Extended drug resistance TB (XDR-TB) shows resistance to second-line drugs fluoroquinolones (FLQ) and aminoglycosides (AMI). It is estimated that 6% of MDR-TB cases are estimated to be XDR-TB. 6
Hence, novel antimycobacterial drugs are urgently required to combat this resistance. Alongside this, better knowledge of the essentiality in the pathogenic organism and larger databases of compounds can contribute to the discovery of new drug molecules. The number of protein structures, X-ray based and modeled, is increasing and now accounts for greater than > 80% of all predicted Mycobacterium tuberculosis proteins, allowing novel targets to be investigated. 1
Methotrexate (MTX) is a DHFR inhibitor that binds to both human DHFR (h-DHFR) and mycobacterium tuberculosis DHFR (mt-DHFR) without any significant selectivity. This inhibition is the basis for the use of MTX in cancer chemotherapy and it has been widely used in the past for the treatment of a variety of malignancies, including breast, head and neck, leukemia, lymphoma, lung, osteosarcoma, bladder, and trophoblastic neoplasms.7,8 Nevertheless, MTX is not selective for mt-DHFR 9. Trimethoprim (TMP) and pyrimethamine (PYR) are selective and powerful inhibitors of protozoan DHFR,9,10 but they have a low affinity for mt-DHFR. 9 The triazine Br-WR99210 has been shown to be a potent inhibitor of malaria DHFR but not mycobacteria. 11
As DHFR is present in both prokaryotes and eukaryotes, 12 special attention must be paid to the selectivity between the two proteins; mt-DHFR and h-DHFR. This difference can be exploited for the design of a new selective inhibitor for mt-DHFR, a glycerol molecule (glycerol A) is found close to the active site in a pocket (GOL pocket) in mt-DHFR. 13 This pocket does not exist in the human protein. 13
The aim of our study is to explore two new specific and selective inhibitors of mt-DHFR and h-DHFR, respectively, that have an affinity better than MTX and the natural ligand by screening a large library of molecules from chemical databases using structure-based drug design (SBDD), focusing on the key player in our workflow study, which is selectivity. Molecular docking and 3D pharmacophore modeling were used in virtual high-throughput screening to discover potential inhibitors of mt-DHFR and to highlight the main features involved in the biological inhibitory activity in the drugs used against the target protein and to examine the potency of the selective hit found and also to explore a new non-toxic inhibitor for h-DHFR protein.
Materials and Methods
Target selection and preparation of DHFR protein
The X-ray crystallographic structure of mt-DHFR (PDB ID: 1DF7) with a resolution of 1.70 Å and h-DHFR (PDB ID: 1OHJ) with a resolution of 2.50 Å were obtained from the Protein Data Bank (PDB) database. 14 The protein structures were prepared using AutoDockTools-V.1.5.7, 15 by removing water molecules and adding polar hydrogens and Kollman United Atom charges then the file was saved in “pdbqt” format.
Construction of ligands data set and filtration
To search for ligands that have structural complementarity with the active site of the target in BindingDB 16 and selleckchem 17 databases. A library of 8 412 molecules was collected and then downloaded under spatial data file (SDF) format with 3D conformation, the elimination of duplicates and the conversion into a PDB file format was carried out by OpenBabel. 18 Drug-likeness score prediction of those compounds was evaluated by calculating the descriptors in Molecular Operating Environment (MOE). From 8 412 molecules, 54 compounds were selected for molecular docking preparation that respects Lipinski’s rule of five (RO5) and Veber rules.
The toxicity assessment of the molecules used in our study was performed using Mcule Toxicity Checker, 19 StopTox,20,21 and Protox-II. 22 Based on this evaluation, only 11 compounds have been selected for molecular docking with the mt-DHFR and h-DHFR, all the ligands were prepared and contain atom types supported by AutoDock tools plus extra records that specify rotatable bonds and then saved in pdbqt format.
Five known approved drugs against mt-DHFR were collected from literature, namely, Bromo_WR99210, methylbenzoprim, MTX, PYR, TMP, and also the dihydrofolic acid as a positive control. From each of them, we searched for their 3D structures using the PubChem database. 23
Docking and scoring
After selecting the active site residues of DHFR protein using AutoDock tools 1.5.7, 24 this allowed the preparation of the grid box, with a spacing of 1 Å and center coordinates fixed at X = 10.16 Å, Y = 37.35 Å, Z = 14.58 Å and a size of X = 18 Å, Y = 18 Å, Z = 18 Å. The grid settings file was saved via the output option of the grid menu.
To select ligands with lower affinity values, AutoDock Vina was used to assess the binding affinity and binding conformation of these selected ligands. The tool is designed for protein–ligand docking, using multiple CPUs simultaneously, making it faster and more accurate, and uses Lamarckian genetic algorithm and semi-empirical free energy field to generate ten ligand poses after docking. 25 Three-dimensional protein–ligand interactions were visualized in PyMol. 26
Three-dimensional pharmacophore building
To build the training set of the five compounds commonly used as inhibitor DHFR protein, pharmacophore query methodology was used, this method is based on searching 3D distances between features like HBD/HBA, hydrogen bond (H-bond) donors/acceptors; PI, positive ionizable; ARO, aromatic ring; Hyd, hydrophobic centers; ML, metal ligand; Cat, cation; Ani, anion. 27 For this purpose, MOE tool was used.
Energy optimization and minimization of these compounds were done and aligned through the flexible alignment tool of MOE, and the generation of the best pharmacophoric model with the lowest S value and with the first common features has been done using the pharmacophore query editor tool.
The test set of the best four of 11 final compounds was created and then screened against the pharmacophore model to find the best matches in terms of root mean square deviation (RMSD) between pharmacophore query features and corresponding ligand points. The compound with the lowest RMSD value (close to 0) was selected as the more potent compound that has the best binding affinity and biological activity. In parallel, we used the scatter plot to highlight the best ligand with the lowest (RMSD), which provides the mapping between features and annotation points of ligands in several conformations.
Molecular dynamics simulations
To analyze protein–ligand interaction energy, MD simulations of 100 ns interval were performed using GROMACSv.2020.4, 28 for the following complexes: the human protein h-DHFR with the best ligand found by docking, human protein h-DHFR with MTX, mycobacterium protein mt-DHFR with the best compound found by docking, and mycobacterium protein mt-DHFR with MTX.
The CHARMM27 force field 29 was used to describe the interactions between the protein and the solvent, while TIP3P 30 was used as the water model. A cubic simulation box with an edge length of 1.0 nm was used to solvate the protein. The system was neutralized by adding an equal number of positive and negative ions to reach a target ion concentration. The system was then minimized using the steepest descent algorithm, and equilibrated at 300 K using V-rescale for 100 ps as NVT ensemble, and finally, production runs were carried out using an NPT ensemble with a time step of 100 ps followed by equilibration at 1 atm pressure using Parrinello–Rahman algorithm. 31 While LINCS algorithm was applied for bond constraints with distance cut-off using Verlet during the simulation. 28
Molecular dynamics analysis
To gain a better understanding of the structural changes that occur during the simulation, The RMSD of atomic coordinates, root mean square fluctuation (RMSF), radius of gyration (Rg), solvent accessible surface area (SASA), and H-bond parameters were calculated using the gmx_rms tool in GROMACS. 32
Results
Data set generation
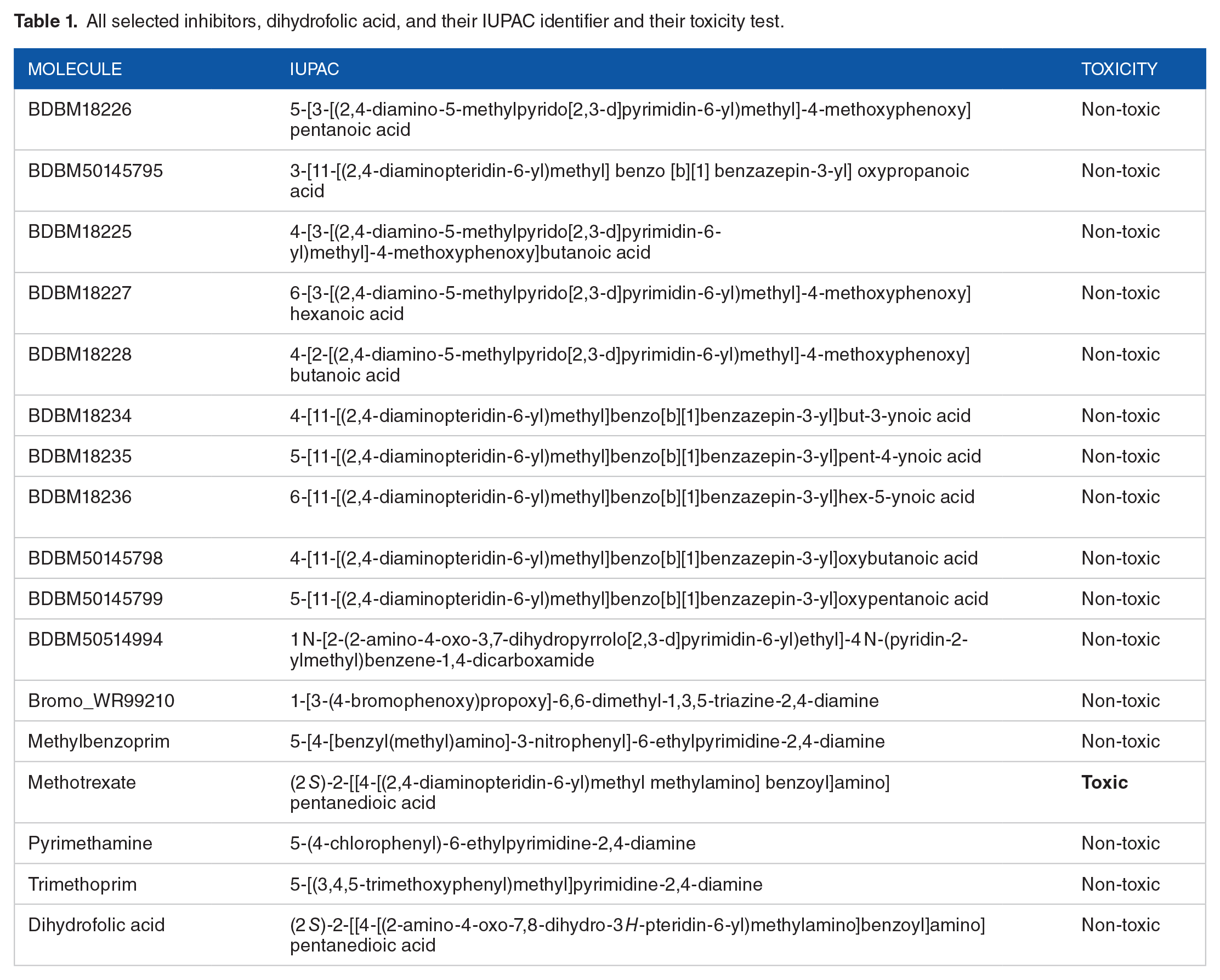
Table 1 depicts the 11 selected potential inhibitors with their toxic/non-toxic report and the five commonly used inhibitors of the protein as a test reference.
All selected inhibitors, dihydrofolic acid, and their IUPAC identifier and their toxicity test.
Docking and scoring
The 11 molecules that passed the RO5, Veber, and toxicity filters were compared with five reference molecules in addition to the natural ligand of the target (dihydrofolic acid). The results showed that MTX is the best reference inhibitor as shown in Figure 1A, with an affinity of –8.7 and an H-bond number of 6.

The results of the docking analysis: (A) The binding affinity of the known inhibitors against mt-DHFR protein. (B) Bar graph representing the binding energy (in kcal/mol) for the 11 ligands and the potent reference ligand; methotrexate with the two targets; h-DHFR in pink and mt-DHFR in purple.
Based on the binding affinity, 11 molecules with the best binding affinity to mt-DHFR ranging from –9.6 to –7.4 kcal/mol were selected. As shown in Figure 1B, ligand 1 and ligand 2 (BDBM18226 and BDBM50145795) were the most important ones as they showed a greater affinity than the rest of the inhibitors with the best affinity (–9.6 kcal/mol) for mt-DHFR and (–7.3 kcal/mol) for h-DHFR, the affinity of the 11 molecules toward the two target proteins, the number of H-bonds are shown in Table 2.
Docking results of mt-DHFR, h-DHFR, and selected molecules.

Abbreviation: h-DHFR, human dihydrofolate reductase.
Figure 2A shows the 3D interaction between the mt-DHFR and ligand 1, the result shows that BDBM18226 binds with the target protein with seven H-bonds; one H-bond to the following residues ILE-5, ARG-32, ARG-60, ILE-94, TYR-100, and two H-bonds with MET-36. While BDBM50514994 binds with the h-DHFR with four H-bonds with four residues; ILE-7, GLU-30, GLN-35, and TYR-121, and an affinity of 9.9 kcal/mol (Figure 2B).
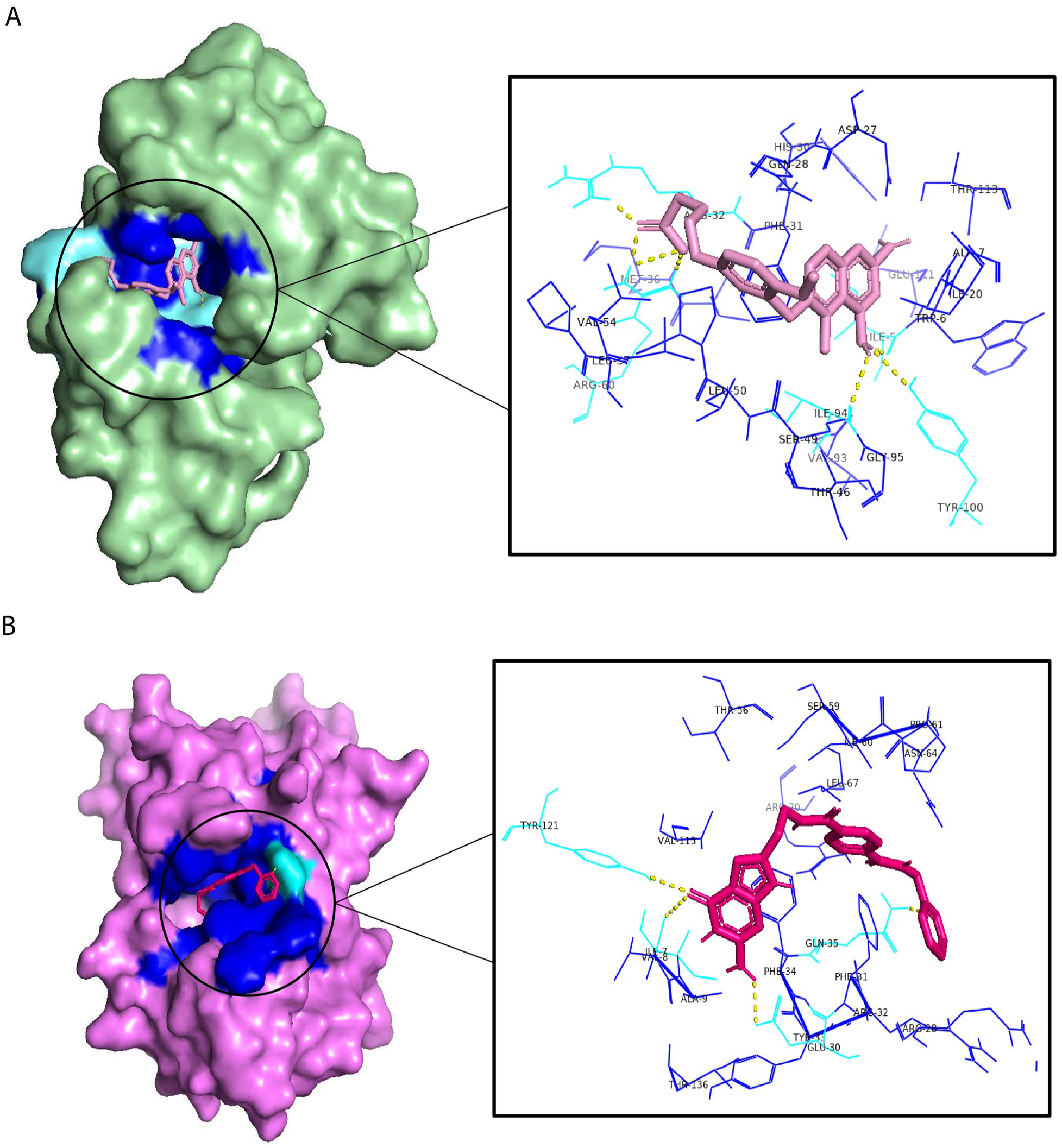
The 3D interactions between the target protein and the best ligand found: (A) The 3D interactions between mt-DHFR–-ligand 1. (B) The 3D interaction between the h-DHFR–ligand 9.
Toxicity profile prediction
The best molecules obtained were compared with the best drug used (MTX) based on affinity score, H-bonds, and toxicity prediction.
The toxicity profile of MTX in comparison with the two best selective molecules found in BDBM18226 and BDBM50514994 shows that MTX clearly ranks in the most dangerous class (Supplemental Figure S1); class 1 with a median lethal dose (LD50) of 3 mg/kg, while the other two molecules rank in less critical classes with an affordable lethal dose (Supplemental Figure S2).
Pharmacophore model result
Based on the five known reference inhibitors with biological inhibitory activity against the DHFR protein, a pharmacophore map was generated to highlight the main features that are involved in this inhibitory effect as shown in Figure 3.
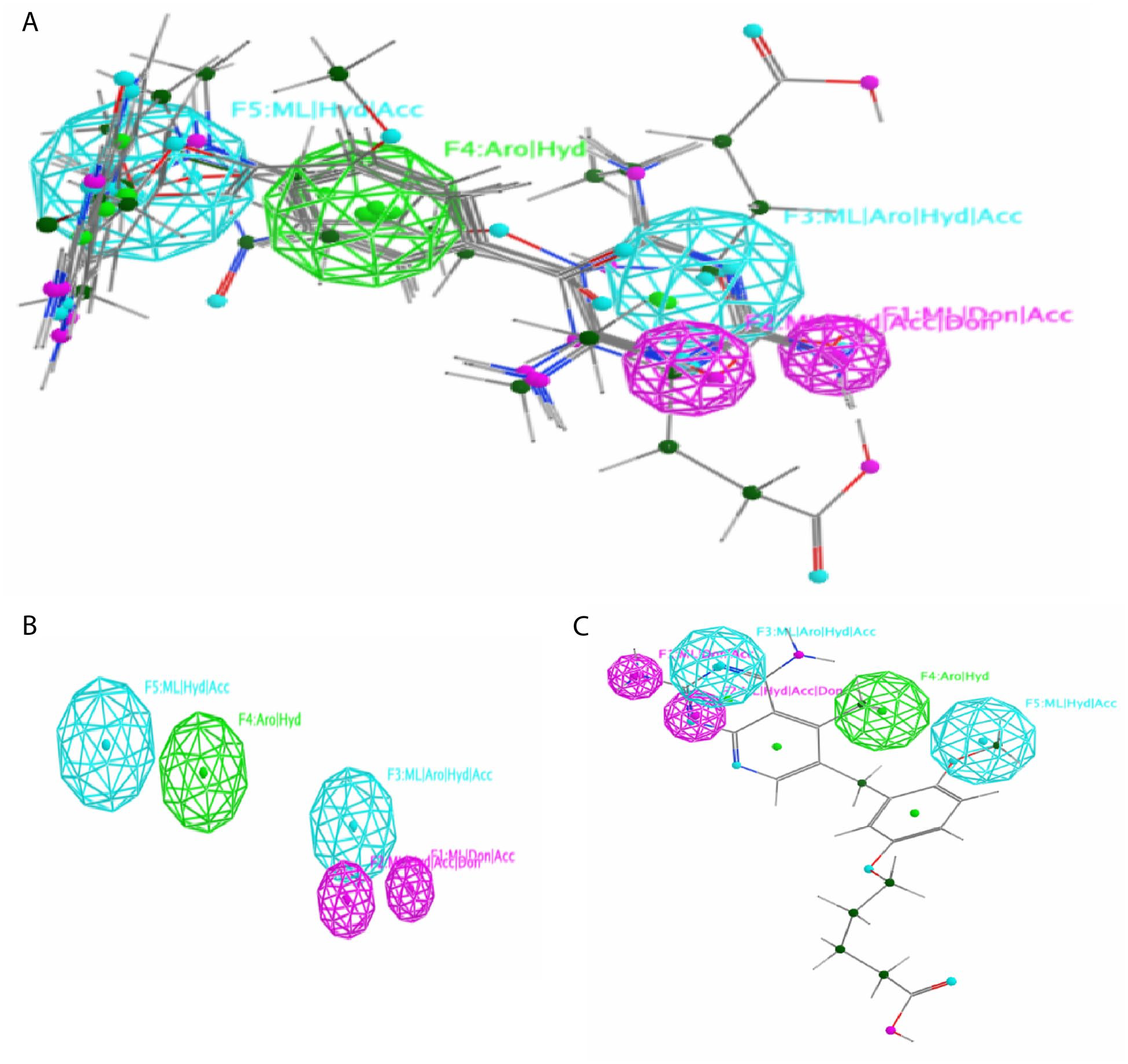
The 3D-pharmacophore results. (A) The pharmacophore map generated after the flexible alignment of the five reference inhibitors, it shows the different features explored in the model structure. (B) A simplified view of the model without ligands. (C) The best molecule obtained after the screening with the reference pharmacophoric map.
The map clearly shows that there are five features: F1: ML|Don|Acc, F2: ML|Hyd|Acc|Don, F3: ML|Aro|Hyd|Acc, F4: Aro|Hyd, and F5: ML|Hyd|Acc, respectively (Figure 3A and B).
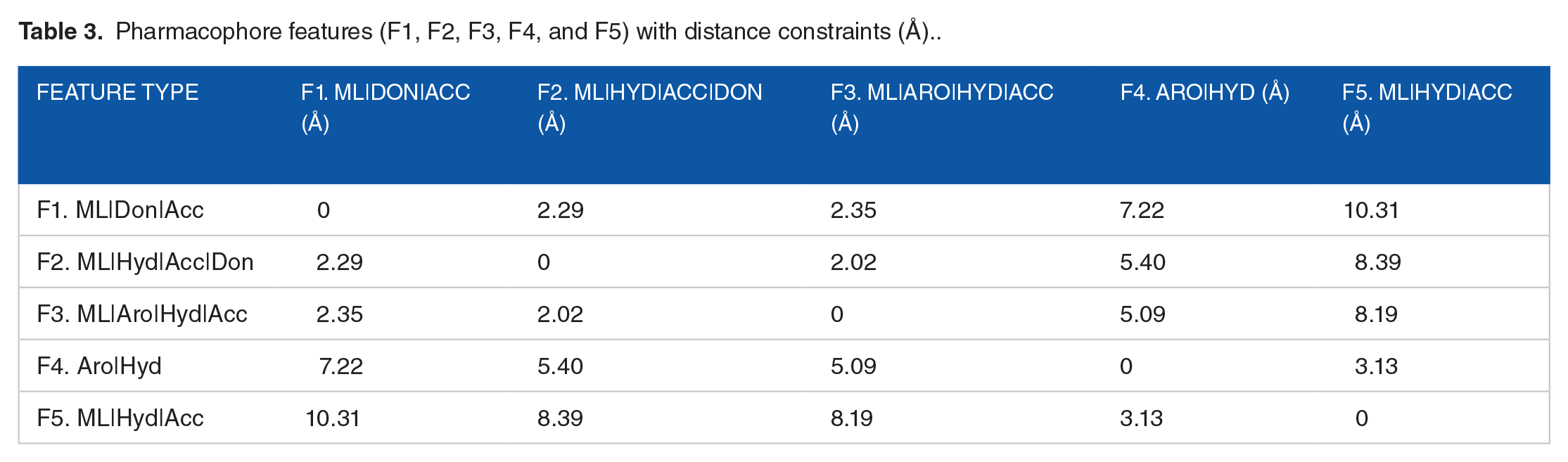
The distances between the pharmacophore features were determined in Table 3, and it represents how the pharmacophoric features are involved in interactions against the DHFR target protein.
Pharmacophore features (F1, F2, F3, F4, and F5) with distance constraints (Å)..
As it was concluded that MTX is the best reference inhibitor, we took ligands that have a better affinity than the reference indicated and which bind with mt-DHFR better than h-DHFR, which are in the order of four molecules. The resulting compounds were then subjected to screening against the pharmacophore model to find which one is close to the reference model, and which has the lowest RMSD close to zero, and corresponding ligand points, Figure 3C shows the best result obtained after the pharmacophore search, the best compound that has a low RMSD and has the characteristics described in the pharmacophore model is ligand 1 with the following ID: BDBM18226. This result was confirmed by calculating the RMSD values obtained from the four molecules in different conformations, Figure 4 shows a scatter plot that clearly describes that BDBM18226 is closer to zero than the other molecules.
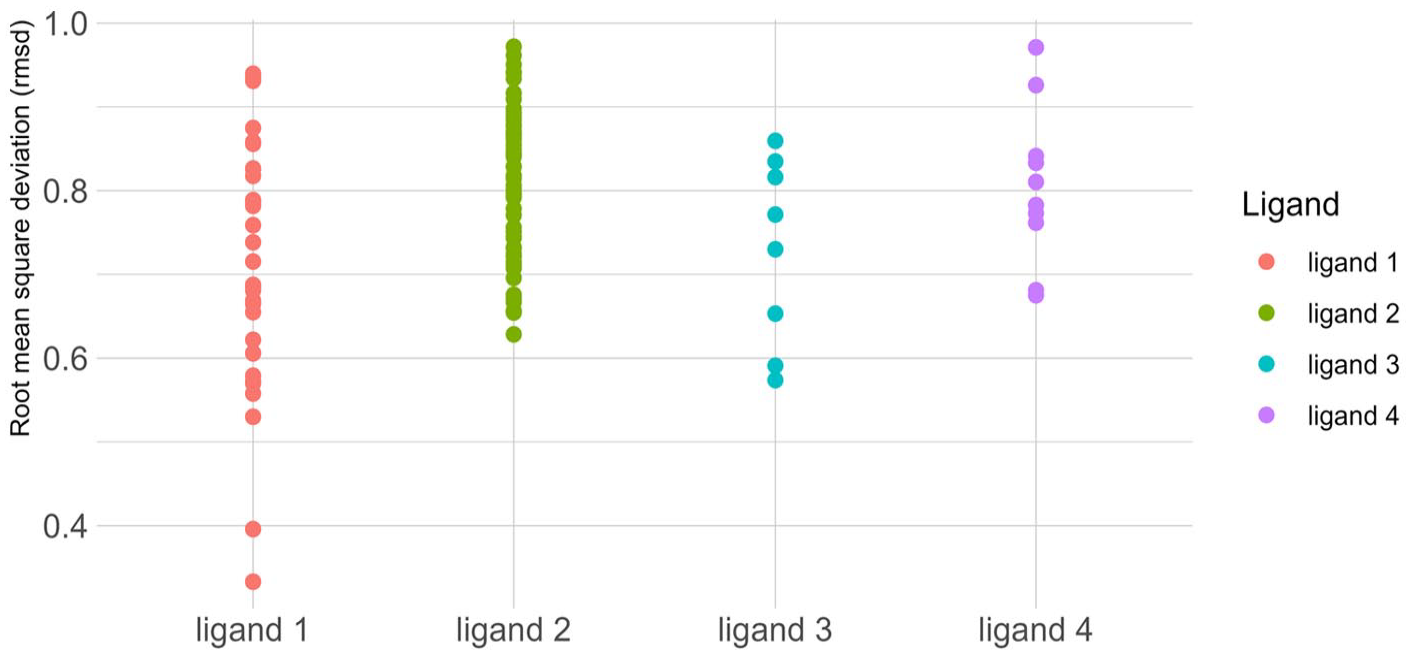
Scatter plot representation that groups all the RMSD values of the four candidate molecules in different conformations. Each point presents a conformation with an RMSD value.
Molecular Dynamic Analysis of Ligand–Protein Complex
mt-DHFR–ligand 1 complex
The dynamic simulation was performed by simulating two different complexes: mt-DHFR–MTX and mt-DHFR–ligand 1.
The result of the simulations was analyzed using RMSD, RMSF, Rg, SASA, and hydrogen bonding analysis, the results are shown in Figure 5 and Table 4 presents the mean and standard deviation. By quantifying the deviation from the simulated complex structure, the RMSD values indicate for the mt-DHFR–ligand 1 complex a closer match to the reference structure with a low RMSD value (0.16 ± 0.02) compared to the mt-DHFR–MTX complex. In addition; by measuring the mobility or flexibility of the residues in the complex; RMSF values showed that mt-DHFR–ligand 1 complex shows low flexibility with an RMSF value of (0.06 ± 0.02) compared to mt-DHFR–MTX complex showing a higher value (0.09 ± 0.05). The Rg values indicated a more compact shape for mt-DHFR–ligand 1 (1.53 ± 0.008) in contrast to mt-DHFR–MTX (1.55 ± 0.001). SASA values revealed a greater solvent-exposed surface area for the mt-DHFR–ligand 1 complex, compared to mt-DHFR–MTX, indicating more potential interactions with its environment. Finally, H-bond analysis, showed a higher number of H-bonds for the mt-DHFR–ligand 1 complex with numbers that can reach 12 H-bonds while the complex mt-DHFR–MTX can reach only six H-bonds, contributing to its stability and target specificity compared its mt-DHFR–MTX.

Protein–ligand molecular dynamics analysis for mycobacterium tuberculosis protein. (A) The RMSD analysis of mt-DHFR–ligand 1 (green) and mt-DHFR–MTX (blue) calculated for 100 ns. (B) The RMSF of each mt-DHFR–ligand 1 and mt-DHFR–MTX complex according to residue numbers. (C) The radius of gyration of the two protein–ligand complexes calculated at the two simulations. (D) The SASA analysis calculated for the mt-DHFR–ligand 1 and mt-DHFR–MTX for 100 000 ps. (E) The hydrogen bonds analysis of the two complexes.
The average of the analyzed parameters of mt-DHFR, h-DHFR, and the ligands 1 and 9.

Abbreviations: h-DHFR, human dihydrofolate reductase; mt-DHFR: mycobacterium tuberculosis dihydrofolate reductase; RMSD, root mean square deviation; RMSF, root mean square fluctuation; SASA, solvent accessible surface area.
H-DHFR–ligand 9 complex
Concerning the h-DHFR–ligands simulations, the results of molecular dynamics between h-DHFR–MTX and h-DHFR–ligand 9 complex showed differences in various parameters, such as RMSD, RMSF, Rg, SASA, and H-bonds. Figure 6 and Table 4 present the mean and standard deviation. The RMSD values indicated a higher degree of structural stability with a good agreement between the h-DHFR protein and the ligand 9 with a low RMSD value of (0.17 ± 0.04) compared to the h-DHFR–MTX complex that has a value of (0.29 ± 0.05). The RMSF value revealed that h-DHFR–ligand 9 interaction reduces flexibility in certain regions of the protein and the model is more stable compared to the h-DHFR–MTX complex. The h-DHFR–ligand 9 complex had also a lower Rg value (1.60 ± 0.1). The SASA results showed an increase in the exposed surface area of the protein in the h-DHFR–ligand 9 complex with a value of (93.98 ± 0.9). Finally, the H-bond analysis showed a higher number of H-bonds formed in the complex h-DHFR–ligand 9 that can reach 14 H-bonds, while the complex h-DHFR–MTX can reach only a number of six H-bonds.

Protein–ligand molecular dynamics analysis for the human protein. (A) The RMSD analysis of h-DHFR–ligand 9 (cyan) and h-DHFR–MTX (pink) calculated for 100 ns. (B) The RMSF values according to residue numbers of the human protein. (C) Radius of gyration (Rg) of the h-DHFR–ligand 9 and h-DHFR–MTX calculated at the two simulations for 100 000 ps. (D) The SASA analysis calculated for the h-DHFR–ligand 9 and h-DHFR–MTX. (E) The hydrogen bonds analysis of the two complexes.
Discussion
Our study treats two topics, the first one is to identify the specific features required to prevent the activity of mt-DHFR protein and to explore a new compound for selective inhibition through a computational approach to solve the resistance to treatments of TB, due to the mutations of this bacterium, and the second one is about the exploration of a new non-toxic h-DHFR inhibitor that can be used as an alternative to MTX in cancer therapy as MTX is a toxic drug and used like medicine that target DHFR protein.
The current study uses the advantages of virtual high-throughput screening approaches to identify molecules targeting mt-DHFR protein. In the extension of the mt-DHFR ligand pocket, there is a small hydrophobic pocket that hosts a glycerol molecule (GOL), this pocket does not exist in the human protein. 13 Based on these data, and considering that DHFR is present in both humans and bacteria, selectivity becomes an important criterion in the design and development of new compounds.
To explore selective inhibitors for mt-DHFR, the study was executed with two target proteins mt-DHFR and h-DHFR. A database of 8 412 compounds from the binding database and selleckchem was created and screened against mt-DHFR, the results obtained were subsequently screened against h-DHFR to study the selectivity. Among the 8 412 molecules, 11 compounds that obeyed Lipinski’s RO5 and Veber’s rules, and are non-toxic were selected and docked against both human and bacterial enzymes to measure the potency as well as the selectivity.
As a positive control, five known and Food and Drug Administration (FDA)-approved inhibitors that have inhibitory activity against the target protein and the natural substrate (dihydrofolate) were selected, all five inhibitors of reference have shown a good affinity toward the enzyme, the most potent one is MTX that showed the lowest value of affinity energy (–8.9 kcal/mol), this molecule is used usually as an anticancer drug, 7 based on the aforementioned information, MTX was taken as the inhibitor of reference in terms of energy score to compare its activity and potency with the 11 selected molecules.
The total of the 11 molecules was docked against the human enzyme (h-DHFR) and the bacterial enzyme (mt-DHFR) with glycerol to see the effect of the extra glycerol pocket on the selectivity of the inhibitors and if we can use this information in the design of specific drugs.
The inhibition of DHFR is the basis for the use of MTX as a cancer chemotherapy drug. 7 In our study, we approved the toxic profile of this drug using in silico approach, MTX was found to be in class 1 in toxicity level with an LD50 of 3 mg/kg, which was approved by another study; a meta-analysis reported across Asian, Caucasian, pediatric, and adult patients that treats three systematic reviews on MTX-induced toxicity, 33 another research on MTX doses described that MTX is administered at doses ranged from 12 mg intrathecally and 20 mg/m2 orally, or intravenously as weekly maintenance chemotherapy for all to doses as high as 33 000 mg/m2, higher given intravenously are defined as high-dose MTX (HDMTX) therapy that can cause significant toxicity, which not only leads to morbidity and occasional mortality but may also interrupt cancer treatment. 34
Searching for a non-toxic alternative drug that has a better affinity than MTX for h-DHFR was the second part of this study. The h-DHFR docking results showed that only BDBM50514994 with (–9.9 kcal/mol) has a better affinity than the reference ligand (MTX) with a binding energy of –8.5 kcal/mol—and the natural ligand itself (dihydrofolic acid) that have an affinity of 9.2 kcal/mol, In regards to the mt-DHFR docking results, showed that only four compounds of 11 have better affinity than the inhibitor of reference (MTX) that have binding affinity energy of –8.9 kcal/mol and the dihydrofolic acid (natural ligand) that have a binding energy of –8.8 kcal/mol, the lowest affinity value registered is (–9.6 kcal/mol) and it is the same shared between ligands 1, 2, and 11, followed by ligand 3 (–9.4 kcal/mol) with 7, 6, 6, 7 H-bonds, respectively. The ligands BDBM18226 (ligand 1) and BDBM50514994 (ligand 9) proved to be the most effective compounds promising to inhibit mt-DHFR and h-DHFR, respectively.
To identify the features involved in the inhibitory activity of the five known inhibitors, a reference pharmacophoric map was generated in our study, the map clearly shows that the five molecules share five features that are: F1: ML|Don|Acc, F2: ML|Hyd|Acc|Don, F3: ML|Aro|Hyd|Acc, F4: Aro|Hyd, and F5: ML|Hyd|Acc, respectively, as was clearly shown in Figure 3, and to evaluate the inhibitory activity of the four best compounds found, the resulting compounds were screened against the pharmacophore model, and a search in the pharmacophoric scaffold was carried out, the best with a lowest RMSD that has all the features found in the map model is the compound 5-[3-({2,4-diamino-5-methylpyrido[2,3-d]pyrimidin-6-yl}methyl)-4-methoxyphenoxy]pentanoic acid, that present an RMSD close to zero, this compound has been identified as a 50% inhibition of growth in the culture at 0.0011 mM for DHFR protein of Bacillus cereus. 35
Based on the structure of these compounds, the pharmacophoric map showed that the hydrophobic Hyd groups and the donor/acceptor region play a vital role in their binding to the mt-DHFR protein. HBA groups enforce the interaction between the ligands and the pocket site of the target protein.36-38
BDBM18226 (ligand 1) and BDBM50514994 (ligand 9) have shown promising potential as inhibitory agents of mt-DHFR and h-DHFR, respectively. To assess the stability and quality of the protein–ligand complex models, we compared these compounds with MTX using molecular dynamics simulations and various parameters including RMSD, RMSF, Rg, SASA, and H-bonds. The h-DHFR/mt-DHFR–MTX complexes were found to be less optimal, as reflected by the higher RMSD and RMSF values and larger Rg values, indicating less stability and compactness. 39 The most significant fluctuations in the RMSF values were observed in the active site residues, particularly Asn19, Glu44, Lys55, Ser59, Lys80, Asp145, Arg150, Ser153, and Gly164 for h-DHFR, and Gly17, Asp70, Glu122, and Ala123 for mt-DHFR. The limited number of H-bonds formed by MTX with the h-DHFR/mt-DHFR protein and the higher SASA values suggest that MTX is less buried in the complex and less protected by the solvent environment, leading to instability. Overall, BDBM18226 and BDBM50514994 showed better stability and specificity compared to MTX, making them promising candidates for inhibitory agents against mt-DHFR and h-DHFR for therapeutic intervention against TB and cancer. 40
The dynamic molecular analysis of mt-DHFR–ligand 1 and h-DHFR–ligand 9 provided valuable insights into the binding behavior of these ligands. The RMSD value for mt-DHFR–BDBM18226 (0.16 ± 0.02 nm) and h-DHFR–ligand 9 (0.17 ± 0.04 nm) was found to be lower than that of the DHFR–MTX complexes, indicating a tighter binding of these ligands to the mt-DHFR/h-DHFR proteins with stable interactions and a strong binding affinity. Moreover, the RMSF values for BDBM18226 and ligand 9 were lower (0.06 ± 0.02 and 0.10 ± 0.03 nm, respectively), indicating a more stable and accurate model with fewer fluctuations. In addition, the Rg and SASA values of the complex models were found to be smaller, indicating that the two ligands induced a more compact structure and had stronger interactions with the mt-DHFR/h-DHFR proteins. This could be attributed to the formation of new H-bonds between the protein and the ligand, as demonstrated in the analysis of H-bonds, which showed that the complexes had a higher number of H-bonds (up to 14 H-bonds). Overall, the dynamic molecular analysis of mt-DHFR–ligand 1 and h-DHFR–ligand 9 revealed important information about their binding behavior, highlighting their strong affinity and stable interactions with the mt-DHFR/h-DHFR proteins.41,42
Based on their stability and specificity, as evidenced by their low RMSD and RMSF values, as well as their ability to induce a more compact shape and a higher number of stable H-bonds, Ligand 1 (5-[3-[(2,4-diamino-5-methylpyrido[2,3-d]pyrimidin-6-yl)methyl]-4-methoxyphenoxy]pentanoic acid) and BDBM50514994 (4-[11-[(2,4-diaminopteridin-6-yl)methyl]benzo[b][1]benzazepin-3-yl]oxybutanoic acid) have the potential to be effective inhibitory agents of mt-DHFR and h-DHFR, respectively. These findings suggest that these compounds could be used for therapeutic intervention against TB and cancer therapy.
Conclusion
The problem of resistance to anti-TB drugs poses a significant challenge in combating TB. The need to explore new alternative hits remains the most effective solution to combat this problem. The novo synthesis pathway of folic acid is a very powerful pathway and a very interesting therapeutic target. Our findings identified two new compounds; (5-[3-[(2,4-diamino-5-methylpyrido[2,3-d] pyrimidin-6-yl) methyl]-4- methoxyphenoxy] pentanoic acid) and (4-[11-[(2,4-diaminopteridin-6-yl) methyl] benzo[b][1] benzazepin-3-yl] oxybutanoic acid) that are selective for mt-DHFR, and h-DHFR, respectively. These compounds could be developed as potential inhibitors for the DHFR protein and could significantly expand the chemical space for new mt-DHFR inhibitors. Our study presents promising opportunities to address the challenge of resistance to anti-TB drugs and provide new avenues for the development of drugs against cancer.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322231171778 – Supplemental material for Selective Non-toxics Inhibitors Targeting DHFR for Tuberculosis and Cancer Therapy: Pharmacophore Generation and Molecular Dynamics Simulation
Supplemental material, sj-docx-1-bbi-10.1177_11779322231171778 for Selective Non-toxics Inhibitors Targeting DHFR for Tuberculosis and Cancer Therapy: Pharmacophore Generation and Molecular Dynamics Simulation by Ghyzlane EL Haddoumi, Mariam Mansouri, Houda Bendani, Mohammed Walid Chemao-Elfihri, Jouhaina Kourou, Hanane Abbou, Lahcen Belyamani, Ilham Kandoussi and Azeddine Ibrahimi in Bioinformatics and Biology Insights
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was carried out under national funding from the Moroccan Ministry of Higher Education and Scientific Research (COVID-19 program) to A.I. This work was also supported by a grant from the Moroccan Institute of Cancer Research and the PPR-1 program to A.I. and Biocodex grant to AI.
Declaration Of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
GEH contributed to the conceptualization, data collection and analysis, and writing the initial draft and revisions. MM, HB, and MWCE contributed to the data curation, methodology, and visualization. JK and HA contributed to the draft revision. LB, and IK supervised, revised, and validated the last draft. AI conceived and supervised the study, contributed to writing revisions and validation. All authors have read and agreed to the published version of the manuscript.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.