
Abstract
Type II diabetes is an endemic disease and is responsible for approximately 90% to 95% of diabetes cases. The pathophysiological distortions are majorly β-cell dysfunction, insulin resistance, and long-term inflammation, which all progressively unsettle the control of blood glucose levels and trigger microvascular and macrovascular complications. The diverse pathological disruptions which patients with type II diabetes mellitus exhibit precipitate the opinion that different antidiabetic agents, administered in combination, might be required to curb this menace and maintain normal blood glucose. To this end, natural compounds were screened to identify small molecular weight compounds with inhibitory effects on protein tyrosine phosphatase 1B (PTP1B), dipeptidyl-peptidase-4 (DPP-4), and α-amylase. From the result, the top 5 anthocyanins with the highest binding affinity are reported herein. Further ADMET profiling showed moderate pharmacokinetic profiles for these compounds as well as insignificant toxicity. Cyanidin 3-(p-coumaroyl)-diglucoside-5-glucoside (−15.272 kcal/mol), cyanidin 3-O-(6ʺ-malonyl-3ʺ-glucosyl-glucoside) (−9.691 kcal/mol), and delphinidin 3,5-O-diglucoside (−12.36 kcal/mol) had the highest binding affinities to PTP1B, DPP-4, and α-amylase, respectively, and can be used in combination to control glucose fluctuations. However, validations must be carried out through further in vitro and in vivo tests.
Keywords
Background
Type II diabetes is widely regarded as a 21st-century epidemic and accounts for approximately 90% to 95% of diabetes cases. 1 There are several factors associated with the disease progression including genetic and environmental factors, and the major pathophysiological features include insulin resistance and impaired insulin secretion in addition to at least 6 other pathophysiological abnormalities, all of which disrupt the control of blood glucose levels.2,3 Diabetes (type II) patients have a high risk for both microvascular and macrovascular complications. 2 To maintain normal blood glucose levels, it is considered that multiple antidiabetic agents, administered in combination, will be required owing to the several pathological irregularities present in type II diabetes. 2
Protein tyrosine phosphatase 1B (PTP1B) is expressed in most cells in the body but overexpressed in cells under diabetic conditions. 4 The protein is a down-regulator of vital metabolic pathways including but not limited to the insulin signaling pathway, and plays a crucial role in diabetes.5-7 Inhibition of PTP1B has shown promising effects in the treatment of diabetes.8-10 Overexpression of PTP1B enhances insulin resistance, but its depletion or inhibition negates insulin resistance, promotes insulin sensitivity, and stimulates insulin-regulated glucose uptake. 11 Hence, PTB1B inhibitors are considered good therapeutic molecules in treating diabetes. 4
Owing to the substantial dysregulation of the expression of dipeptidyl peptidase-4 (DPP-4), DPP-4 inhibitors have risen to the limelight as a novel class of antidiabetic agents that show promising results in decreasing glucose fluctuations in diabetic patients. 12 Interestingly, the inhibition of DPP-4 causes the plasma concentration of glucagon-like peptide-1 (GLP-1) to increase and also stimulate the secretion of insulin in response to increased glycemia. 13
Alpha-amylase (α-amylase) is a lytic enzyme that degrades complex molecules of sugar into constituent glucose molecules in the body thereby elevating the sugar levels in the blood and resulting in hyperglycemia. 14 The brain solely depends on sugar as biofuel for various metabolisms. However, the complex molecules of sugar in food cannot be absorbed and would not cross the blood-brain barrier. To overcome this hurdle, alpha-amylase degrades the polymer into smaller constituents to enable the entry of the molecules into the brain. 15 This is done specifically by overseeing the catalytic cleavage of the α-(1,4)-D-glycosidic bonds present in the polymeric chain. 14 Alpha-amylase is an eminent therapeutic target for the treatment and maintenance of hyperglycemia, a predominant symptom of diabetes mellitus. Inhibitors of α-amylase delay the degradation of polymers of sugars and reduce the postprandial blood sugar level. 16 Acarbose and mitigol are commonly used inhibitors of α-amylase, but side effects including flatulence, diarrhea, and abdominal discomfort have been reported. 17 Interestingly, flavonoids, a group of chemically similar bioactive compounds which includes anthocyanins have a wide range of molecular interactions that ultimately lead to the inhibition of α-amylase. 18
Anthocyanins like cyanidin and its widespread derivatives that are naturally occurring constituents of common foods have shown beneficial effects in the treatment and prevention of diabetes, but the mechanism of action is not yet fully understood.19,20 Therefore, it is of interest to study the inhibitory effect of a wide range of anthocyanins on different molecular targets of therapeutic intervention in diabetes.
Structure-based virtual screening of small molecular weight compounds including but not limited to molecular docking and molecular mechanics generalized Born surface area (MM/GBSA) calculations scores and ranks compounds on the basis of their complementary binding to the binding site of the target protein and the result obtainable can be employed in recommending compounds for experimental testing at a relatively lower cost.21,22 Herein, an integrated computational workflow including molecular docking, MM/GBSA calculation, pharmacophore modeling, and ADMETox screening was employed to screen a library of anthocyanins to specifically identify compounds with individual inhibitory effects against 3 molecular targets in the treatment of diabetes namely, PTP1B, dipeptidyl-peptidase 4 (DPP4), and α-amylase.
Methodology
Ligands and protein targets
A library of 118 anthocyanins was obtained from phytohub database in SMILES format and converted to 2D (sdf) format using Marvin sketch. 23 The compounds were incorporated into Maestro (2017 release) and subsequently prepared using the Ligprep functional tool. Similarly, the 3D structures of the protein targets (depiptidyl peptidase IV PDB ID: 3VJM, pancreatic alpha-amylase PDB ID: 4GQR, and PTP1B PDB ID: 1KAV) were obtained from an online repository (rcsb.org). The files were incorporated into Maestro and subsequently prepared using the protein preparation wizard. Furthermore, a grid box was generated with respect to the position of the co-crystallized ligand in preparation for molecular docking.
Molecular docking
The molecular docking protocol was carried out using Glide script in Schrödinger suite. The procedure was done across the 3 levels of precision with the protein targets treated as rigid bodies and the ligands, flexible. Initially, high throughput virtual screening (HTVS) was employed to briefly score and rank the compounds; this is the least rigorous level of the scoring function. Around 70% of the docked compounds were subsequently subjected to a more rigorous standard precision (SP). Thereafter, two-thirds of the compounds were subjected to the most rigorous extra precision (XP). The binding poses of the protein-ligand complexes were further analyzed and the docking scores were exported.
MM/GBSA calculation
The relative binding energy calculations, in which the ligand

This can be represented by several interactions’ contributions,

where the changes in the gas phase molecular mechanics ∆EMM, solvation Gibbs energy ∆Gsol, and conformational entropy
Rotamer search techniques from Prime were used in conjunction with the OPLS3 force field and the VSGB solvent model to accomplish this procedure.
Pharmacophore modeling
The receptor-ligand complex of the top-scoring compound against each protein target was analyzed and an E-pharmacophore was generated in each case using phase graphical user interface in Schrodinger’s suite. This is done to identify or highlight the major molecular interactions that substantially contribute to the firm binding of the ligands to the active sites of the protein targets.
ADMET profiling
The pharmacokinetic properties and drug-likeness of the test compounds were profiled using SwissADME and Pro-tox II online servers. The major properties analyzed include physicochemical properties such as lipophilicity, water solubility, molecular refractivity, topological surface area, and the molecular weight of the compounds. SwissADME Consensus log P which is the arithmetic mean of 5 models was adopted as the partition coefficient of n-octanol to water, and water solubility was predicted using the ESOL model. 27 Furthermore, other pharmacokinetic descriptors were also analyzed including cytochrome P450 inhibition, blood-brain barrier permeation, and skin permeability. An extensive toxicity study was carried out on the test compounds using Pro-tox II online server whose toxicity study is classified into 5 different categories, namely oral acute toxicity, organ toxicity, toxicological and genotoxicological endpoints, toxicological pathways, and stress response pathway. 28 The oral toxicity predictive model consists of 6 different classes ranging from fatal to non-toxic. The organ-toxicity-predictive model predominantly includes hepatotoxicity. Furthermore, the genotoxicological predictive model includes cytotoxicity, mutagenicity, and carcinogenicity prediction.
Results and Discussion
A total of 118 anthocyanins were screened to investigate their individual binding affinities to 3 molecular targets of therapeutic intervention in diabetes. These compounds were docked against the identified active sites of the protein targets to reveal the degree of firmness of the protein-ligand complexes using the scoring function. Subsequently, the Glide-generated poses of the complexes were subjected to an MM/GBSA protocol for rescoring. The docking scores and MM/GBSA calculations of the top-scoring compounds are presented in Table 1.
Docking scores and MM/GBSA calculattions of test compounds.

Abbreviations: DPP-4, dipeptidyl peptidase-4; MM/GBSA, molecular mechanics generalized Born surface area; XP, extra precision.
The scoring function is used for 2 crucial purposes: (1) to identify the correct binding orientation and conformation (docking pose) of each small molecular weight ligand out of a wide range of several modes and (2) to rank different ligands based on their estimated binding affinity. 29 Cyanidin 3-(p-coumaroyl)-diglucoside-5-glucoside, Malvin, Nasunin, cyanidin 3-O-xylosyl-rutinoside, and cyanidin 3-O-rutinoside are the highest scoring compounds against DPP4 with docking scores in the range of −10.813 to −15.272 kcal/mol. Cyanidin 3-(p-coumaroyl)-diglucoside-5-glucoside showed the highest binding affinity to the protein with an impressive docking score of −15.272 kcal/mol and exhibited different molecular interactions with the amino acid residues in the active site of DPP4, such as H-bond, Pi-pi stacking, Pi cation, and Salt bridge (Table 2). Dipeptidyl peptidase-4 inhibitors control hyperglycaemia by stimulating insulin secretion. Cyanidin and its derivatives are widespread metabolites occurring naturally in foods and are considered to have potential benefits for the treatment of diabetes. 19
The molecular interactions of the top-scoring compounds with the target proteins.

Abbreviations: DPP-4, dipeptidyl peptidase-4; PTP1B, Protein tyrosine phosphatase 1B.
Furthermore, cyanidin 3-O-(6ʺ-malonyl-3ʺ-glucosyl-glucoside), delphinidin 3-O-(6ʺ-malonyl-glucoside), cyanidin 3-O-(6ʺ-malonyl-glucoside), cyanidin 3-(sinapoyl)-diglucoside-5-glucoside, and delphinidin 3,5-O-diglucoside showed the highest binding to PTP1B. With docking scores in the range −8.944 to −9.691, the compounds exhibited hydrogen bonds and formed a salt bridge with the active site of the protein target. Inhibition of PTP1B by these compounds will potentially increase insulin sensitivity.
However, delphinidin 3,5-O-diglucoside, Malvin, Nasunin, cyanidin-3-(6-acetylglucoside), and cyanidin 3-O-xylosyl-rutinoside were obtained as the top-scoring compounds against the third molecular target namely, alpha-amylase hydrogen bonding and pi-cation are the general forms of interaction observed in the binding of the ligands to the active site of the protein. Delphinidin 3,5-O-diglucoside, with a binding affinity of −12.36 kcal/mol had the highest binding to this protein. Interestingly, Malvin, Nasunin, and cyanidin 3-O-xylosyl-rutinoside showed robust binding to both DPP4 and alpha-amylase. This could open up a window of possible exploration of dual inhibitors of both DPP4 and alpha-amylase in diabetes chemotherapy. Similarly, delphinidin 3,5-O-diglucoside exhibited impressive binding to both PTP1B and alpha-amylase. However, in all cases, the test compounds showed higher docking scores than the standard inhibitors of the target proteins.
The glide-generated poses of the ligands bound to the active site of the protein targets were subjected to an MM/GBSA protocol for rescoring. The protocol of using Glide for pose generation and an MM/GBSA protocol for rescoring has shown promising results in its application to structure-based lead optimization of small molecular weight inhibitors. 30
The binding energy (∆Gbind) gives an understanding of the spontanieity of receptor-ligand complexation. Interactions with more negative ∆Gbind are expected to be more spontaneous. The calculations suggest that the reaction between DPP-4 and the highest scoring compound, cyanidin 3-(p-coumaroyl)-diglucoside-5-glucoside is expected to be the most spontaneous. This is because this interaction exhibited a ∆Gbind value of −88.945 kcal/mol. This is followed by cyanidin 3-O-rutinoside which had a binding energy of −78.402 kcal/mol. Intiguingly, all the reported anthocyanins had higher binding energy tham the standard compound (Saxagliptin). The spontaneity of the reactions is predicted to be in the order cyanidin 3-(p-coumaroyl)-diglucoside-5-glucoside > cyanidin 3-O-rutinoside > Malvin > cyanidin 3-O-xylosyl-rutinoside > Nasunin > Saxagliptin.
Also, per the MM/GBSA calculations, the spontaneity of complexation of the test compounds with PTP-1B is expected to be in the order cyanidin 3-O-(6ʺ-malonyl-3ʺ-glucosyl-glucoside) (−72.034 kcal/mol) > delphinidin 3-O-(6ʺ-malonyl-glucoside) (−68.434 kcal/mol) > cyanidin 3-O-(6ʺ-malonyl-glucoside) (−68.34 kcal/mol) > cyanidin 3-(sinapoyl)-diglucoside-5-glucoside (−63.864 kcal/mol) > Ertiprotafib (−45.018 kcal/mol) > delphinidin 3,5-O-diglucoside (−37.071 kcal/mol). Finally, the spontaneity of the interaction between the reported compounds and α-amylase is predicted to be in the order cyanidin-3-(6-acetylglucoside) (−70.251 kcal/mol) > delphinidin 3,5-O-diglucoside (−66.095 kcal/mol) > Acarbose (−62.835 kcal/mol) > Malvin (−55.313 kcal/mol) > Nasunin (−45.345 kcal/mol) > cyanidin 3-O-xylosyl-rutinoside (−40.144 kcal/mol).
Intermolecular interactions have gained wide recognition from experimental and theoretical standpoints primarily because of their basic roles in shaping the 3D structures of several biomolecules and in controlling the molecular recognition, binding, and interaction between 2 complementary molecules. 31 Molecular drug design identifies and optimizes the chemical architecture in the binding platform of small molecular weight drug candidates and their complementary molecules, primarily proteins. Molecular identification in living systems relies on appropriate interactions between 2 complementary molecules. The predominant aim of drug design is the identification and optimization of ligand-protein interactions. 32 The potential optimization of these molecules requires prior knowledge of the chemical nature, structure, and strength of the interactions between the molecules. 33 The molecular interactions of the test compounds with the active sites of the target proteins are presented in Figures 1 to 3 and Table 2. The interactions observed include hydrogen bonding, pi-pi stacking, pi cation, and salt bridge. Above all, hydrogen bonding is the major form of interaction observed.
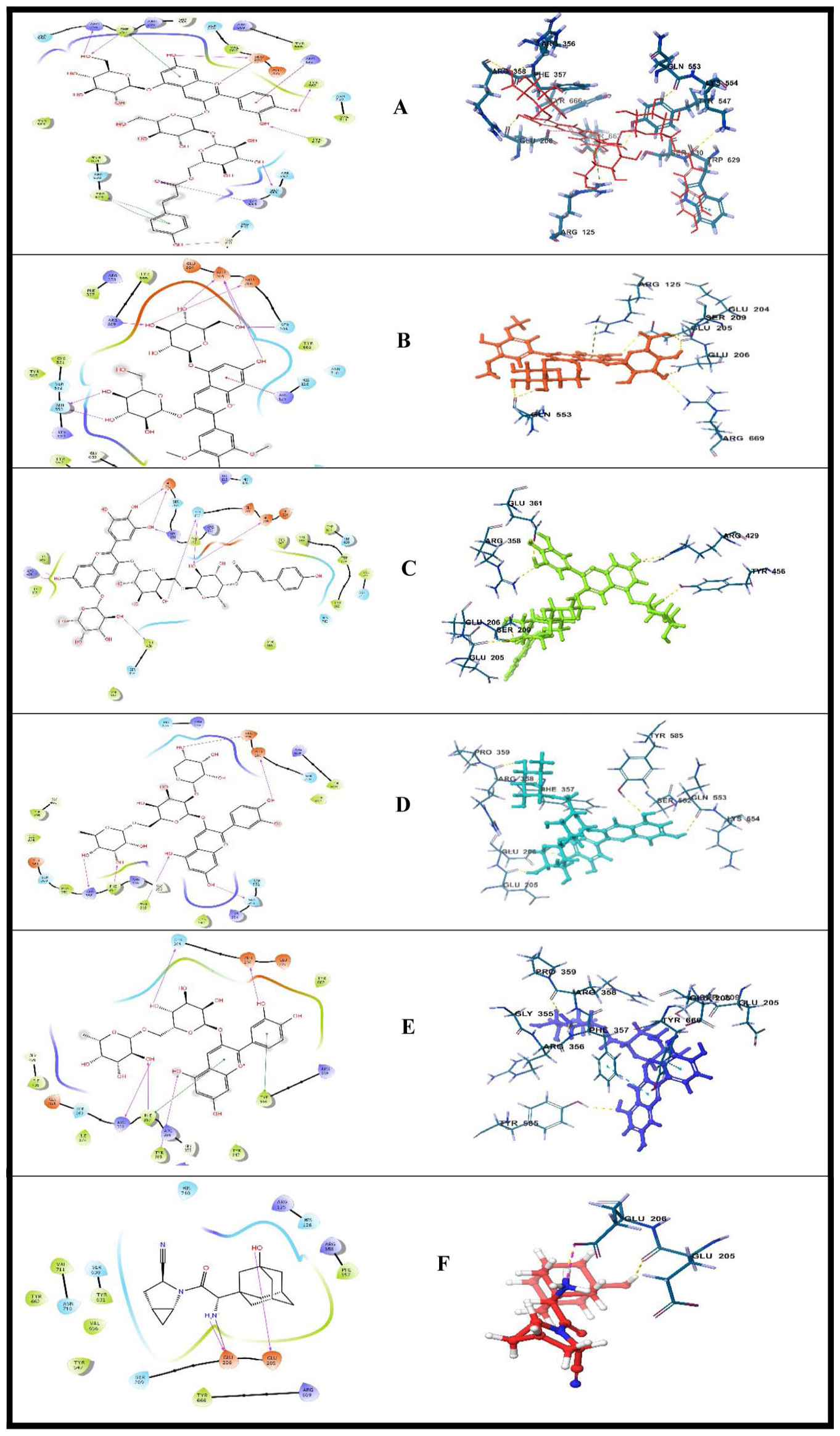
Interactions of the lead ligands with the ligand binding site of DPP-4: (A) cyanidin 3-(p-coumaroyl)-diglucoside-5-glucoside, (B) Malvin, (C) Nasunin, (D) cyanidin 3-O-xylosyl-rutinoside, (E) cyanidin 3-O-rutinoside, and (F) standard (Saxagliptin).

2D amino acid interaction of top-scoring compounds with PTP1B: (A) cyanidin 3-O-(6ʺ-malonyl-3ʺ-glucosyl-glucoside), (B) delphinidin 3-O-(6ʺ-malonyl-glucoside), (C) cyanidin 3-O-(6ʺ-malonyl-glucoside), (D) cyanidin 3-(sinapoyl)-diglucoside-5-glucoside, (E) delphinidin 3,5-O-diglucoside, and (F) standard (Ertiprotafib).
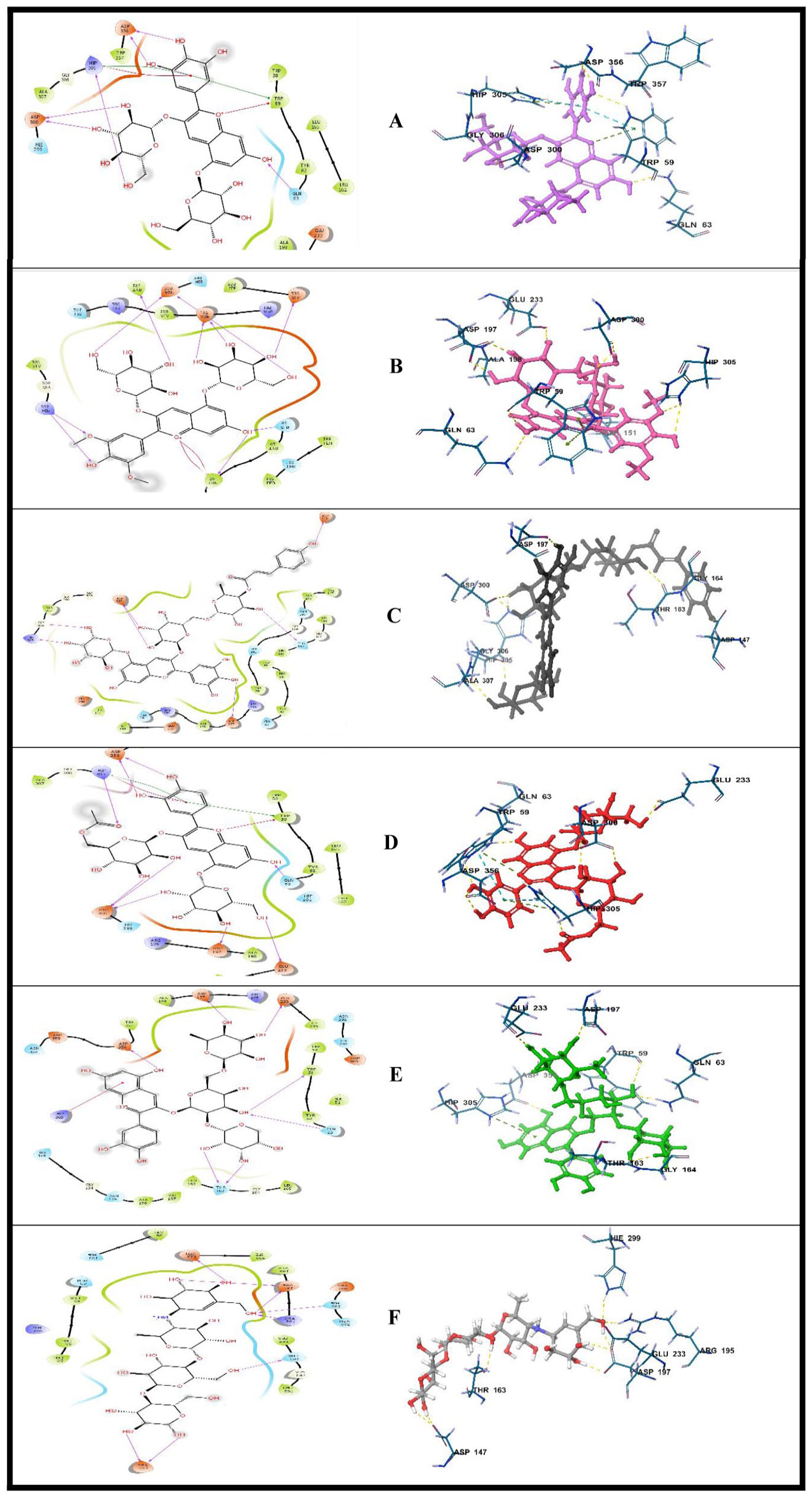
2D amino acid interactions of top-scoring compounds with α amylase: (A) delphinidin 3,5-O-diglucoside, (B) Malvin, (C) Nasunin, (D) cyanidin-3-(6-acetylglucoside), (E) cyanidin 3-O-xylosyl-rutinoside, and (F) standard (Acarbose).
Cyanidin 3-(p-coumaroyl)-diglucoside-5-glucoside, Malvin, and cyanidin 3-O-rutinoside interacted with Arg358 via hydrogen bond in the active pocket of DPP-4. Recently, attention has been drawn to the interaction with Arg358 in the design of DPP-4 inhibitors. Sitagliptin was found to interact with the guanidine moiety of Arg358, which interacts via its trifluoromethyl substituent, and this interaction was suggested to increase its activity. 34
All the test compounds and Saxagliptin (Standard) made hydrogen bond contact with at least one of Glu205 and Glu206. In addition to this, cyanidin 3-(p-coumaroyl)-diglucoside-5-glucoside formed a salt bridge with Glu206. Previously, the design of some quinoline and isoquinoline derivates showed that the most potent derivative (8 g) formed a salt bridge with Glu205 and Glu206. 35
Cyanidin 3-O-rutinoside, cyanidin 3-O-xylosyl-rutinoside, and cyanidin 3-(p-coumaroyl)-diglucoside-5-glucoside made hydrogen contact with Phe357. Cyanidin 3-(p-coumaroyl)-diglucoside-5-glucoside and cyanidin 3-O-rutinoside formed additional pi-pi stacking interaction between an aromatic ring in its structure and aromatic amino acid residues Trp629. Another pi-pi stacking was observed in the binding of cyanidin 3-(p-coumaroyl)-diglucoside-5-glucoside to the binding pocket of DPP-4 via Phe357. Similarly, cyanidin 3-O-rutinoside formed a second pi-pi stacking interaction with Tyr666.
In the interaction with PTP-1B, cyanidin 3-O-(6ʺ-malonyl-3ʺ-glucosyl-glucoside), delphinidin 3-O-(6ʺ-malonyl-glucoside), cyanidin 3-O-(6ʺ-malonyl-glucoside), and cyanidin 3-O-(6ʺ-malonyl-glucoside) formed hydrogen bond contacts with Asp48, Arg221, Ala217, Ser216, and Cys215 as well as a salt bridge with Arg221. Cyanidin 3-(sinapoyl)-diglucoside-5-glucoside and delphinidin 3,5-O-diglucoside formed interactions with Asp181, Lys116, and Gln262. In addition, an interaction was seen with Asp48 in both compounds; cyanidin 3-(sinapoyl)-diglucoside-5-glucoside through salt bridge and delphinidin 3,5-O-diglucoside through hydrogen bond. The standard (Ertiproafib) had hhydrogen bond interactions with Arg24 and Gln262 in the binding pocket of PTP-1B.
Delphinidin 3,5-O-diglucoside, Malvin, Nasunin, and cyanidin-3-(6-acetylglucoside) established contacts with His305 and Asp300 in the active pocket of alpha-amylase. All but Nasunin had hydrogen bond contact with Gln63 and formed a pi-cation interaction with at least one of His305 and Trp59. In addition, all the lead compounds except delphinidin 3,5-O-diglucoside had interacted with Asp197. Likewise, cyanidin 3-O-xylosyl-rutinoside made additional contacts with Glu233, Asp356, Trp59, and Thr163, cyanidin-3-(6-acetylglucoside), with Asp 336 and Glu233, Nasunin, with Asp147 and Gly306, Malvin with Tyr151 and Glu233, and delphinidin 3,5-O-diglucoside with Asp356. In the same vein, Acarbose formed hydrogen bond interactions with residues Glu233, Asp197, His209, Arg195, Thr163, and Asp147 while interacing with the target enzyme.
Of all interactions, hydrogen bond is one of the most stabilizing and most specific interactions in living systems. 36 The major effects and responsibilities of hydrogen bond interaction in ligand binding include but are not limited to ligand-protein binding recognition and protein affinity for ligands. 36 Salt bridges are also another form of noncovalent interaction between 2 charged molecules. In ligand-receptor interactions, the proton may move from a carboxylic acid side chain to an amine portion (or the amino acid functional group) or vice versa. 31 π-stacking interactions between aromatic rings contribute massively to ligand binding and play an essential role in medicinal chemistry.33,37 The distinct π-electron cloud over and below the rings offer different interaction prospect and give room for overcoming particular problems in target recognition.37,38
Pharmacophore model
The pharmacophore model generated from the molecular interactions of the compounds with the target proteins showed that hydrogen bonds (acceptor and donor) and aromatic rings are the major features that contribute to the strong binding (Figure 4). The precise details of the molecular architecture of the functional groups that actively contribute to the binding of ligands to the binding pockets of the target proteins were obtained. Although the accuracy of a pharmacophore model based on experimentally active compounds is deemed to be higher, predictive models are also significant. 39

Pharmacophore model of the top-scoring ligands: (A) DPP4-cyanidin 3-(p-coumaroyl)-diglucoside-5-glucoside complex, (B) PTP1B—cyanidin 3-O-(6ʺ-malonyl-3ʺ-glucosyl-glucoside) complex, and (C) alpha-amylase—delphinidin 3,5-O-diglucoside complex.
Structure-based ADMET profiling
The predicted physicochemical properties of the lead compounds are presented in Table 3. The molecular weight of the compounds ranges from 491.42 to 979 g/mol with C11 (cyanidin-3-(6-acetylglucoside)) having the least molecular weight and C9 (cyanidin 3-(sinapoyl)-diglucoside-5-glucoside) the highest. Furthermore, the predicted consensus log P which is an arithmetic mean of 5 different models ranges from −3.53 to −0.75. The log P value is a measure of the lipophilicity of small molecular weight compounds. A high log P value represents a high degree of lipophilicity and vice versa.40,41 Oral drug candidates are expected to have a sufficient level of lipophilicity to enable them to cross the intestine into the systemic circulation.42,43
SWISSADME and Pro-Tox II predicted physicochemical properties.

Abbreviations: C1, cyanidin 3-(p-coumaroyl)-diglucoside-5-glucoside; C2, Malvin; C3, Nasunin; C4, cyanidin 3-O-xylosyl-rutinoside; C5, cyanidin 3-O-rutinoside; C6, cyanidin 3-O-(6ʺ-malonyl-3ʺ-glucosyl-glucoside); C7, delphinidin 3-O-(6ʺ-malonyl-glucoside); C8, cyanidin 3-O-(6ʺ-malonyl-glucoside); C9, cyanidin 3-(sinapoyl)-diglucoside-5-glucoside; C10, delphinidin 3,5-O-diglucoside; C11, cyanidin-3-(6-acetylglucoside); TPSA, total polar surface area; ESOL, estimated solubility.
Also, the Extimated solubility (ESOL) model of water solubility showed that the compounds have an ESOL Log S value in the range of −4.11 to −1.4 with C4 (cyanidin 3-O-xylosyl-rutinoside) being the most soluble. As a result, C2, C4, C5, C7, C8, and C10 are predicted to be very soluble. However, C1, C3, C6, and C11 are soluble and C9 is predicted to be moderately soluble. The ESOL Log S value is a technical representation of the extent to which a compound is water soluble. In addition to a sufficient level of lipophilicity, a drug candidate should have a promising degree of water solubility to aid its movement in the hydrophilic condition of the systemic circulation. However, the log P value of a compound has an inverse relationship with the log S value.44,45 Theoretically, the log P value increases with decreasing log S value. This implies that the more lipophilic a compound is, the less water soluble it becomes.
The drug-likeness prediction and bioavailability score of the test compounds are shown in Table 4. Two rule-based filters are adopted for the drug-likeness prediction, namely the Lipinski rule of 5 and the Egan rule-based filter. All the compounds violated 3 of the Lipinski rule-based filter except C11 which violated 2. The major violation which is general to almost all the compounds is a molecular weight of more than 500 g/mol. The rule states that drug candidates should have a molecular weight less than that value. However, lead optimization may be carried out to reduce the bulkiness of the compounds and still retain the chemical architecture that contributed to the binding. Furthermore, the bioavailability score of the compounds is mostly 0.17 except for C7 which has a score of 0.11. Abbot Bioavailability Score is the likelihood of a compound having greater than 10% bioavailability in rats or measurable Caco-2 permeability. 46 Precisely, a bioavailability score of 0.17 suggests that the compound has a 17% chance of having greater than 10% bioavailability in rats or measurable Caco-2 permeability.
Drug-likeness and bioavailability.

Abbreviations: C1, cyanidin 3-(p-coumaroyl)-diglucoside-5-glucoside; C2, Malvin; C3, Nasunin; C4, cyanidin 3-O-xylosyl-rutinoside; C5, cyanidin 3-O-rutinoside; C6, cyanidin 3-O-(6ʺ-malonyl-3ʺ-glucosyl-glucoside); C7, delphinidin 3-O-(6ʺ-malonyl-glucoside); C8, cyanidin 3-O-(6ʺ-malonyl-glucoside); C9, cyanidin 3-(sinapoyl)-diglucoside-5-glucoside; C10, delphinidin 3,5-O-diglucoside; C11, cyanidin-3-(6-acetylglucoside).
The pharmacokinetic profiles of the test compounds are presented in Table 5. The result showed that none of the compounds has a structural orientation that will enable it permeate the blood-brain barrier. Except the target of a drug is the brain, permeation of the blood-brain barrier by the drug is capable of inducing an adverse drug reaction. Furthermore, C1, C3, C4, and C9 are predicted to be substrates of permeability glycoprotein. Substrates of permeability glycoprotein are actively effluxed out of the cell by the protein, and bioaccumulation is reduced in the process. Interestingly, none of the compounds showed an inhibiting potential against the cytochrome P450 isoforms predominantly involved in phase I biotransformation. Inhibition of any of these isoforms is likely to elicit a drug-drug interaction. 47
Predicted pharmacokinetic properties f the test compounds.
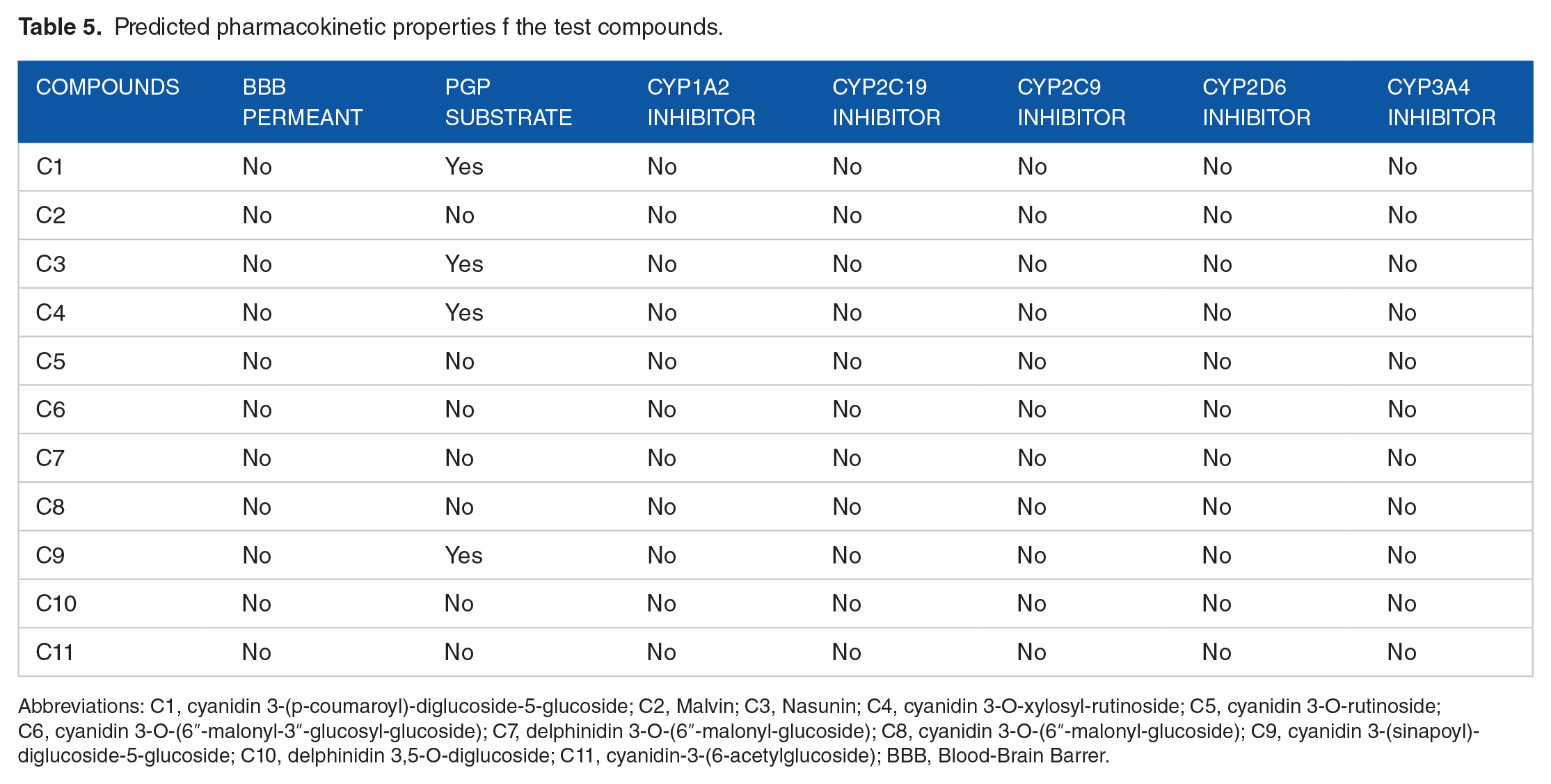
Abbreviations: C1, cyanidin 3-(p-coumaroyl)-diglucoside-5-glucoside; C2, Malvin; C3, Nasunin; C4, cyanidin 3-O-xylosyl-rutinoside; C5, cyanidin 3-O-rutinoside; C6, cyanidin 3-O-(6ʺ-malonyl-3ʺ-glucosyl-glucoside); C7, delphinidin 3-O-(6ʺ-malonyl-glucoside); C8, cyanidin 3-O-(6ʺ-malonyl-glucoside); C9, cyanidin 3-(sinapoyl)-diglucoside-5-glucoside; C10, delphinidin 3,5-O-diglucoside; C11, cyanidin-3-(6-acetylglucoside); BBB, Blood-Brain Barrer.
The predicted oral LD50 (mg/kg), Pro tox II toxicity, and the accuracy (%) of the predictive model are presented in Table 6. All the test compounds have an Oral LD50 value of 5000 mg/kg and fall into the fifth class (Class V: may be harmful if swallowed (2000 < LD50 ⩽ 5000)) of the pro-tox II toxicity index.
Oral acute toxicity predictive model.

Abbreviations: A, 5000 mg/kg; C1, cyanidin 3-(p-coumaroyl)-diglucoside-5-glucoside; C2, Malvin; C3, Nasunin; C4, cyanidin 3-O-xylosyl-rutinoside; C5, cyanidin 3-O-rutinoside; C6, cyanidin 3-O-(6ʺ-malonyl-3ʺ-glucosyl-glucoside); C7, delphinidin 3-O-(6ʺ-malonyl-glucoside); C8, cyanidin 3-O-(6ʺ-malonyl-glucoside); C9, cyanidin 3-(sinapoyl)-diglucoside-5-glucoside; C10, delphinidin 3,5-O-diglucoside; C11, cyanidin-3-(6-acetylglucoside).
The 5 different categories namely of Pro tox II toxicological study; oral acute toxicity, organ toxicity, toxicological, and genotoxicological endpoints, toxicological pathways, and stress response pathway are presented in Table 7. The result showed that none of the compounds is hepatotoxic and carcinogenic. However, C6 (cyanidin 3-O-(6ʺ-malonyl-3ʺ-glucosyl-glucoside)) was found to be mutagenic and C2 (Malvin), cytotoxic ; LD, Lethal Dose.
Organ toxicity, toxicological, and genotoxicological endpoints, toxicological pathways and stress response pathway activities.

Abbreviations: A, hepatotoxicity; B, carcinogenicity; C, mutagenicity; C1, cyanidin 3-(p-coumaroyl)-diglucoside-5-glucoside; C2, Malvin; C3, Nasunin; C4, cyanidin 3-O-xylosyl-rutinoside; C5, cyanidin 3-O-rutinoside; C6, cyanidin 3-O-(6ʺ-malonyl-3ʺ-glucosyl-glucoside); C7, delphinidin 3-O-(6ʺ-malonyl-glucoside); C8, cyanidin 3-O-(6ʺ-malonyl-glucoside); C9, cyanidin 3-(sinapoyl)-diglucoside-5-glucoside; C10, delphinidin 3,5-O-diglucoside; C11, cyanidin-3-(6-acetylglucoside); D, cytotoxicity; E, Aryl hydrocarbon receptor (AhR); F, androgen receptor (AR); G, androgen receptor ligand binding domain (AR-LBD); H, estrogen receptor alpha (ER); I, estrogen receptor ligand binding domain (ER-LBD); J, heat shock factor response element (HSE); K, mitochondrial membrane potential (MMP); L, phosphoprotein (tumor supressor) p53; +, active; –, inactive.
In the same vein, all the compounds showed no activity against the studied stress response pathway such as Heat shock factor response element, mitochondrial membrane potential, and phosphoprotein (tumor suppressor) p53, and may not give rise to reactive oxygen species (ROS).48-50 Adaptive stress response pathways are signaling pathways that ultimately result in the transcriptional activation of genes that protect the integrity of a cell under stressful conditions. 50
Similarly, none of the compounds showed activity against the tested nuclear receptor (NR) signaling or toxicological pathway descriptors namely; Aryl hydrocarbon receptor (AhR), androgen receptor (AR), androgen receptor ligand binding domain (AR-LBD), estrogen receptor alpha (ER), and estrogen receptor ligand binding domain (ER-LBD). Nuclear receptor signaling is activated to maintain development, cellular growth, inflammation, and metabolism. 51
Conclusion
Anthocyanins on several occasions have been considered a class of compounds with numerous health benefits. In this study, the antidiabetic potential of anthocyanins was predicted using an integrated computer-aided approach. The reported compounds showed promising binding affinity to the protein targets examined. Further ADMETox screening anticipates no significant toxic activity against several toxicity descriptors. The reported compounds could be explored as antidiabetic agents with individual and/or combined activity to mitigate or eradicate glucose fluctuations; however, further analyses must be carried out to validate these findings.
Footnotes
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration Of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
PAA, SOO—study concept and design; Adebowale AA, Aderemi AA—Target retrieval/Data collection; PAA, Adebowale AA, SOO—Software and analysis; OLO, AOA, APE—Original Draft; SAS, OYA—Supervision, Review & Editing. All authors reviewed the results and approved the final version of the manuscript.