
Abstract
The Nipah virus (NiV) belongs to the Henipavirus genus is a serious public health concern causing numerous outbreaks with higher fatality rate. Unfortunately, there is no effective medication available for NiV. To investigate possible inhibitors of NiV infection, we used in silico techniques to discover treatment candidates in this work. As there are not any approved treatments for NiV infection, the NiV-enveloped attachment glycoprotein was set as target for our study, which is responsible for binding to and entering host cells. Our in silico drug design approach included molecular docking, post-docking molecular mechanism generalised born surface area (MM-GBSA), absorption, distribution, metabolism, excretion/toxicity (ADME/T), and molecular dynamics (MD) simulations. We retrieved 418 phytochemicals associated with the neem plant (Azadirachta indica) from the IMPPAT database, and molecular docking was used to ascertain the compounds’ binding strength. The top 3 phytochemicals with binding affinities of −7.118, –7.074, and −6.894 kcal/mol for CIDs 5280343, 9064, and 5280863, respectively, were selected for additional study based on molecular docking. The post-docking MM-GBSA of those 3 compounds was −47.56, –47.3, and −43.15 kcal/mol, respectively. As evidence of their efficacy and safety, all the chosen drugs had favorable toxicological and pharmacokinetic (Pk) qualities. We also performed MD simulations to confirm the stability of the ligand-protein complex structures and determine whether the selected compounds are stable at the protein binding site. All 3 phytochemicals, Quercetin (CID: 5280343), Cianidanol (CID: 9064), and Kaempferol (CID: 5280863), appeared to have outstanding binding stability to the target protein than control ribavirin, according to the molecular docking, MM-GBSA, and MD simulation outcomes. Overall, this work offers a viable approach to developing novel medications for treating NiV infection.
Introduction
The Nipah virus (NiV) is responsible for causing encephalitis and severe respiratory illnesses, which have a significant mortality rate. The NiV is classified as an enveloped, negative-sense, nonsegmented, single-stranded RNA paramyxovirus belonging to the genus Henipavirus. This virus is preeminently found in Southeast Asia, which is considered endemic.1,2 Large fruit bats, often known as flying foxes belonging to the genus Pteropus, were determined to be the natural reservoir for NiV based on epidemiological and virological findings. 3
According to the previous study, pigs, dogs, cats, goats, horses, and rodents can contract the NiV infection, an increasingly dangerous zoonotic illness that can infect humans. 4 These animals can serve as intermediate hosts for humans in the event of an infection. 2 Human NiV infection was initially identified during a significant outbreak on the Malaysian Peninsula from September 1998 to May 1999. The virus was mostly transferred from fruit bats, its principal reservoir, to pigs, then transmitted to people, with a 38% death rate reported. 5 Prolonged death is associated with significant brain-stem involvement, denoted by coma, aberrant pupils, an abnormal doll’s-eye reflex, vomiting, tachycardia, and segmental myoclonus. 6 However, the NiV was initially found to be the source of an encephalitis outbreak in Bangladesh’s Meherpur District in 2001. 7 Since 2001, there have been multiple outbreaks in Bangladesh that have shown that NiV can spread both human-to-human and foodborne (through ingestion of raw date palm sap) when in close contact with the respiratory and body fluids of infected individuals.8,9 Several outbreaks linked to the same NiV transmission pattern as Bangladesh since 2001 have also been reported in India, owing to the historical proximity of these 2 countries. 10 Among the individuals infected with NiV in those respective nations, fever, changed mental status, cough, and respiratory symptoms were the most prevalent symptoms.11,12 Although fewer NiV infections have been recorded in Bangladesh recently, an outbreak in 2023 led to 11 cases, 8 deaths, and a 72% case fatality rate. 13 So, according to case studies of several epidemics, the Philippines, Bangladesh, Singapore, India, Malaysia, and Cambodia are the main countries where NiV is circulated. 14 However, very little sequence information from earlier NiV epidemics has been compiled. Based on these data, NiV has been genetically divided into 2 separate genotypes, NiV-Malaysia and NiV-Bangladesh. NiV-Bangladesh is more virulent than NiV-Malaysia because of its greater death rate. 15
The RNA genome of NiV is made of the following 6 genes: long polymerase, phosphoprotein, nucleocapsid, fusion glycoprotein, matrix, and attachment glycoprotein. 16 The successful attachment, membrane fusion, and penetration of the NiV virus into a host cell necessitate the presence of 2 different membrane-anchored glycoproteins, namely, a fusion glycoprotein and an attachment glycoprotein. The stalk domain and the head domain comprise the G glycoprotein present on the surface of the viral particle. The head domain controls binding to the host cell receptor, either ephrin-B2 or ephrin-B3 l, while the stalk domain anchors the G glycoprotein to the viral membrane. The contact between the receptor and NiV-G triggers a conformational cascade in NiV-F, leading to the fusing of viral and/or cell membranes.17,18 An essential target in pursuing vaccine and antiviral development is the NiV’s attachment glycoprotein. In a novel ferret model, it has been shown that the monoclonal antibody of human m102.4, which specifically targets the G glycoprotein, is advantageous for treating and preventing NiV infection. 19
The only medication used in clinical settings to treat NiV patients thus far is the broad-spectrum nucleoside analogue, Ribavirin. Research conducted on the 1998-1999 outbreak in Malaysia found that treating 140 individuals with Ribavirin significantly reduced the number of fatalities and neurological impairments by 36%. 20 In the more contemporary context of the 2018 outbreak in Kerala, India, it was seen that Ribavirin was used as a treatment strategy, resulting in a survival rate of only 2 individuals out of the overall 23 reported cases. Both survivors from a subgroup of 6 patients who were administered ribavirin therapy had orally ingested the medicine. 21 Nevertheless, it was shown that Ribavirin did not demonstrate any therapeutic efficacy in hamsters infected with henipavirus. In addition, its impact on NiV infection and death in subsequent animal challenge models was temporary.22,23 As well as in African green monkeys, Ribavirin did not stop the progression of henipavirus illness; rather, it simply postponed it by 1 or 2 days. 24 Due to this rationale, extensive efforts have been dedicated to developing and exploring innovative therapeutic strategies to mitigate NiV infection. These endeavors mostly concentrate on studying the fusion and entrance phases of the viral infection mechanism. 17 This is because NiV infection has an extreme pathogenic capacity in humans.
Thus, medicinal plants can act as a valuable source of therapeutic compounds for treating NiV infectious diseases when particular antivirals and vaccines are unavailable. A member of the Meliaceae family, neem (Azadirachta indica) has recently received worldwide recognition for its diverse medical properties. 25 Astringent, insecticidal, anthelmintic, antiviral, anti-inflammatory, antiseptic, antifungal, and antinociceptive qualities are only a few of its many qualities. 26 Moreover, neem contains several chemical components, including nimbin, nimbiol, polyphenolic flavonoids, 17-hydroxyazadiradione, 7-desacetyl-7-benzoylgedunin, and azadirachtin. 27 One study claimed that administering an extract from neem leaves could decrease the reproduction of the dengue virus. Hepatitis B virus DNA polymerase was also demonstrated to be inhibited following the injection of neem leaf extract in another investigation. 28 A separate investigation discovered that administration of neem leaf extract to mice significantly reduced the number of splenocytes activated by nipah disease virus (NDV), reducing it to a level comparable with that of the uninfected control group. It can be deduced from this that the extract possesses anti-NDV capabilities. 29 Neem extracts and compounds have been demonstrated to inhibit the replication of several viruses, including flaviviruses like Dengue virus. The NiV, also belonging to the flavivirus family, could potentially be susceptible to neem’s antiviral effects. 30 Furthermore, findings suggest that this particular plant possesses potential therapeutic properties for treating ailments caused by poliovirus, duck plague virus, bovine herpesvirus type 1, and herpes simplex virus type I. 31 Because of its established anti-microbial properties and minimal toxicity, neem is a suitable plant to harvest and design possible antiviral components.
Integrating several computational tools and software applications is frequently used to pursue the intended outcome in computational drug design, a prominent strategy in contemporary preclinical drug development. 32 The utilization of contemporary CADD approaches such as molecular docking, post-docking molecular mechanism generalised born surface area (MM-GBSA), absorption, distribution, metabolism, excretion, and toxicity (ADMET), and MD simulation has been shown to expedite the identification of lead compounds in comparison with the conventional drug design process. 33
With CADD’s assistance, several medicines have been approved. But as far as we know, there has yet to be any report on in silico analysis of phytochemicals from A indica against the NiV. Considering the above discourse, the present investigation was formulated to examine the phytochemicals of A indica in relation to the attachment glycoprotein of the NiV, using diverse in silico methodologies.
Materials and Method
Target protein preparation
The protein structure of the 2VSM (NiV envelope glycoprotein) was retrieved from the RCSB Protein Data Bank (PDB) at https://www.rcsb.org/. The x-ray diffraction technique, which has a resolution value of 1.80 Å, was used to determine the atomic and molecular structure of the crystal. The 416 amino acids (AA) long and anticipated molecular weight (MW) of the NiV envelope glycoprotein, with the PDB ID 2VSM, is 46.84 kDa. 34 The protein’s A chain was generated by removing the extra chains, co-crystallized ligands, and solvent molecules for further study. The protein structure was prepared using Schrödinger 2020-3’s protein preparation wizard. With the default setting, the protein’s bond ordering has been imposed, hydrogens and missing side chains have been added, and the protein’s water has been removed. For this protein preparation, we used the OPLS3e force field. 35
Retrieval and preparation of phytochemicals
At first, we selected the neem plant (A indica) for the identification of potential phytochemicals using the IMPPAT database at https://cb.imsc.res.in/imppat/. The IMPPAT database is an extensive collection of phytochemical substances derived from therapeutic plants. It includes details about the substances’ pharmacological characteristics, biological activities, and chemical structure. Phytochemical substances from the leaves, seeds, roots, fruits, flowers, and bark of the A indica plant are listed in IMPPAT. All of the IMPPAT-enlisted phytochemical compounds of the neem plant (A indica) were downloaded from the IMPPAT database and preserved in 3-dimensional SDF format for virtual screening and subsequently prepared by the LigPrep module of maestro v11.4. The optimization of chemical formation and protein was conducted via the OPLS3e force field.
Study of molecular docking
Molecular docking studies can uncover the binding relationships between particular proteins and particular phytochemicals. Using the Schrödinger Desmond software, GLIDE v8.8 and maestro v12.5.139 were used to determine the binding pose of the target protein with specific phytochemicals. 36 The OPLS3e force field was used in standard precision mode. This approach uses an energetic grid-based ligand docking method to find the desirable contact between one or more small phytochemical compounds and the receptor protein. A receptor grid is a sort of grid that shows where a protein binds. After that, the target protein and the control ligand (Ribavirin) bound positions were examined and selected to create a receptor grid. Based on binding site atoms, the receptor grid determined the box with coordinates (X, Y, Z) of (17.087, 64.413, –18.104). The research revealed insights into the energetics of ligand-protein interaction. The Maestro viewer showed several kinds of chemical bonds and ligand-binding residues.
Post-docking MM-GBSA analysis
The technique MM-GBSA was used to look at the free binding energies of the protein and ligand complexes. 37 The chosen compounds that exhibit the lowest energy binding were analyzed and visualized using both Maestro v-12.5.139, and Glide v-8.8; this approach served as a post-docking validation tool. Following identifying the binding site atoms for the target protein using the native ligand active site, the binding site position was represented by a grid box. Binding free energy, residues, and interactions were all displayed in the Maestro v-12.5.139’s interacting plane.
Safety analysis of the compounds
Greek terms “pharmakon” (drug) and “kinetikos” (movement) are combined to form the term “PK,” which is primarily concerned with the dynamic entry of a tiny molecule candidate into the body and the observation of the ADME (absorption, distribution, metabolism, and excretion) features of a compound that resembles a therapeutics. 38 It generates a model and provides message about the time course of ADME of exotic substances in the body using several mathematical equations. In the current drug development method, toxicity prediction is a crucial stage that aids in identifying the adverse impacts of a particular molecule on people, plants, animals, or the environment. The toxicity of substances is assessed using various animal studies in conventional drug design procedures, which is expensive, time-consuming, and ethically problematic. 39 Swissadme (http://www.swissadme.ch/) and the PkCSM (http://structure.bioc.cam.ac.uk/pkcsm) server evaluated the phytochemical compounds’ pharmacological and toxicity properties. The entrance mechanism for all phytochemical substances was their canonical smiles, serving as a distinctive identifier that can be used to represent it in a wide range of software applications.
Molecular dynamics simulation
The structural strength of the ligand-protein complex was examined by an MD simulation method in a particular physiological condition. For analyzing the ligand-protein stability, the chosen compound in binding with the apoprotein was examined via MD simulations. 40 By using the Desmond package offered by the Schrödinger suit, a 100 ns MD simulation was completed. 41 A protein preparation wizard was used to preprocess the complicated structure after it had been created from molecular docking investigations of ligand-protein complexes. 42 To keep a particular volume of the systems, an orthorhombic periodic boundary box shape with an interval (10 × 10 × 10 Å3) has been used for every complicated simple point-charge (SPC) water model that was performed to analyze the system. To keep the salt concentration at 0.15 M, Cl– and Na+ ions were selected and filled randomly in the solvated method. 43 The OPLS3e force field mitigated and relaxed the system. 35 At last, the constant pressure-constant temperature (NPT) ensemble was executed at 1.01325 bar pressure and 300.0 K temperature. 25 The system for every complex was initially relaxed, and then the final production cycle was conducted with 100 picosecond recording intervals and an energy of 1.2. 44 In addition, data for root mean square deviation (RMSD), solvent accessible surface area (SASA), root mean square fluctuation (RMSF), radius of gyration (rGyr), the protein-ligand interaction analysis, and ligand-protein interaction were computed to measure these complexes’ stability and dynamic features. 45
Results
Retrieval and preparation of phytochemicals
The IMPPAT database has been primarily explored to get accessible molecules of the required plant. Using cheminformatic techniques to enhance natural product-based therapeutics development, IMPPAT consist of more than 1742 Indian plants having medicinal properties and over 9500 phytochemical compounds. 46 A total of 418 phytochemical substances from the neem plant (A indica) were found in the database and are listed in Supplementary File 1.
Molecular docking study
A molecular docking investigation was carried out to comprehend the molecular interactions and binding affinities between the target protein and particular phytochemical substances. 45 Docking analysis was 45 performed to detect the binding score between the drugs and the investigated Nipah-enveloped attachment glycoprotein. In the molecular docking approach, all the phytochemicals produced 692 confirmations during preparation with the LigPrep function of Maestro v11.8. The binding score was found to be distributed between 6.876 and −7.118 kcal/mol following the phytochemical compound’s molecular docking. Twelve compounds identified have a higher outstanding docking score than the control ligand (Ribavirin). Depending on the docking score and ADMET, the top 3 compounds (CIDs: 5280343, CID: 9064, and CID: 5280863) have been chosen. These compounds had a superior binding interaction than the control, with cut-off energy scores of −7.118, –7.074, and −6.894 kcal/mol (Table 1). Because Ribavirin (control) has been shown to have an inhibitory effect against the NiV in the past and has a binding interaction of −6.152 kcal/mol.
List of PubChem identity, compound name, 2D structure, and biological use of selected top 3 phytochemicals and control (Ribavirin) with their binding affinity.

Protein-ligand interaction evaluation
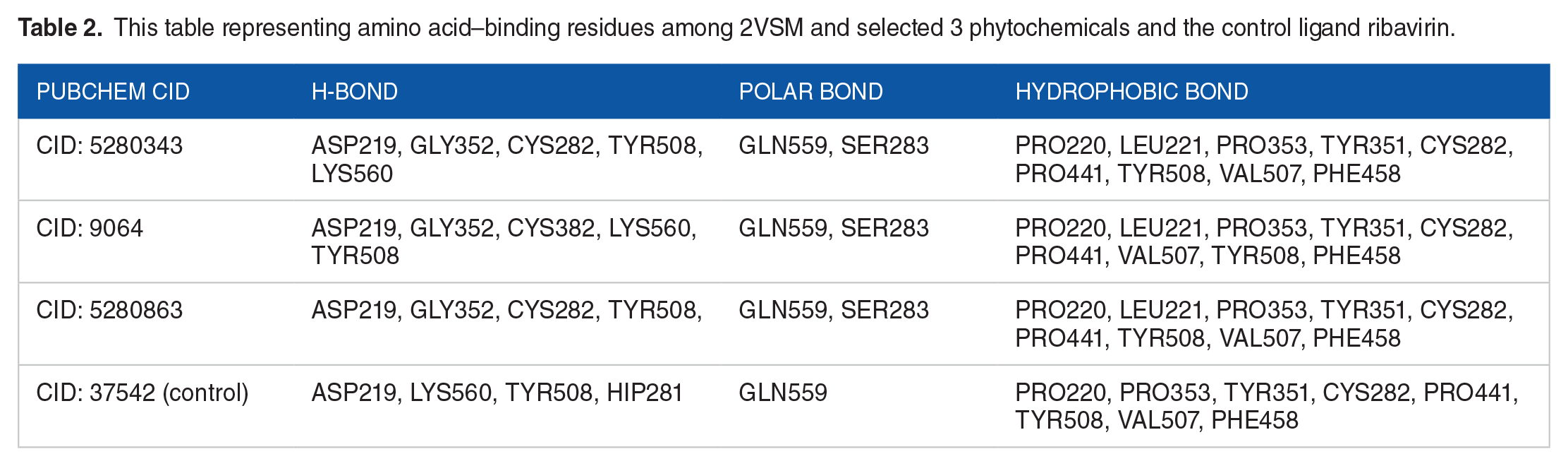
The highest docking score, comprising 3 chosen compounds, has been obtained for additional examination, and the Maestro module available in the Schrodinger suite, illustrated in Figure 1, has enabled the visualization of molecular interactions. Numerous interactions have been found between phytochemicals and receptor proteins, including hydrophobic, electrostatic, and hydrogen bonding. Throughout molecular docking, the best 3 compounds (CIDs: 5280343, 9064, and 5280863) and control ligand interacted with the common AA residues (ASP219, LYS560, TYR508, GLN559, PRO220, PRO353, TYR351, CYS282, PRO441, TYR508, VAL507, and PHE458) of the protein. This suggests that each ligand compound and the control ligand bind in the protein’s common active site. Each of the 3 compounds interacted with a separate AA, depicted in Figure 1 and Table 2.

Interaction between the 2VSM and selected 3 phytochemicals with control (A) CID 5280343, (B) CID: 9064, (C) CID: 5280863, and (D) CID: 37542 (control) representing in 3D and 2D format in the active site of the protein.
This table representing amino acid–binding residues among 2VSM and selected 3 phytochemicals and the control ligand ribavirin.
Post-docking MM-GBSA analysis
The ligand-protein complex’s endpoint binding free energy has been determined by using the MM-GBSA techniques. According to an analysis of MM-GBSA, all 3 chosen compounds, CID: 5280343, 9064, and 5280863, showed larger net negative binding free energy scores when they came into contact with apoprotein than the control compound, CID: 37542 (Table 3). Negative binding free energies of −47.56, –47.3, –43.15, and −21.55 kcal/mol, were found for the compounds CID: 5280343, 9064, and 5280863 as well as the control CID: 37542 following the molecular docking. As a result, compared with the reference compound (CID: 37542), the 3 chosen compounds (CID: 5280343, CID: 9064, and CID: 5280863) had a more persistent interaction with the protein. As shown in Table 3, the results for the chosen 3 compounds also showed a notable contribution from the following factors: dG Bind Packing, dG Bind Solv GB, dG Bind Lipo, dG Bind Coulomb, and dG Bind vdW. The preceding finding suggests that the 3 chosen compounds can attach to the protein-binding site for an extended length of time and inhibit the target protein.
Calculated binding free energies from post-docking MM-GBSA of selected compounds [kJ/mol].

Safety analysis of the compounds
Pharmacokinetic properties, which comprise ADME (absorption, distribution, metabolism, and excretion) properties, are influenced by the drug’s chemical qualities, the patient’s characteristics, and how the drug’s bodily organs work. These characteristics suggest that medications will succeed in clinical trials for avoidable causes that have yet to be thoroughly investigated. Consequently, the chosen substance’s properties of pharmacokinetics have been assessed. Based on several ADME evaluations, the compounds (CID: 5280343, CID: 9064, and CID: 5280863) are listed in Table 4. Good pharmacokinetic parameters for every molecule chosen for the study suggest little probability of drug failure during clinical trials. Toxicity assessment aids in determining and reducing a new drug’s possible dangers to human health. This is significant because adverse effects can occur from medications, even those that are beneficial in treating an illness. The toxicity properties of all 3 selected compounds (CIDs: 5280343, 9064, and 5280863) were good, as shown in Table 5.
Pharmacokinetic properties of selected 3 phytochemicals and control ligand.

Toxicity properties of the selected 3 phytochemicals and control ligand.

MD simulation
The examination and assessment of ligand-protein complex binding stability was conducted by MD simulations. During the orientation period, the MD simulation collected information on intermolecular affinity. Molecular dynamics (MD) simulation is an excellent and unique method to use to examine the stability of a ligand to a specified target protein. Using a 100 ns MD simulation, the ligand-protein complexes’ stability was evaluated. Based on the radius of gyration (Rg), RMSD, RMSF, and SASA, the MD simulation result was examined. During the simulation period, the protein-ligand interaction and ligand-protein interaction were also analyzed.
RMSD analysis
The RMSD of a ligand-protein complex aids in calculating the mean distance caused by a selected residue’s dislocation during a given session of time compared with an alluded time. To detect the changes in protein structure relative to the initial point, the RMSD of the 3 lead compounds and control has been evaluated. The protein’s equilibration status can also be established through the flattening of the RMSD graph. The simulation’s steady fluctuation and narrower RMSD range indicate a stable backbone of the protein. In contrast, a greater RMSD and/or a considerable change from the native structure imply that the ligand-protein pair is fragile. A value higher than the required parameter denotes a considerable structural change in the protein. It is deemed entirely acceptable to consider the mean value alteration between a specific and a reference frame encompassing a parameter of 1-3 Å. Therefore, to evaluate the protein structure stability throughout a 100 ns simulation period, the RMSDs of the Cα atoms for the compounds CIDs 5280343, 9064, 5280863, and 37542 (control) ligand-protein complexes have been calculated and observe the alterations in the order depicted in Figure 2, the RMSD of the complex structures of the selected 3 compounds with control including CIDs: 5280343, 9064, 5280863, and 37542 (control) were compared with the apoprotein structure. The average RMSD for all of the molecules was in the range of 1.52 Å to 1.85 Å, which was completely permissible. The largest fluctuations generated for the compound CID: 37542 (control) were between 10 to 20 ns and 80 to 100 ns but kept constant fluctuation with apoprotein between 20 and 80 ns during the simulation period, and the average RMSD was 1.81 Å. Because of a little high fluctuation, this compound underwent a little structural shift throughout the simulation in Figure 2, to exhibit better stability for a minimum amount of time. The other 3 compounds, including CID: 5280343, CID: 9064, CID: 5280863, exhibit extremely optimal fluctuations than control ligand, and their average RMSD values were 1.66 Å, 1.69 Å, and 1.45 Å, respectively, and the fact that they fell within the intended range demonstrated the structural stability of the ligand-protein complex in Figure 2. Most of the time, the selected 3 compounds maintained constant fluctuation with apoprotein compared with the control. The maximum RMSD values of the selected 3 compounds with control ligand bearing CID: 5280343, CID: 9064, CID: 5280863, and CID: 37542 (control) were 1.936 Å, 2.021 Å, 1.836 Å, and 2.173 Å, respectively, while their lowest values were 1.027 Å, 0.986 Å, 0.975 Å, and 0.891 Å, respectively.

The apoprotein (deep blue) complexed with the following 3 selected compounds and control (A, B) shows all the RMSD and RMSF for all the compounds CID: 5280343 (red), 9064 (green), 5280863 (violet), and 37542 (control) (light blue) and the protein, (C, D) The 100 ns simulation was used to calculate the Rg, and SASA of the ligand-protein complex. The 3 selected compounds, CID 5280343, 9064, 5280863, and 37542 (control), have Rg values that are represented by the colors blue, red, green and violet, respectively, when they are in combination with the apoprotein.
RMSF analysis
The prediction of local variations in protein chain residues and alterations in the positioning of ligand atoms can be accomplished through the utilization of the RMSF method. The stability and alteration level of AA residues in a complex system are determined by the RMSF value of the AA atoms. A compound may have attained greater stability if its AA residues have low RMSF values, whereas a complex may have attained less stability if its AA residues have higher RMSF values. In this RMSF graph analysis, the lead 3 compounds with control, including CID: 5280343, 9064, 5280863, and 37542 (control), bear average RMSF value 0.753 Å, 0.755 Å, 0.734 Å, and 0.792 Å, respectively. In the RMSF investigation, the compounds CID: 5280343 and 5280863 represent the best stability than the control ligand as shown in Figure 2. The RMSF showed minimum 55 AA to 130 AA and 255 AA to 385 AA residues index presenting the most stable secondary structural components, such as β-strands and α-helices. All the lead compounds exhibit a peak area of the protein at the sites of the THR206, SER239, ASP257, SER325, LYS386, ASP420, ASP446, and ASP582 residuals that flutter the greatest throughout the simulation. Due to the presence of α-helix, β-sheets, and the N- and C-terminal, most of the fluttering is noticed at the starting and ending of the protein. The highest RMSF values of the lead 3 compounds, CID: 5280343, 9064, 5280863, and 37542 (control), were 5.869 Å, 4.971 Å, 5.025 Å, and 5.836 Å, respectively, while their respective lowest values were 0.345 Å, 0.361 Å, 0.327 Å, and 0.367 Å.
The radius of gyration
The positioning of atoms in a ligand- protein complex along its axis can be considered the Rg. The Rg of the ligand-protein complex has been examined to ascertain the mobility and stiffness of the protein. Thus, the stability of CID: 5280343, 9064, 5280863, and 37542 (control) in complex with the target protein was also examined in terms of Rg throughout the 100 ns simulation duration depicted in Figure 2. The mean Rg for the compounds CID: 5280343, 9064, and 5280863 was determined to be 3.77 Å, 3.65 Å, and 3.63 Å, respectively, and they kept constant fluctuation throughout the simulation time. In contrast, with random fluctuation, the CID: 37542 (control) average Rg value was found to be 2.99 Å. Therefore, it indicates the active site of protein did not influence great conformational dislocation after binding these 3 lead compounds than CID: 37542 (control).
Solvent accessible surface area
The SASA was introduced to understand the structure and function of biological macromolecules. A protein’s surface AA residues typically function as active sites and/or interact with other molecules. The SASA analysis helps to determine the solvent-like nature (hydrophobic or hydrophilic) of a protein molecule as well as protein-ligand complexes. Consequently, the SASA value of the protein-ligand complex with the compounds including CID: 5280343, 9064, 5280863, and 37542 (control) was computed, and the mean SASA value was 85.29 Å2, 59.99 Å2, 88.28 Å2, and 57.56 Å2, respectively, as shown in Figure 2. The average SASA score identified for the intricate system ranged from 55 Å2 to 90 Å2, demonstrating an AA atom’s extensive exposure to the intricate systems’ selected compounds.
Protein-ligand contact analysis
Protein interactions with the lead 3 compounds, including CID: 5280343, 9064, 5280863, and 37542 (control), have been observed for 100 ns simulation period via the simulation interactions diagram (SID). The stacked bar charts (Figure 3) depict the hydrophobic, ionic, hydrogen bonds, and water bridge affinity that were generated by the MD simulation time. It was monitored that each compound established numerous interactions via water bridges, hydrogen, hydrophobic, and ionic bonds. These interactions persisted until the ending of the simulation, promoting the construction of a durable bond with the selected protein. The compound CID: 5280343 showed multiple interactions at the residues ASP219, HIS281, CYS352, GLY352, TYR508, GLN559, and LYS560, and their interaction fractions (IFs) were 0.09, 1.4, 0.82, 1.4, 0.63, 0.68, 0.09, and 0.95, respectively. Multiple interactions of the similar subtype with the ligand are made throughout the simulation to maintain the specific interaction (Figure 3A). Following simulation time, the compound CID: 9064 established numerous interactions at the residues ASP219 (0.34), PRO220 (0.54), ARG248 (0.1), HIS281 (0.83), CYS282 (1.2), TYR351 (1.2), TYR508 (0.8), GLN559 (0.5), and LYS560 (0.89) (Figure 3B). The compound CID: 5280863 formed several interactions at the residues HIS281 (0.52), CYS282 (0.68), TYR351 (0.8), GLY352 (0.61), TYR508 (0.8) GLN559 (0.75), LYS560 (1.75), and GLU579 (0.19), and CID: 37542 (control) also generated a few interactions at the residues ASP219 (0.52), PRO220 (0.69), HIS281 (0.99), CYS282 (0.6), TYR351 (0.9), TYR508 (0.75), LYS560 (1.75), and GLU579 (0.23) during the entire simulation time (Figure 3C and D). The apoprotein in Figure 3 interacts well with the compound CID: 5280343 and CID: 5280863 with more interaction fraction with the help of hydrogen and other bonds.

Protein-ligand contact analysis of selected 3 compounds CID: 5280343 (A), CID: 9064 (B), CID: 5280863 (C), and CID: 37542 (D)(control) depicting H-bonds (green), hydrophobic (light violet), and water bridges (blue) in the different color indicator.
Ligand-protein contact analysis
Throughout the simulation interaction diagram (SID), affinity has been analyzed when the 3 selected compounds with the control ligand CID: 5280343, CID: 9064, CID: 5280863, and CID: 37542 (control) complexed with the apoprotein. During the simulation time, the compounds produced multiple interaction residues. The specified interaction is assessed by the multiple interactions of the similar subtype with the ligand (Figure 4). The compounds CID: 5280863 and CID: 9064 are more stable in ligand-protein interaction than the compound CID: 37542 (control), and the compound (CID: 5280343) was more stable than the other 2 selected compounds (CID: 9064, CID: 5280863) when compared with control (CID: 37542).
Protein-ligand contact analysis of selected 3 compounds CID: 5280343 (A), 9064 (B), 5280863 (C), and 37542 (D) (control
Discussion and Conclusion
The COVID-19 pandemic brought attention to the threat of emerging infectious diseases, and NiV, a deadly biosafety level 4 pathogen, poses a significant challenge.55,56 The NiV’s attachment glycoprotein, NiV-G, has a key contribution in infection by attaching to the host receptors. Targeting this protein could lead to promising anti-Nipah drugs, as no licensed treatments exist. Previous attempts with Ribavirin and acyclovir were insufficient. 57 Supportive care is the only kind of therapy that can be administered to infected people.58,10
Phytochemicals like flavonoids, terpenoids, and phenols are known well for their antiviral features.59,60 The phytochemicals, or potential medication, from the A indica plant chosen for this investigation had antiviral, immunomodulatory, and antioxidant qualities. However, how these plant compounds work as a Nipah virus therapy is still uncertain. To escape the fatal condition that resulted from NiV, the study intends to uncover possible glycoprotein inhibitors that impede the attachment of the NiV using computational methods. Computational methods such as computer-aided drug design (CADD) are an efficient, rapid, and economical method of screening many drug candidates using bioinformatics techniques. 61 In the present research, the virus’s G protein (2VSM) was targeted for uncovering a possible therapeutic candidate. Furthermore, protein preparation, ligand development, and molecular docking research yielded 3 compounds with higher binding affinities than CID: 37542 (control) and other phytochemicals CID: 5280343, CID: 9064, and CID: 5280863. The docking scores of these compounds were −7.118, –7.074, and −6.894 kcal/mol, respectively. Besides, these 3 best compounds, control ligand CID: 37542 (control), have a docking value of −6.152 kcal/mol. The analysis of the binding interaction verified the high affinity between the ligand and protein. The post-docking MM-GBSA analysis was used to validate the docking results. The compounds CID: 5280343, CID: 9064, and CID: 5280863, as well as the control CID: 37542, have negative binding free energies of −47.56, –47.3, –43.15, and −21.55 kcal/mol, according to the complex analysis of MM-GBSA.
After that, the pharmacokinetics and toxicity of these 3 selected phytochemicals and the control ligand were assessed. The selected phytochemicals did not provide any toxicological risks, and all had good pharmacokinetics and Lipinski criteria (RO5) maintenance. When the numbers of hydrogen bond donors < 5, acceptors < 10, molecular mass < 500 daltons, and logP value < 5 were examined, the chosen compounds showed a favorable response in the Lipinski rule of five. In addition, the compounds’ MWs for CIDs 5280343, 9064, and 5280863 were 302.24, 290.27, and 286.24 g/mol, respectively. This was advantageous for heat molecules to consider, as compounds with greater MWs break the Lipinski rule. For CIDs 5280343, 9064, and 5280863, the hydrogen bond donors are 5, 5, and 4, and the hydrogen bond acceptors are 7, 6, and 6, respectively. The drug molecule’s absorption into the body is contingent upon the logP value. Based on Lipinski criterion (RO5), the chosen 3 compounds exhibited acceptable ranges of logP values.
As a result, if these compounds exhibit possible binding stability with the targeted protein, as determined by MD simulation, they can be used as a potential therapeutic candidate for NiV infection treatment. However, it should be examined for in vivo and in vitro analyses before starting with clinical trials. By MD simulation, the stiffness and stability of ligand-protein complexes can be determined outside of their natural context, such as the human body. The selected compounds were put through a 100 ns simulation to determine how stable their ligand-protein complexes were. Using MD simulation, we evaluate SASA values, RMSD, Rg, RMSF, protein-ligand contact, and ligand-protein contact analyses of these 3 drugs and the control ligand. In MD simulation, the mean distance occurred by the dislocation of a selected atom for a certain time frame with respect to a reference time frame is measured using RMSD. 62 RMSD was calculated for 3 compounds and a control ligand using an MD simulation, and it turned out that the lead 3 compounds, CID: 5280343, 9064, and 5280863, have the lowest fluctuations when compared with the CID: 37542 (control). Protein chain residues and ligand atom locations can be predicted using RMSF. During the RMSF study, all 3 compounds demonstrated the highest stability compared with the control ligand. The Rg of a ligand-protein complex structure specifies how the atoms are organized along its axis. The region of a biomolecule that a solvent can contact is referred to as its SASA. In MD simulation, optimal Rg and SASA values are considered favorable as lower Rg and SASA values indicate that the compounds with the target protein, AA atoms, and water molecules are tightly constricted in the complex. The mean Rg for the compounds CID: 5280343, 9064, and 5280863 is higher than the control ligand, according to the analysis. CID: 9064 and CID: 37542, alternatively, have lower SASA scores than the other 2 compounds. The 2 compounds CID: 5280343 and CID: 5280863 continue constant fluctuation than the other compound CID: 9064. However, the control ligand exhibits some fluctuation as well. Furthermore, in ligand-protein and ligand-protein contact analyses, all 3 compounds produced identical findings, except compound CID: 9064, which showed a reduced chemical interaction fraction with the help of hydrogen and other bonds in the ligand-protein contact study. The control ligand did not do well in any analysis compared with the lead compounds.
CID: 5280343 known as Quercetin 63 is a flavonoid compound characterized by the presence of 5 hydroxy groups, which are located at the 3-, 3'-, 4'-, 5-, and 7-positions. This particular flavonoid is highly prevalent in a variety of consumable vegetables, fruits, and wines. The compound possesses various functions, including acting as an antibacterial agent, a protein kinase inhibitor, an antineoplastic agent, an antioxidant, an EC 1.10.99.2 inhibitor, a plant metabolite, a phytoestrogen, a radical scavenger, an Aurora kinase inhibitor, a chelator, and a geroprotector.64,65 Clinical research does not support the use of quercetin extracts for the medication or prevention of a wide variety of medical situations, including cardiovascular disease, hypercholesterolemia, infections, rheumatic diseases, and cancer. Despite the deficiency of knowledge regarding its precise mode of action, in vitro studies have demonstrated the subsequent effects of this drug: induction of suppression of heat shock protein synthesis, G1 phase cell cycle arrest, and downregulation of expression of mutant p53 protein and p21-ras oncogene. When coupled with chemotherapeutic medications, this chemical has been shown to display synergy and reversal of the multidrug resistance phenotype, according to studies conducted in vitro.47,66,67
CID: 9064 known as Cianidanol is a type of flavonoid that acts as an antioxidant and is found primarily in the (+)-catechin and (-)-epicatechin (cis) forms in woody plants. The effects of (+)-catechin (Cianidanol), one of the polyphenols found in green tea, on animal models of hepatitis and in human clinical investigations have been investigated. Results in mice reveal that this chemical is an effective immune stimulant, increasing the number of cytotoxic T lymphocytes, activated macrophages, and natural killer cells. (+)-catechin has been shown to be useful in the treatment of viral hepatitis in a number of clinical studies. Some patients have hemolysis after taking pure (+)-catechin, which may be due to the stimulation of antibody production against (+)-catechin that may cross-react with red blood cells.68,69
CID: 5280863 known as Kaempferol is classified as a tetrahydroxyflavone due to the fact that its 4 hydroxy groups can be found at positions 3, 5, 7, and 4'. It is being studied as a potential treatment for cancer because it acts as an antioxidant, lowering oxidative stress. 70 Onions, broccoli, chives, tea, grapes, tomatoes, and strawberries all have significant concentrations of the antioxidant kaempferol. 71
In conclusion, all 3 selected compounds performed tremendously in molecular docking, post-docking MM-GBSA and MD simulation analysis. As a result, if the preclinical and clinical trials are successful, all 3 compounds can be used as possible therapeutic candidates for treating NiV sickness. However, experimental studies must be conducted in vitro and in vivo to prove the compounds’ efficacy against the target protein and secure the required approval for their medication usage.
Footnotes
Acknowledgements
The authors acknowledge the Noakhali Science and Technology University Research Cell (NSTURC) and R&D Project, Ministry of Science and Technology, Bangladesh for providing the support to the researchers.
Author Contributions
Conceptualization: OS and NHS; Data curation: OS, RA, and NHS; Formal analysis: NHS, RA, and UPB; Investigation: OS and NS; Methodology: OS, NS, UPB, RA, and NHS; Resources: OS and NHS; Software: OS and NHS; Supervision: OS, NS, and KFS; Validation: NHS, RA, MSA, and SMLRR; Visualization: AS and TAS; Writing—original draft: OS, NHS, RA, and NS; Writing—review & editing: OS, FA, NHS, MMR, and FH. All authors have read and agreed to the published version of the manuscript.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Data Availability Statement
Data is contained within the article and Supplementary Material.
Supplemental Material
Supplemental material for this article is available online.