
Abstract
Vibrio parahaemolyticus, an aquatic pathogen, is a major concern in the shrimp aquaculture industry. Several strains of this pathogen are responsible for causing acute hepatopancreatic necrosis disease as well as other serious illness, both of which result in severe economic losses. The genome sequence of two pathogenic strains of V. parahaemolyticus, MSR16 and MSR17, isolated from Bangladesh, have been reported to gain a better understanding of their diversity and virulence. However, the prevalence of hypothetical proteins (HPs) makes it challenging to obtain a comprehensive understanding of the pathogenesis of V. parahaemolyticus. The aim of the present study is to provide a functional annotation of the HPs to elucidate their role in pathogenesis employing several in silico tools. The exploration of protein domains and families, similarity searches against proteins with known function, gene ontology enrichment, along with protein-protein interaction analysis of the HPs led to the functional assignment with a high level of confidence for 656 proteins out of a pool of 2631 proteins. The in silico approach used in this study was important for accurately assigning function to HPs and inferring interactions with proteins with previously described functions. The HPs with function predicted were categorized into various groups such as enzymes involved in small-compound biosynthesis pathway, iron binding proteins, antibiotics resistance proteins, and other proteins. Several proteins with potential druggability were identified among them. In addition, the HPs were investigated in search of virulent factors, which led to the identification of proteins that have the potential to be exploited as vaccine candidate. The findings of the study will be effective in gaining a better understanding of the molecular mechanisms of bacterial pathogenesis. They may also provide an insight into the process of evaluating promising targets for the development of drugs and vaccines against V. parahaemolyticus.
Introduction
Litopenaeus vannamei and Penaeus monodon are two of the most economically important species of farmed shrimp grown in Asia. Unfortunately, the emergence of various viral, bacterial, and fungal infections continues to wreak havoc on shrimp productivity.1-3 Vibriosis is one of the most prevalent illnesses in Asia, causing severe morbidity in farmed aquatic products (ie, shrimp, fish, and shellfish). 4 Vibrio harveyi, V. anguillarum, V. alginolyticus and V. parahaemolyticus are examples of opportunistic Vibrio pathogens with particularly virulent strains. 5 In 2009, acute hepatopancreatic necrosis disease (AHPND) had a quick, devastating effect on early stages of shrimp during an initial outbreak elsewhere in southwest of China. 6 Since then, it has expanded all over the world, posing a severe threat to the shrimp industry in several Asian countries. 7
When shrimp are 1 month old or post larvae are approximately 20 to 30 days old, AHPND can potentially result up to 100% morbidity. 8 V. parahaemolyticus infection provokes an atrophied, pale hepatopancreas, severe sloughing off of the hepatopancreatic epithelial cells, destruction of the brush boundary of the anterior midgut of the shrimp and hemocyte infiltration.1,8-10 V. parahaemolyticus is a facultative anaerobic, gram-negative bacterium that could be found in estuarine, marine, and coastal ecosystems across the world, including in shrimp aquaculture.11,12 Ever since discovery, it has been associated to human gastrointestinal illness, septicemia, and wound infections and is considered a causative agent of foodborne illness over the world.12-14
Only the strains of opportunistic marine pathogen V. parahaemolyticus (named as VPAHPND) carrying a 69-kbp virulent pVA1 plasmid encoding binary toxin genes homologous to the Photorhabdus insect-related (Pir) toxin, PirA and PirB, were found to produce AHPND.6,15,16 VPAHPND lacks its potential for causing AHPND when the pVA1 plasmid has been removed or the Pir genes are knocked down selectively. 16 The plasmid also contains conjugative transfer genes as well as transposons, implying that the plasmid might be mobilized into some other strains or even into other species.1,16 Interestingly, after being reported in V. parahaemolyticus, the pVA1 plasmid was also reported in a variety of Vibrio species, including V. campbelli, V. owensii, V. punensis, and V. harveyi, all of which have the potential to cause AHPND. 1 Even though the plasmid-encoded binary toxins pirA and pirB have been identified as the leading cause of AHPND in shrimp, additional virulence factors reported in V. parahaemolyticus might play a key role during infection.17,18
The shrimp production has been severely impacted by the AHPND causing strain of V. parahaemolyticus, VPAHPND. 19 The bacteria are transmitted orally and subsequently accumulates inside the gastrointestinal tract of the shrimp, where it produces and secretes binary toxins (ie, PirAVP and PirBVP) which induces tissue destruction and invalidism of the hepatopancreas of the shrimp digestive system.16,20 Farmers sought to prevent AHPND epidemics by eradicating and renovating their ponds, but they have been unable to stop outbreaks once the infection emerged repeatedly in farmlands. 5 To eliminate AHPND, the etiologic agent, V. parahaemolyticus, has to be extensively characterized.
Two isolates of V. parahaemolyticus (strains MSR16 and MSR17) were isolated from cultured shrimp (P. monodon) in the southwest region of Bangladesh, and their genomes had been sequenced and published, way that allows genomics and proteomics analyses to gain a detailed understanding of microbial virulence mechanisms and underlying pathogenesis. 18 Nonetheless, gaining a better understanding of these mechanisms remains a challenge. During proteomic studies, functional annotation is essential to determine the function of proteins. 21 In the meanwhile, the function of a large number of coding sequences still remains unknown. Pathogenesis and virulence determination have been challenging to fully understand due to a lack of comprehensive proteome data due to coding sequences without a proper prediction of functions. The term “hypothetical protein” (HP) is used to describe these molecules. The majority of such proteins are thought to play a significant role to play in the cell; therefore, proper annotation can potentially lead to new insights into their structures, functions, and pathways.22,23
Considering wet lab approaches are often time-consuming, expensive, and labor intensive for unraveling functions of desired proteins, in silico approaches have emerged as significant methods for predicting or identifying the functions of hypothetical proteins. Due to the obvious similarity with known proteins, homology-based functional annotation may be used to assign functions to HPs.21,24-26 Bioinformatics approaches, particularly protein-protein interactions, can aid in the proper characterization of the biological processes in which HPs are involved. 27 The objective of this study was to assign functions to the hypothetical proteins encoded within the genomes of the V. parahaemolyticus isolates to identify novel proteins that might aid to better understand the pathogenesis and virulence mechanism of the bacteria as well as identify new therapeutic targets employing computational approaches.
Materials and Methods
The overview of methodology is illustrated in Figure 1.

Workflow for functional annotation of V. parahaemolyticus hypothetical proteins.
Retrieval of protein sequence data
The genomes of two isolates of V. parahaemolyticus (strains MSR16 and MSR17) isolated from cultured shrimp (P. monodon) in the southwest region of Bangladesh were retrieved and analyzed in this study (accession numbers: RPDA00000000.1 and RPDB00000000.1, respectively) from the National Center for Biotechnology Information (NCBI). 28 The strain MSR16 genome encoded a total of 5479 genes, 1403 of which were annotated as HP, whereas the strain MSR17 genome encoded a total of 5187 genes, 1228 of which were annotated as HP. 18 Using an in-house python script, the coding sequences (CDS) annotated as hypothetical proteins were retrieved from those genomes (Supplementary Table 1).
Functional annotation of hypothetical proteins
Gene ontology prediction
To annotate the function of the HPs, the protein sequences were initially analyzed using the GO FEAT 29 and PANNZER 30 tools to obtain a preliminary gene ontology (GO) prediction with an e-value of 1e-03. For functional annotation of protein-coding genes, the GO vocabulary is used. 31 Protein sequences with GO IDs predicted by both servers were carefully chosen, and domain and function were further investigated with a number of bioinformatics tools.
Family and domain prediction
Multiple databases were scanned for identification of conserved domains to predict protein function based on domain structure. HP sequences were initially evaluated using the HMMER tool, which offers fast screening against frequently used sequence databases, employs profile hidden Markov model libraries for functional annotation of the HP sequences as well as protein families and domains and enables protein homology search algorithms within the HMMER 3.3.2 software suite. 32 For significant e-values, the cutoff was set at 0.01. The Pfam 33 and Superfamily 34 databases were used to identify protein families, while the Gene3D 35 database was used to identify protein domains. The InterProScan 36 tool, which scans the InterPro 37 database for matches, was then used to perform functional analysis of proteins by categorizing them into families as well as identifying domains and essential sites encoded by the HPs. The NCBI Batch CD-Search tool was used to compare query HP sequences to databases of conserved domain models using RPS-BLAST. 38 The CDD – 58235 PSSMs database was searched with the NCBI Batch CD-Search tool, with a threshold of 0.01.
HP functions have been predicted up to this point based on domains and families identified by screening databases like as SUPERFAMILY, Pfam, Gene3D, InterPro, and CDD – 58235 PSSMs. InteractiVenn 39 was used to identify HPs with predicted functions from three or more tools.
Finally, the annotated homologous proteins from related organisms were identified using the Basic Local Alignment Search Tool (BLAST). 40 The non-redundant (nr) database of the NCBI was searched for homologs with an identity of more than 90% and an e-value of less than 1e-03.21,41,42 REVIGO 43 was implemented to enrich the GO terms of the annotated HPs and visualize data.
Prediction of protein physiochemical properties
Physical and chemical properties of the virulent HPs were determined using Expasy’s ProtParam 44 tool in Linux operating system implemented in Julia (version 1.5.1) 45 with package BioSequences (v2.0.5).
Determination of subcellular localization
PSORTb v3.0 46 and CELLO v.2.5 47 were used in the study to explore the subcellular locations of the HPs using default parameters for gram-negative bacteria. The presence of transmembrane helices as well as the topology of the HPs were predicted using TMHMM 2.0 48 and CCTOP 49 with default parameters. SignalP 6.0, 50 which employs a neural network design using a conditional random field, was used in predicting the presence of signal peptides specific for the secretory (Sec) and the twin-arginine translocation (Tat) pathways, as well as the location of signal peptide cleavage sites.
Virulent HP detection
To identify virulence factors from annotated HPs, they were initially evaluated with the MP3 tool, 51 which uses an SVM and HMM approach to accurately estimate virulent proteins present within genomic and metagenomic data. VirulentPred, 52 another SVM-based virulence prediction tool, was also used to classify pathogenic proteins in bacteria. Furthermore, using the BastionX prediction method, BastionHub 53 was used to predict substrates for several secretion systems found in gram-negative bacteria (System I-IV, VI). The HPs that were identified to be virulent by all three tools were then identified and further studied.
Predictions of antigenicity, allergenicity, and toxicity index
VaxiJen v2.0 54 server and ANTIGENpro server 55 were used to predict the antigenicity of the vaccine peptide. The ToxIBTL server 56 and the AllerCatPro v. 2.0 57 server were used to predict the peptide’s toxicity and allergenicity, respectively.
Analysis of Protein-Protein Interaction (PPI)
Finally, the PPIs for the hypothetical proteins from the V. parahaemolyticus MSR16 and MSR17 strains were constructed using the String 11.5 database to validate the functions of the annotated HPs and their interactions with other proteins. 58 Only interactions having score values exceeding 0.700 (high confidence) as well as high FDR stringency (1%) were used to provide the most accurate and reliable PPIs. 21 The P value for PPI enrichment was <10−16.
In the String 11.5 search, the V. parahaemolyticus RIMD 2210633 strain was selected here as the strain with highest similarity. The interolog mapping approach was used to translate the identified interactions to V. parahaemolyticus MSR16 and MSR17 strains, which implies that when two proteins interact, their orthologous partners would also interact.59-61 STRING, as described in eggNOG, exploits hierarchically organized orthologous group relations to retrieve association between relevant species.62-64
To perform a more in-depth analysis and understanding of the interactions of the potentially virulent HPs with other proteins and among themselves, Cytoscape 3.9.0 65 was employed. PPI networks were validated using the network analyzer plugin 66 in the Cytoscape 3.9.0 program. Protein molecules were allocated to nodes in Cytoscape, whereas molecular interactions were given to edges.
Results
Function annotation of hypothetical protein to both strains
All of the protein sequences were evaluated employing the GO FEAT and PANNZER tools, which facilitated the prediction of GO annotation. The MSR16 strain of V. parahaemolyticus produced a total of 1403 HPs, and GO terms were predicted for 564 of those HPs. On the other hand, the MSR17 strain encoded 1228 HPs, and 514 of those encoded HPs indicated hits on the GO database. For the identification of protein domains and/or families, this pool of 1078 proteins was thoroughly investigated using the HMMER, Pfam, Superfamily, Gene3D, and InterPro, NCBI Batch CD-Search tools. The results obtained from using those tools were analyzed to determine the appropriate functions to refer to HPs. Proteins with similar function predictions from three or even more programs were functionally annotated with high confidence. Consequently, the functions of 656 HPs (338 HPs from MSR16 strain and 318 HPs from MSR17 strain) have been annotated with a high level of confidence (Supplementary Table 2). The NCBI BLASTp program was used to validate the annotated functions of these HPs based on homologous proteins, indicating the accuracy of the annotation.
Analysis of GO terms of annotated HPs
In this study, the GO terms of 656 HPs with functional annotation were evaluated to identify their association with any of the following GO categories: Biological Process (BP), Cellular Components (CC), and Molecular Functions (MF). As far as the biological process is considered, there were 224 proteins identified with 78 GO keywords in total (Supplementary Table 3). This analysis revealed a cluster of transport proteins as well as the protein clusters essential for growth (Figure 2A). A total of 187 proteins were classified as cellular components using 18 different GO keywords. Surprisingly, 89 proteins were found to be integral component of the membrane (Figure 2B, Supplementary Table 4). In terms of molecular function, 370 proteins with 134 distinct GO terms were identified, and after analysis, a cluster of metal ion binding proteins was observed (Figure 2C, Supplementary Table 5). These findings imply that HPs might have a role in the development and pathogenesis of the organism, and the identified groups were investigated further.

Classification of the HPs based on gene ontology data: (A) enriched biological processes, (B) enriched cellular components, and (C) enriched molecular function. The logSize of the enzyme number is represented both by the diameters of the circles and the colors used. HP indicates hypothetical protein.
Enzymes related with growth and survival
Within the set of annotated HPs, multiple enzymes that are essential to the growth and development of bacteria were observed. To have a complete grasp of the host-pathogen interaction, it is necessary to have knowledge about these enzymes. MSR16_hyp_1391 and MSR14_hyp_815 were annotated to be a radical SAM (S-adenosylmethionine) protein, whereas MSR16_hyp_11, MSR16_hyp_148, MSR16_hyp_1211, MSR17_hyp_124, MSR17_hyp_539, MSR17_hyp_768 and MSR17_hyp_797 were found to be SAM-dependent methyltransferase. It is well established that radical SAM proteins play a critical role in organism survival, and it has also been demonstrated that inhibiting these enzymes has been effective in preventing serious infections.67,68 The phasin protein (MSR16_hyp_676 and MSR17_ hyp_1221) was identified in the genome of the both strain. Phasins are the major polyhydroxyalkanoate (PHA) granule-associated proteins. They have both structural as well as regulatory functions, and they have the ability to affect the accumulation of PHA within the bacterial cell and to mediate protein folding, which in turn stimulates the growth of bacteria.69,70
The ubiquinone biosynthesis genes, which were encoded by both of the bacterial strains (MSR16_hyp_1380, MSR16_hyp_1387, MSR17_hyp_151, MSR17_hyp_161 and several others), were considered to be an additional significant cluster of genes that have been identified as a part of this study. Ubiquinone, commonly known as Coenzyme Q (CoQ), is a component of the respiratory chain in many prokaryotic organisms and is crucial for energy production as well as a variety of other intracellular activities. 71 Surprisingly, ubiquinone was found to be linked to bacterial pathogenicity in Francisella novicida and Xanthomonas campestris.72,73
Gram-negative bacteria have such a cell envelope that is made up of two membranes known as the inner membrane and the outer membrane (OM), along with an enclosed chamber that is known as the periplasm. The bacterial cell envelope is of particular interest because it serves the dual purpose as both a structural component as well as a permeability barrier.74-76 These unique dual characteristics make the cell envelop a target of exhaustive study. Several HPs were identified in this study that may have a function in cell envelop biogenesis. OmpA (MSR16_hyp_1006 and MSR17_hyp_41) is a well-studied protein which is a major virulence factor that mediates the formation of bacterial biofilm, infection of eukaryotic cells, antibiotic resistance, and immunomodulation. 77
For the synthesis of peptidoglycan in bacterial cell walls, the glycosyltransferase (MSR16_hyp_1127) is essential. This enzyme transfers the disaccharide-peptide from the lipid II onto the expanding glycan chain. 78 The peptidoglycan binding protein (MSR16_hyp_157, MSR17_hyp_1035, and MSR17_hyp_864) may also play a part in cell wall biogenesis. 79 MSR16_hyp_229 and MSR17_hyp_548 were identified as BamA, which play a major role in the biogenesis of outer membrane (OM) proteins in bacteria. 80 An additional protein (MSR16_hyp_875), characterized as TolA, is engaged in the process of ensuring the integrity of the outer membrane. 81 To survive, it is extremely crucial for bacteria to monitor and preserve the integrity of the cell envelope in the presence of agents and situations that can destabilize the envelope.82,83
Interestingly, we found several potential therapeutic target proteins within the annotated HPs. Shikimate kinase (MSR16_hyp_748) is a possible therapeutic target that can be used against both of the strains that are being explored in this study. Studies are currently being conducted with the goal of identifying shikimate kinase inhibitors that are effective against methicillin-resistant Staphylococcus aureus and Mycobacterium tuberculosis.84-86 The phosphoribosyl-ATP pyrophosphohydrolase (MSR16_hyp_1190), an enzyme that is involved in the histidine biosynthesis pathway, is another possible therapeutic target. 87 It has been experimentally validated that extracts from tropical plants have the ability to block aminoacyl-tRNA hydrolase (MSR16_hyp_1287 and MSR17_hyp_580), an enzyme that is essential to the process of protein biosynthesis.88,89 Another possible target for novel selective antimicrobial drugs is the enzyme GTP cyclohydrolase-2 (MSR16_hyp_1336 and MSR17_hyp_1153). GTP cyclohydrolase-2 is involved in the process of flavin biosynthesis, and an inhibition of the flavin biosynthesis pathway may result in the death of the bacteria. 90
When it comes to function or stability of a protein, the incorporation of disulfide bonds into proteins can be absolutely necessary. The periplasmic enzyme DsbA is essential for the incorporation of disulfide bonds into a large number of extra-cytoplasmic proteins and is associated with bacterial virulence.91-94 We predicted that MSR16 hyp 605 is the DsbA oxidoreductase, and it has the potential to be a good therapeutic target. 95 The biosynthesis pathway for thiamine (vitamin B1) is yet another significant metabolic pathway in bacteria that possesses the potential to be exploited as a drug target. 96 The thiamine-phosphate synthase enzyme was observed to be present in both of the bacterial strains studied (MSR16_hyp_1301, MSR17_hyp_22, and MSR17_hyp_966). This enzyme can be targeted for drug development against the pathogens.96,97
Iron binding protein
During the process of pathogenesis, the ability of bacteria to accumulate iron and other essential metal ions from the surroundings becomes a major determining factor during the severity of the infection. This is due to the fact that hosts inevitably employ a strategy of nutritional immunity for minimizing the accessibility of metal ions to the bacteria. 98 Gram-negative bacteria comprise a variety of iron uptake systems that are capable of capturing iron-containing substrates to circumvent this situation.99,100 One of the most interesting host-pathogen interactions to be examined is the competition for iron that takes place between pathogenic bacteria and the organisms that they infect.
Our pathogenic bacterial strains also encoded multiple proteins related to iron uptake and regulation, as expected. We found that bacterial strains in this study encoded the enzyme known as siderophore ferric iron reductase (MSR16_hyp_1279 and MSR17_hyp_934). Siderophores have a high affinity for ferric ions (Fe3+), whereas they have only a moderate affinity for ferrous ions (Fe2+). 101 Ferric iron reductase is an enzyme that catalyzes the reduction of ferric iron to ferrous, which is a form of iron that has a lower affinity and is highly soluble.102,103 In addition, we observed that the pathogens encoded proteins essential for heme biosynthesis (MSR16_hyp_1339, MSR16_hyp_1340, MSR17_hyp_328 and MSR17_hyp_1130). To maintain the stability of the iron homeostasis within the bacterial cell during an infection, the synthesis of heme is a vital step. 104
Antibiotic resistance genes
We identified several proteins which are associated with antibiotic resistance. A large cluster of MFS transporters was identified within the genome of both strains (for example- MSR16_hyp_1321, MSR16_hyp_1322, MSR16_hyp_1324, MSR17_hyp_162, MSR17_hyp_326, MSR17_hyp_475, MSR17_hyp_734, and few more). MFS transporters are capable of transporting a wide range of substrates across the cell wall, leading to the development of multidrug resistance (MDR) strains.105,106 PACE efflux transporter (MSR16_hyp_1198) is a distinct type of transporter protein that is encoded by pathogens. Like other types of transporter proteins, it transports antimicrobial compounds out of the cell. 107
GNAT family N-acetyltransferase proteins were also identified (MSR16_hyp_832, MSR16_hyp_878, MSR16_hyp_1039, MSR16_hyp_1067, MSR16_hyp_1109, MSR16_hyp_1326, and MSR16_hyp_1327 were encoded by MSR16 strain while MSR17_hyp_139, MSR17_hyp_200, MSR17_hyp_526, MSR17_hyp_573, and MSR17_hyp_1100 were encoded by MSR17 strain). GNAT family N-acetyltransferase proteins have prominent roles across a wide variety of biological processes, one of which is the development of aminoglycoside antibiotic resistance.108,109
We further identified the antibiotic resistance protein VanZ (MSR16_hyp_778 and MSR17_hyp_703), which reduces the binding of lipoglycopeptide antibiotics to cell wall components, resulting in resistance to teicoplanin and vancomycin antibiotics.110,111
Toxin proteins and toxin transporters
The thermostable direct hemolysin (TDH) and the TDH related hemolysin (TRH) are both regarded to be key virulence factors in pathogenic strains of the bacteria V. parahaemolyticus.112,113 Along with them, we identified an additional hemolysin protein called enterohemolysin EhxA (MSR16_hyp_1044 and MSR17_hyp_988) that is encoded in the genome of the bacteria and contributes to the pathogenesis of the bacteria.114-116 In addition to this, we found an RTX toxin (MSR16_hyp_560) that is capable of acting as a synergistic virulence factor.117-119 We also identified Hemolysin D (HlyD) (MSR16_hyp_510) and the outer membrane protein TolC (MSR16_hyp_929), which are involved in the hemolysin secretion system. The HlyD protein forms a continuous channel by docking to the protein TolC which forms a part of HlyA specific type I secretion system (T1SS).120-122 We also identified two types of phospholipase; zinc-dependent phospholipase C (MSR16_hyp_651 and MSR16_hyp_988) and patatin-like phospholipase (MSR16_hyp_706, MSR17_hyp_54, and MSR17_hyp_854). Phospholipase C, commonly known as alpha-toxin, can bind to eukaryotic cell membranes and hydrolyze membrane lipid moieties (ie, phosphatidylcholine and sphingomyelin), leading to cell lysis.123,124 Again, genomes of bacterial pathogens, particularly gram-negative species, encodes patatin-like PLA2 enzymes which act as effector molecules to target host cellular membranes, suggesting a role in host-pathogen interaction.125,126 We also identified two other virulence genes in this study: VcgC (MSR16_hyp_127 and MSR17_hyp_103) and transcriptional activator HlyU (MSR16_hyp_114), which are commonly found in other Vibrio species also.127,128
Secretion system and associated proteins
It is widely known that gram-negative bacteria possess nine distinct types of secretion systems (type I-IX).95,129 Out of them, the first six types (type I-VI) are the most prevalent and are linked to the virulence of the bacteria. 130 In our study, we also found several effector proteins as well as proteins involved in formation of secretion system.
We identified a large number of protein involved in the formation of type VI secretion system (eg, MSR16_hyp_1341, MSR16_hyp_1344, MSR16_hyp_1345, MSR17_hyp_1036, MSR17_hyp_1140, MSR17_hyp_1043, MSR17_hyp_1196 and several others). In gram-negative bacteria, Type VI secretion systems are associated with the machinery that is required for injecting effector proteins into eukaryotic cells to exert virulence, symbiosis, as well as antibacterial activity.130-132 We also identified S-type Pyocin (MSR16_hyp_89 and MSR17_hyp_256), an effector of the type VI secretion system that has the potential to mediate antibacterial toxicity.133,134
V. parahaemolyticus also encodes genes for the formation of the type III secretion system, and we identified Type III secretion chaperone CesT among the hypothetical proteins (MSR16_hyp_195, MSR16_hyp_1271, MSR17_hyp_116, and MSR17_hyp_745). CesT chaperone is a multi-effector chaperone and is required for secretion of several effector proteins.135-137 Another annotated HP which might be involved in bacterial virulence is the type II secretion system (T2SS) pilot lipoprotein GspSβ (MSR16_hyp_82 and MSR17_hyp_414). This protein is essential for the formation of the T2SS, thus facilitating the pathogenesis of the bacteria.138,139
Physicochemical parameters and subcellular localization of the annotated HPs
In this study, the amino acid sequences of all 657 annotated HPs were examined to determine their physicochemical properties (Supplementary Table 6). It is well recognized that in silico analysis could reveal molecular relevance since several parameters are related to protein stability and function. According to the ProtParam results, the length of the analyzed sequences ranged from 53 to 1578 amino acids. The theoretical pI of the proteins ranged from 3.68 to 10.55. This parameter refers to the point at which the amino acid can no longer tolerate liquid charge and the mobility of the ampholyte sums to zero. 140 The direction of protein migration on the gel during electrophoresis is determined by the charge. As a result, proteins can be separated in a gel based on their pI. 141 The molecular weights ranged from 5910.23 to 176889.35 Da. In laboratorial experiments, 2D gel electrophoresis visualization can be accompanied by a combination of pI and molecular weight, allowing new proteins to be identified and their relative abundance measured between comparative samples. 142
For the HPs, the extinction coefficient, which is crucial for determining the accurate concentration of a protein, was also predicted. With respect to the concentration of cystine, tryptophan, and tyrosine amino acid residues in the protein sequences, the extinction coefficients of the proteins ranged from 125 M-1cm-1 to 235185 M-1cm-1 at 280 nm. High extinction coefficient occurred in some HPs because of the presence of high concentration of the cystine, tryptophan, and tyrosine residues while extinction coefficient was not predicted for some HPs because of the absence of these residue in their protein sequence.
The aliphatic index and instability index were predicted to evaluate the stability of HPs. The aliphatic index is directly related to the molecular fraction of aliphatic amino acids (ie, alanine, valine, isoleucine, and leucine) and is related to the thermostability of the protein; the higher the aliphatic index, the greater the thermostability. 143 Values for aliphatic index ranged from 44.86 to 155.24. The instability index was used to estimate an assumption of protein stability in a test tube. Proteins with instability index less than 40 are regarded as stable proteins. 144 In the study, instability index for the annotated HPs ranged from 10.19 to 98.97, indicating that 367 out of 656 proteins should be stable in a test tube.
Finally, the GRAVY (Grand Average of Hydropathy) value of the proteins was predicted. This value ranged from −0.535 to 0.434 in the study, with 543 proteins having a score of less than 0 and 113 proteins having a score of greater than 0. Proteins that have GRAVY values that are lower than 0 are regarded as relatively hydrophilic, whereas proteins that have GRAVY scores that are higher than 0 are regarded as relatively hydrophobic. 145 This information is useful for identifying proteins by categorizing them as globular or membrane-bound proteins.145,146
Since the function of a protein is typically associated with the location of the protein, the subcellular localization of a protein can provide helpful insights regarding the functions of proteins.147,148 Among the annotated HPs, 60% (390) of them were predicted to be localized at the cytoplasm. The number of HPs present at the inner membrane 17.41% (90) and 2.52% (81) are present at outer membrane. It is predicted that 14% (57) HPs are present in periplasm and 5.89% (38) in extra cellular matrix (Figure 3A, Supplementary Table 7).

Annotated HPs classified on the basis of (A) subcellular localization and (B) signal peptide. HP indicates hypothetical protein.
Signal peptides are the key players in determining the transport of proteins to the target location. Hence, prediction of signal peptide is essential to learn about the transport system of the specific proteins and the cleavage sites. We predicted the presence of signal peptide sequences in 110 HPs out of 656 (78 contains Sec/SPI and 32 HPs contain Sec/SPII) (Figure 3B, Supplementary Table 7). Presence of transmembrane helices were also analyzed using the protein sequences, and it was predicted that transmembrane helices were present among 120 HPs (Supplementary Table 7).
Identification of virulent proteins
MP3, VirulentPred, and BastionHub were employed to accurately predict virulence factors with a high level of confidence. Only those proteins that had been projected to be pathogenic by each of the three different prediction tools were selected and studied further. There were a total of 656 annotated HPs, of which 92 were predicted to be virulent. This included 47 HPs from the MSR16 strain and 45 HPs from the MSR17 strain (Supplementary Table 8).
Virulent HPs with therapeutic potential
The antigenicity of the pathogenic HPs was analyzed, and the results indicated that 56 of them have the ability to produce an antigenic response, and out of those 56, 36 proteins have been projected to be non-allergenic and non-toxic toxic (Supplementary Table 8). These results indicate that these proteins could be used as vaccine candidates. Further analysis of these proteins identified that 27 of the candidate proteins either are extracellular proteins or are localized at the outer membrane or periplasm of the bacterium. This implies that these proteins may have the potential to be used in the development of a vaccine that is effective against the pathogen (Table 1).
List of virulent HPs with therapeutic potential.
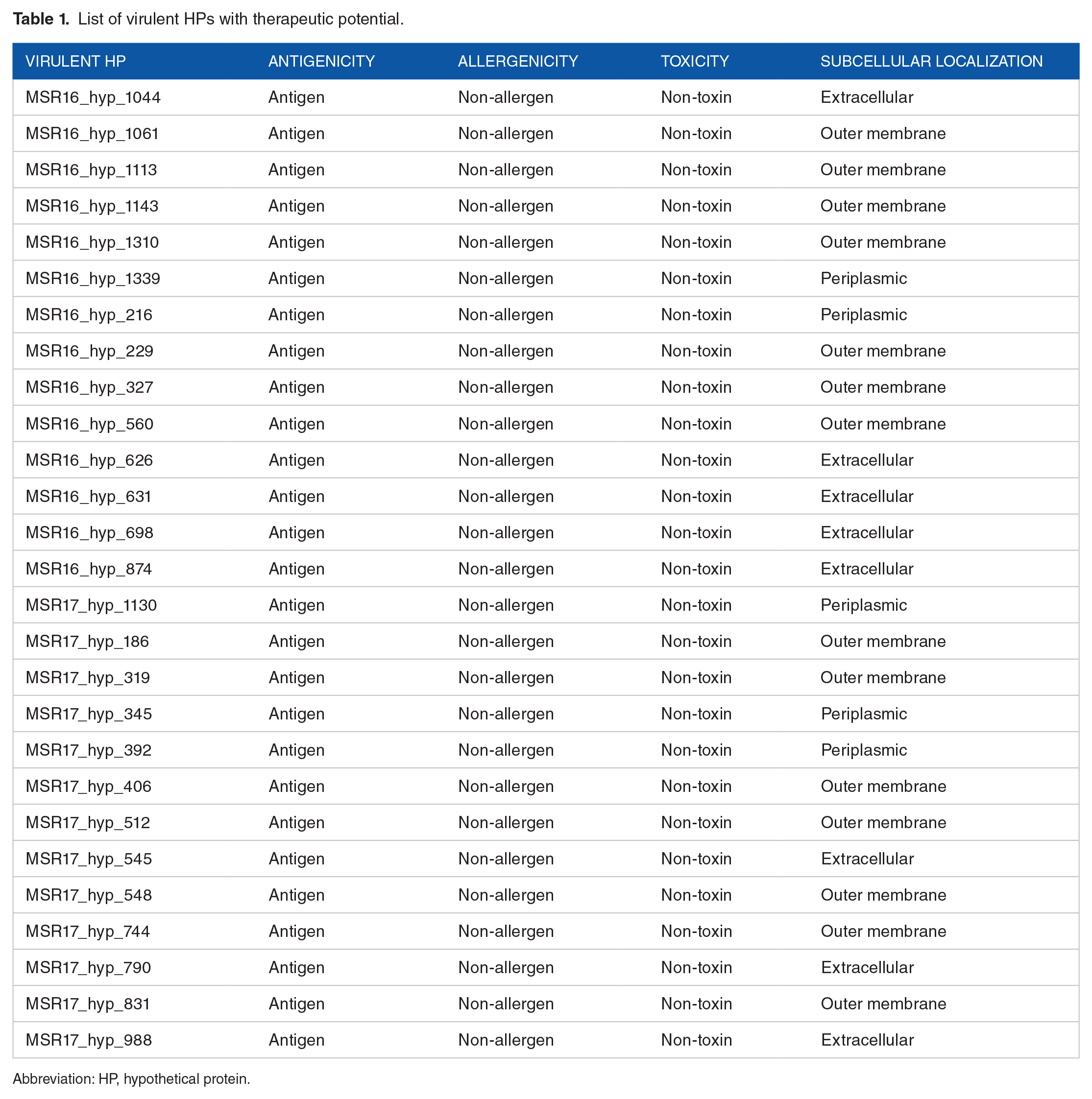
Abbreviation: HP, hypothetical protein.
PPI network analysis
On the annotated HPs, we performed a PPI analysis, which is a method that can further validate the annotation and also suggest potential roles for these proteins. We had to exclude 161 HP from a total of 656 due to low confidence interactions with other proteins. In terms of the remaining 495 HPs, 65 of them either have domains with an unidentified function or the protein itself has not been defined. As a result, 430 proteins have been identified and clustered according to their functions (Supplementary Table 9). These functions include DNA mismatch repair enzymes, DNA/RNA binding molecules, transcription factors, multidrug resistance protein, nitrogen fixation protein, and several other. These proteins have the potential to play a major role in the growth and survival of the pathogen. Target HPs are indicated by homologous proteins in V. parahaemolyticus RIMD 2210633 strain (Supplementary Table 9).
We performed a comprehensive analysis of the protein clusters to identify proteins that play a significant role in the development and pathogenesis of the pathogen. Within the scope of this study, we were able to reveal an interconnected clusters of proteins that play essential roles in the biosynthesis of small compounds (Figure 4A). These compounds include thiamine, heme, riboflavin, and folic acid. Three of these identified pathways, which are encoded by the pathogen of interest, do not exist in mammals. Since mammals do not have the enzymes required for thiamine biosynthesis, the enzymes that are involved in this process could be a viable target for the development of drugs to treat this infection. 149 In addition to thiamine, riboflavin is an essential biomolecule for the continued survival of an organism. However, like thiamine, mammals do not encode the enzymes necessary for the production of riboflavin, which demonstrates their suitability for use in the discovery of new drugs. 150 The potential druggability of the folic acid biosynthetic pathway for the development of therapeutic intervention has been experimentally validated by the use of drugs inhibiting folic acid biosynthesis.151,152

Protein-protein interaction analysis of the annotated HPs. The identified protein clusters include proteins involved in (A) biosynthesis of small compound, (B) DNA binding, and (C) biofilm formation. HP indicates hypothetical protein.
A protein cluster that can bind to DNA was also observed (Figure 4B). Proteins necessary for DNA synthesis, DNA mismatch repair, and homologous recombination have been found in this cluster. Virulence proteins play a pivotal role in pathogenesis. There are two ways that virulence proteins can be transported: they can either be bound to the membrane or secreted through the secretion system. In this study, we were able to identify proteins that are necessary for the production of biofilms, in addition to components of the type VI secretion system (T6SS) (Figure 4C).
Discussion
A significant portion of the proteomes of both prokaryotic and eukaryotic organisms are made up of hypothetical proteins (HP). 153 Hypothetical proteins are proteins that are assumed to be expressed from an open reading frame (ORF), although they have no experimental evidence of translation. Given the significance of understanding the underlying molecular mechanisms that are present in a variety of species, notably pathogenic bacteria, a number of studies have emphasized in past few years on the annotation of proteins that have not yet been assigned a function.154,155 The wet lab approaches that are commonly employed for unraveling desired genes and proteins are time-consuming and expensive. As a result, the in silico methods have arisen as crucial tools for identification of the hypothetical proteins and assigning their function.21,156-158 Wet lab experiments have been used to validate the reliability of the in silico functional annotation approach. 154 An integrated in silico and in vivo method was used to functionally annotate the hypothetical proteins, which led to the identification of the proteins of Pseudomonas sp. Lz4 W involved in cold adaptation 158 and also the high arsenic resistance genes from Exiguobacterium antarcticum strain B7. 21 These evidences have provided the impetus for the development of computational tools and techniques that have a higher degree of accuracy in elucidating the functions of HPs.
The purpose of this study was to annotate the HPs with the aim of filling the information gaps of the genome sequence analysis through the use of computational approaches. Identification of protein family and domain, subcellular localization prediction, secretome analysis, and protein-protein interaction analysis were among the approaches used. The development of new computational tools and the increased availability of existing ones have both had a favorable impact on the understanding of the correlation between the structure and function of proteins. In addition, these tools provided a basis of in vitro assays for the characterization of proteins. The urgency of properly understanding the roles that the hypothetical proteins play in the pathogenesis of V. parahaemolyticus and refining the functional annotation for future research makes this study essential.
To improve the accuracy of computational predictions, we devised a functional annotation pipeline based on sequence analysis coupled with the implementation of other approaches such as PPI (Figure 1). The conservation of structural properties may imply a function for some HP, which can then be annotated according to that function. These structural properties were identified through the integration of data that was available from multiple databases, evaluating conserved domains, families, and superfamilies. As a conclusion, using the pipeline, the functions of 656 of the 2631 HP have been annotated (Supplementary Table 2). We found several proteins that could be exploited as therapeutic and vaccine targets among the annotated HPs.
We discovered a varied array of proteins in the HPs by analyzing their GO keywords, ranging from enzymes essential for survival to structural components of the secretion system critical for virulence (Figure 2). These proteins are essential for a variety of cellular activities, and they also have the ability to play a role in the growth and survival of the organism. Enzymes are an essential component for the continued survival of an organism. In the case of bacteria, enzymes are not only important for promoting the growth and development of bacteria within their host by providing the necessary nutrients, but they are also responsible for the pathogenesis of infections. Enzymes have the ability to alter the local environment in such a way that it becomes favorable for the growth of bacteria as well as the metabolism of compounds that are available within the host. 159
Thiamin, also known as vitamin B1, is an essential cofactor required for the metabolism of carbohydrates as well as branched-chain amino acids when it is present in its active form, thiamin diphosphate.160,161 Although the majority of bacteria, as well as fungi and plants, are capable of de novo synthesis of thiamin, animals are incapable of thiamine synthesis and therefore must obtain essential thiamin entirety through their diet. 162 Recent research has pointed to the thiamin biosynthesis pathway of Plasmodium falciparum, the etiological agent causing tropical malaria, as a potential source of therapeutic targets.163,164 It stands to reason that the enzymes involved in the biosynthesis of thiamine and the regulatory network that underlies this process should be good candidates for use as therapeutic targets in the development of novel antibacterial agents. We were able to identify the enzyme known as thiamin phosphate synthase (ThiE), a potential drug target which is responsible for catalyzing the coupling of thiazole with pyrimidine. 165 In a positive context, we identified another enzyme, GTP cyclohydrolase-2, that is essential for the survival of bacteria. This enzyme is involved in the pathway leading to the biosynthesis of riboflavin (Vitamin B2) and is a prominent antimicrobial drug target since animals do not possess this enzyme.90,166
During the characterization of these two strains, it was surprising to find that both of them were resistant to a wide range of antibiotics.167,168 Both of the strains that were identified were able to demonstrate resistance to a wide variety of antibiotics, which may be explained by the presence of a large cluster of MFS proteins as well as GNAT family N-acetyltransferase proteins within their genome. This observation may shed light on how the strains gained this resistance. It can be hypothesized that the presence of a large number of multidrug resistance proteins and their expression have facilitated the evaluation of the pathogen to adopt mechanisms to demonstrate broad spectrum antibiotic resistance.169,170
In addition to the TDH and TRH, there were a few other hemolysin toxin proteins found in this study. This is an important observation that warrants more investigation. Enterohemolysin EhxA and phospholipase C were found to be present, and both of these proteins are essential for evading the tissue system of the host.115,171-174
Since virulence factors facilitate the colonization of the pathogen at the cellular level of the host and cause disease by evading the host defense mechanisms, it is necessary to have an understanding of the biological function and mechanism of virulence factors to comprehend the role they play during the pathogenesis of bacteria.175,176 Also, in the case of bacterial infections, virulent factors (Supplementary Table 8) have the potential to be used as drug target.177,178 When combined with antibiotics, annotated virulent HPs can contribute to the development of a more effective target-based drug discovery approach and facilitate the treatment of bacterial infections. In the event that these virulence factors are able to circumvent the host’s immune system, the final outcome will be either the sufficient multiplication to establish an infection or persistence of the microbe within the host tissue. It is possible that this will either cause considerable damage to the tissue of the host or enable transmission of the pathogen to other susceptible hosts. 179 Small molecule inhibitors and antibodies are two examples of the types of strategies that can be employed to suppress the activity of the virulence factors. 180 Consequently, the development of a vaccine that targets the virulence factors of these pathogens might be an additional strategy for tackling them.180,181 Throughout the course of our research, we were able to identify a number of virulence factors that hold promise as future areas of concentration for vaccine research (Table 1). The characterization of these proteins warrants for additional research to be conducted.
In the current study, functional and physicochemical characteristics of the HP that are encoded by two pathogenic strains of V. parahaemolyticus were able to be elucidated. This allowed for a better understanding of the role that the HPs play during the growth and pathogenesis of the bacteria. It is conceivable, in context of the molecular mechanisms underlying bacterial pathogenesis, that the virulent proteins reported in this study induce hemolysis and cell damage, which result in AHPND and shrimp mortality.
The findings of this study have provided significant information about proteins that are present within the two strains of this pathogen but roles were previously unknown. Our findings lead to the possibility of further research involving these targets, particularly to explore the role of these proteins in vivo and/or in vitro, as well as their potential to play a role in pathogenesis and their application for the development of novel therapeutic targets against V. parahaemolyticus.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322221136002 – Supplemental material for Functional Analysis of Hypothetical Proteins of Vibrio parahaemolyticus Reveals the Presence of Virulence Factors and Growth-Related Enzymes With Therapeutic Potential
Supplemental material, sj-docx-1-bbi-10.1177_11779322221136002 for Functional Analysis of Hypothetical Proteins of Vibrio parahaemolyticus Reveals the Presence of Virulence Factors and Growth-Related Enzymes With Therapeutic Potential by Sazzad Shahrear, Maliha Afroj Zinnia, Md. Rabi Us Sany and Abul Bashar Mir Md. Khademul Islam in Bioinformatics and Biology Insights
Footnotes
Acknowledgements
We acknowledge high performance computing facility support from Center for Bioinformatics Learning Advancement and Systematics Training (cBLAST), University of Dhaka. We also acknowledge support of Biomolecular Research Foundation (BMRF), Dhaka, Bangladesh.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
ABMMKI conceived the project. SS collected the data. SS, MAZ, and ABMMKI performed the analyses. SS, MS, and ABMMKI wrote the manuscript. The manuscript was reviewed and approved by all authors.
Data and Software Availability
All data are added in the table, figures, and supplementary file and supplementary tables. In this research work, publicly available, free, mostly online and few offline software/tools were used. Necessary link and reference of the software/tools are provided in the method section.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.