
Abstract
Enterobacter cloacae B13 strain is a rod-shaped gram-negative bacterium that belongs to the Enterobacteriaceae family. It can cause respiratory and urinary tract infections, and is responsible for several outbreaks in hospitals. E. cloacae has become an important pathogen and an emerging global threat because of its opportunistic and multidrug resistant ability. However, little knowledge is present about a large portion of its proteins and functions. Therefore, functional annotation of the hypothetical proteins (HPs) can provide an improved understanding of this organism and its virulence activity. The workflow in the study included several bioinformatic tools which were utilized to characterize functions, family and domains, subcellular localization, physiochemical properties, and protein-protein interactions. The E. cloacae B13 strain has overall 604 HPs, among which 78 were functionally annotated with high confidence. Several proteins were identified as enzymes, regulatory, binding, and transmembrane proteins with essential functions. Furthermore, 23 HPs were predicted to be virulent factors. These virulent proteins are linked to pathogenesis with their contribution to biofilm formation, quorum sensing, 2-component signal transduction or secretion. Better knowledge about the HPs’ characteristics and functions will provide a greater overview of the proteome. Moreover, it will help against E. cloacae in neonatal intensive care unit (NICU) outbreaks and nosocomial infections.
Introduction
Enterobacter is a genus of gram-negative, facultative anaerobic, and rod-shaped bacteria. It is a member of the family Enterobacteriaceae. 1 Bacteria which belong to the Enterobacteriaceae are most often isolated from soil, water, or different clinical specimens. 2 Enterobacter cloacae is an integral part of microflora in the human and animal intestinal tracts. 3 E. cloacae complex are largely observed in nature, although they can act as pathogens. 4 E. cloacae has emerged as a significant pathogen to study because of its several outbreaks including neonatal intensive care units (NICU).5 -8 It has become a problematic pathogen for healthcare institutes at a global level as it tends to contaminate various hospital devices. 4 Up to 5% of all hospital-acquired sepsis and nosocomial pneumonia, 4% of nosocomial urinary tract infections (UTIs), and 10% of postsurgical peritonitis cases are caused by E. cloacae.9,10 This microorganism’s transmission to neonates can be accompanied through contaminated intravenous fluid or medical equipment along with the possibility of inpatients acting as a reservoir. 5 In general, they contact mostly through the GI tract and skins in humans.3,11
E. cloacae’s mechanism of pathogenesis is multifactorial and complicated as it involves a couple of virulence factors, whose role in the disease development is still unclear. 4 Production of enterotoxins such as cytotoxin similar to Shiga-like toxin II after their adhesion to epithelial cells, type III secretion system (TTSS) with several virulence factors, and phagocytes destroying capability might contribute to E. cloacae’s pathogenicity.12 -14 Furthermore, induced apoptosis of HEp-2 cells might be a primary strategy of this microorganism to destruct tissues, spread and cause infection or disease. 15 Some studies also pointed to the colony formation ability of Enterobacteriaceae which mediates their binding to host proteins, resulting in cell adhesion and invasion. 16 Curli genes expression and curli fimbria demonstrated a correlation between the biofilm formation and morphology of E. cloacae. 17 In addition, E. cloacae complex has exhibited multidrug-resistance phenotype with their intrinsic β-lactam resistance and genes encoding antibiotic resistance, such as carbapenemase genes.18,19 This multidrug-resistance ability of E. cloacae can emerge as a global threat. 18
The recent NGS technology can produce a huge amount of genomic data for a wide array of bacteria. However, the lack of complete proteome data because of coding sequences without a proper prediction of functions has made it difficult to understand pathogenesis and virulence determination. These molecules are labeled as hypothetical proteins (HP).2,20,21 Nearly 30% to 40% genes of most bacterial genomes are classified as unknown or hypothetical. 22 These HPs are the translated nucleic acid sequences based on sequence similarity, but their biochemical and functional characterization evaluation is necessary for the experimental existence. 23 Therefore, the functional annotation of many hypothetical proteins has become an important focus in bioinformatics. 24 Homology-dependent gene annotation can assign functions to HPs based on their correlation with known proteins, providing the knowledge of new structures, functions, interactions, and pathways.23 -27 A well and precise annotation of E. cloacae HPs can bring additional protein pathways and cascades to our understanding, thus decreasing the gap of knowledge between genome data and protein functions. 27 The identification of proteins and their roles in bacterial growth and pathogenesis could be aided by in silico functional annotation of hypothetical proteins. In vitro and in vivo studies have verified the reliability of the in silico functional annotation approach. The proteins of Pseudomonas sp. Lz4W involved in cold adaptation, as well as the high arsenic-resistance genes from Exiguobacterium antarcticum strain B7, were identified using an integrated in silico and in vivo approach to functionally annotate the hypothetical proteins.20,28
As E. cloacae’s pathogenic ability has already helped it to cause multiple NICU outbreaks, deciphering its HP functions in the genome is essential to completely understand the mode of pathogenesis.2,5 -8 Moreover, novel HPs from E. cloacae can be used as markers and pharmacological targets for drug designing and screening, helping to prevent outbreaks.29,30
Many proteins from E. cloacae B13 strain are still uncharacterized, which might contain crucial functions in their life. In this study, several in silico approaches were utilized to predict the function of the HPs from this organism. Identifying the functions of the HPs will contribute to a better proteome knowledge of the bacteria and its contribution to virulence. This will help to fight against E. cloacae as an emerging threat in nosocomial infections and hospital outbreaks.
Materials and Methods
The methodology overview flowchart is presented in Figure 1.

Methodology overview flowchart for the functional annotation and analysis of E. cloacae B13 strain hypothetical proteins (HP).
Extraction of genomic data
E. cloacae B13 genome was used in this study. This strain was isolated from human urine sample in Bangladesh and its genome size is 4 963 112 bp and encodes 4707 proteins.
The entire sequence of E. cloacae strain B13 was downloaded from NCBI sequence set browser with accession PRJNA472680. 31 Among 4707 proteins, sequences of the HPs were retrieved using the fasta_extract tool in Galaxy. 32
Gene ontology prediction
To determine the HP’s functions, Blast2GO with an E-value 1e−03 was used. Blast2GO is a bioinformatics tool which can be used for high-throughput functional annotation of DNA or protein sequences based on the Gene Ontology (GO) vocabulary. 33 The protein sequences with GO IDs were selected and various bioinformatics tools were used for further analysis of their domain and functions.
Family and domain prediction
The conserved domains and protein functions were searched based on the structure of domains. So, Simple Modular Architecture Research Tool (SMART) was used in general mode to identify and annotate genetically mobile domains of signaling, extracellular and chromatin-associated proteins. 34 Furthermore, NCBI Batch CD-Search was applied that allowed to search multiple protein sequences using RPS-BLAST to compare query HP sequences against databases of conserved domain models. 35 NCBI Batch CD-Search tool searched against CDD—58235PSSMs database, and threshold was set at 0.01.
The HMMER website, allows protein homology search algorithms within the HMMER 3.3.2 software suite and uses profile hidden Markov model libraries to annotate the HP sequences with protein families and domains. 36 The cut off was set at 0.01 for significant e-values. Superfamily and Pfam association of the HPs from HMMER was searched. SUPERFAMILY 2.0 database contains superfamily domain annotations for millions of distinct proteins obtained from UniProtKB and NCBI. 37 In addition, annotated protein families of Pfam database are represented by Multiple Sequence Alignment (MSA) and hidden Markov Model (HMMs). 38 We searched for InterPro IDs for the selected HPs using Blast2GO utilities. InterPro uses predictive models, provided by several databases to provide functional analysis of proteins by classifying families and domains.33,39 So far, the functions of HPs with tools like Blast2GO, NCBI Batch CD-Search, SMART, SUPERFAMILY, Pfam, and INTERPRO were predicted and the HPs with predicted functions by 3 or more tools were identified with the help of InteractiVenn. 40 Finally, the Basic Local Alignment Search Tool (BLAST) against the NCBI nonredundant (nr) database was used to identify the annotated homologous proteins from related organisms, with similarity at ⩾90%.20,41
Subcellular localization determination
In the study, PSORTb v3.0 and CELLO v.2.5: Subcellular Localization Predictor were used to determine the cell locations of the HPs using default parameters for gram-negative bacteria.42 -44 PSORTb database contains information obtained from both laboratory experiments and computational prediction. 45 A 2-level support vector machine (SVM) is used in CELLO, which involves 4 SVM classifiers and the final assignment is determined by using the jury votes from these classifiers.20,43
Determination of transmembrane proteins
TMHMM 2.0 and HMMTOP 2.0 at default parameters were performed for the prediction of transmembrane helices and topology of the HPs in the study.46,47 SignalP 5.0 helped to predict the presence of signal peptides and cleavage site location, which performs through a neural network architecture involving a conditional random field. 48
Physicochemical prediction parameters
To compute several physical and chemical parameters of the HPs, such as molecular weight, theoretical pI, amino acid composition, instability index, aliphatic index, extinction coefficient, and grand average of hydropathicity (GRAVY), ProtParam tool in Expasy was used. 49
Virulent HP detection
MP3 is a tool which can accurately predict virulent proteins in genomic and metagenomic data using SVM and HMM approach. 50 DeepVF uses a deep learning-based hybrid framework to identify virulence factors more accurately by relying on machine learning. 51 Blast search tool in the Virulence Factor Database (VFDB) identified various virulent factors from the submitted HPs. VFDB contains information about virulent factors from several bacterial pathogens. 52 Finally, PHI-base was used for virulent factors detection as it contains curated information on pathogen-host interaction affecting genes based on research articles. Only lethal and hypervirulence proteins were selected after completing a blast search (PHIB-BLAST) in Phi-base against PHI-base 4.12 protein sequences. 53 Virulent HPs predicted by 2 or more tools were then identified and further analyzed.
Predictions of antigenicity, allergenicity, and toxicity index
The antigenicity of the virulent proteins was predicted using the VaxiJen v2.0 server 54 and the ANTIGENpro server. 55 Toxicity and allergenicity of those proteins were predicted using the ToxIBTL server 56 and the AllerCatPro v. 2.0 server, 57 respectively.
Protein-protein interaction
String 11.5 database was utilized to predict the protein-protein interactions (PPIs) for the proteins from E. cloacae B12 strain. 58 String database uses all publicly available PPI information to computationally predict direct (physical) and indirect (functional) forecast. To ensure the most reliable PPIs, only the interactions with score values above 0.700 (high confidence) and high FDR stringency (1%). 20
String 11.5 search for E. cloacae was completed against E. cloacae ATCC 13047 as the strain with highest similarities. Here, the identified interactions were transferred to E. cloacae by the interolog mapping method, which assumes that when 2 proteins interact, their orthologous pairs will interact too.42,59 -61 So, hierarchically arranged orthologous groups relations are applied by STRING to transfer association between applicable organisms as described in eggnog.62 -64
The network analyzer plugin in Cytoscape 3.9.0 program was utilized for the validation of PPI networks.65,66 Cytoscape 3.9.0 was used to obtain a better visualization of the potential virulent HPs with other proteins and among themselves. In Cytoscape, protein molecules are assigned to nodes and molecular interactions to edges. Furthermore, network analyzer tool can compute multiple network topological parameters with details of node degrees, edges, neighbor interactions, and network characteristics.
Results and Discussion
Functional annotation of E. cloacae HPs reveals their association with several biological, molecular, and cellular processes
A total of 604 proteins out of 4707 (12.83%) were labeled as HP in the E. cloacae B13 strain. Functional annotation of this large portion of proteins encoded by the bacterial genome was performed using various bioinformatic tools.
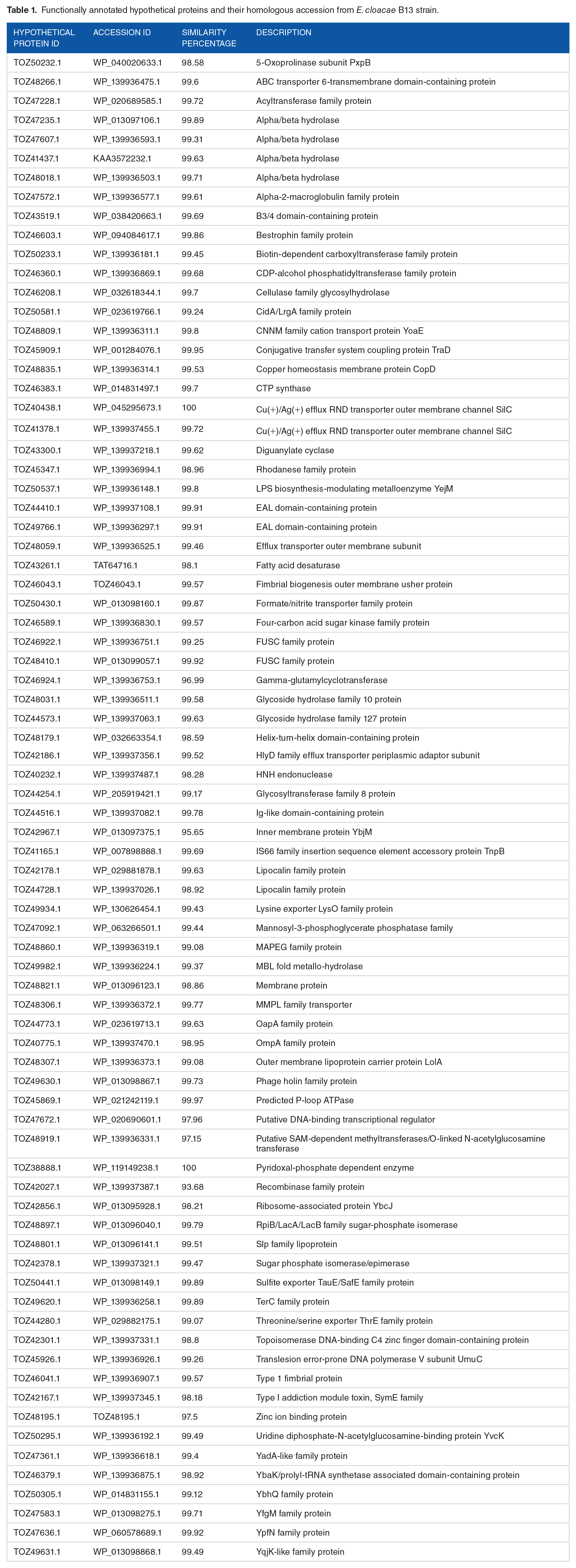
BLAST2GO was utilized to perform a primary prediction of the HPs, which returned 214 HPs with known protein domains or families along with their GO IDs (Supplementary Table 1). Further analysis of the pool of 214 HPs with NCBI Batch CD-Search, SMART, SUPERFAMILY, Pfam, and INTERPRO tools was performed to assign the functions (Supplementary Table 2). Among 214 HPs, functional characterization with strong confidence was possible for 78 HPs as they demonstrated similar functions predicted by 3 or more tools. NCBI BLASTp tool was used to manually annotate the functions of these 78 HPs according to their homologous proteins (Table 1). Multiple tools increase the reliability of the functional prediction. Moreover, as domains are protein’s fundamental unit of structure, folding and function, domain identification is crucial for annotating biological functions of a protein. 67
Functionally annotated hypothetical proteins and their homologous accession from E. cloacae B13 strain.
GO function analysis
Analysis of predicted GO terms for 78 HPs revealed their association in different GO categories: biological process, cellular components, and molecular functions (Figure 2). For biological process, 34 proteins were identified with distinct GO terms. About 12 of them were involved in protein transport and 18 proteins had functions in metabolic process. The cellular component category had 47 different GO terms, among which 38 were an intrinsic part of the membrane. Finally, among 53 GO terms in molecular functions, 33 were enzymes and 22 proteins were binding proteins.

GO categories distribution of the HPs from E. cloacae B13 strain. The figure shows the detailed categories, number and percentage of proteins in (A) molecular functions (B) cellular components (C) biological processes. GO indicates Gene Ontology; HP, hypothetical proteins.
Enzymes
Enzymes produced by gram-negative bacteria play a significant role in their host as they provide support and nutrients for growth, ensure favorable growth by modifying local environment, conduct the pathogenesis of several infections and help in metabolism. 68 A total of 33 proteins were characterized as enzymes, among which 15 proteins are hydrolase and 11 proteins are transferase.
Analysis of several infections by gram-negative anaerobes, involving tissue invasion and inflammation, necrosis, or suppuration, has revealed that hydrolytic enzymes have roles in pathogenesis of infection. 68 Furthermore, study of different hydrolases has supported their potential role in pathogenesis.69 -72 Four proteins TOZ47235.1, TOZ47607.1, TOZ41437.1 and TOZ48018.1 were identified as the α/β hydrolase that are likely to be involved in the immune system evasion and modulation, detoxification, and metabolic adaptation. 73 The α/β hydrolases have also been found to play a major role as virulence factors in Mycobacterium tuberculosis and Staphylococcus aureus.73,74 TOZ40232.1 was predicted as endonuclease protein which functions by stopping the invasion of foreign DNA. 75 TOZ47235.1 was also predicted to have oxidoreductase activity, which are critical for bacterial virulence and pathogenesis. 76 Bromoperoxidase A2 protein is a metal-ion-free oxidoreductase from Streptomyces aureofaciens, which has both hydrolase and oxidoreductase activity and, has been found to contain an alpha/beta hydrolase fold. 77
Similarly, 11 proteins were identified as transferase enzymes. They are necessary for lipoprotein biosynthesis, spore germination, and aid the full virulence of bacteria. 75 TOZ44254.1 was predicted as glycosyl transferase protein. Glycosyl transferase family proteins can alter extracellular polysaccharide and lipopolysaccharide synthesis upon mutation, resulting in the reduction in disease symptoms.78,79 TOZ46360.1 and TOZ50295.1 were predicted to be CDP-alcohol phosphatidyl-transferase family protein and UDP-GlcNAc. Both of the families are associated with lipid biosynthesis.80 -82 Alteration of the synthesized phospholipid has a crucial role in virulence and several human diseases.83,84
TOZ38888.1 and TOZ41165.1 were predicted as lyase enzymes. Lyase enzymes have essential functions for the virulence of pathogenic gram-negative bacteria in host. 68 TOZ38888.1 is pyridoxal-phosphate (PLP)-dependent enzyme, which are a ubiquitous class of biocatalysts. In several free-living prokaryotes, PLP-dependent enzymes are encoded by almost 1.5% of all genes. 85 PLP-dependent enzymes with desulphydrase activity help in amino-acid metabolism, adaption to nutrient sources in a new environment, and sometimes can function as virulence factors.86,87
TOZ48897.1 was annotated as RpiB/LacA/LacB family sugar-phosphate isomerase. This family of proteins takes part in the lactose catabolism pathway. 88
Binding proteins
There are 22 proteins characterized as binding proteins, among which 5 proteins were DNA binding, 3 were RNA binding and 5 were ATP binding ones. HPs with DNA-binding function can contribute to the virulence by altering the expression of virulence factors, which have been observed during S aureus infection. 89 TOZ48179.1 was characterized as helix-turn-helix domain-containing protein. HTH domain containing protein has a large range of functions, such as, DNA repair and replication, RNA metabolism, PPI and 2-component signaling pathway, while 2 component signal transduction system (TCS) are largely used as a target for antimicrobial therapy.90 -92 TOZ45926.1 was a translesion error-prone DNA polymerase and TOZ42027.1 was a recombinase family protein, both of which might have functions in DNA repair.93,94
TOZ46383.1 was found to be a CTP synthase, which converts UTP to CTP, a necessary step in pyrimidine metabolic pathway in community-acquired respiratory tract infection (RTI) causing bacteria. 95 In addition, TOZ48266.1 was identified as an ABC transporter 6-transmembrane domain-containing protein, which are considered to have roles in nutrient uptake and drug resistance. Moreover, evidence of ABC transporters being directly or indirectly involved in the bacterial virulence has been found. 96 Furthermore, TOZ50233.1 was characterized as a biotin-dependent carboxyltransferase protein. They have roles in fatty acid, amino acid and carbohydrates metabolism.97 -100 Furthermore, their activity plays important role in the virulence of organisms like Listeria monocytogenes and Candida albicans.101 -103
Transporter proteins
Eight proteins were characterized to have transmembrane transporter activity. TOZ50430.1 was characterized as formate/nitrite transporter (FNT) protein. Bacterial FNTs monitor the transport of small monoacids. 104 In addition, FNTs can perform as a virulence factor in Salmonella species by helping the bacteria to evade killing from activated macrophages in host. 105 TOZ40438.1, TOZ41378.1, and TOZ48059.1 are efflux transmembrane transporter proteins, and the first 2 are Cu(+)/Ag (+) efflux RND transporters. RND transporters are necessary for the multidrug resistance in several pathogens. 106 Furthermore, RND superfamily transporters are organized as tripartite efflux complexes and span inner and outer membrane of cell envelope. 107 Moreover, RND transporters specific to heavy metals in E coli have been found to raise resistance to copper(I) and silver(I) ions. 108
Regulatory proteins
Regulatory process is a complex network system in bacteria that helps in various gene expression and maintain bacterial pathogenesis, growth, and survival. 2 TOZ43300.1 was identified as a diguanylate cyclase which has functions in cellular process regulation and signal transduction. Interestingly, diguanylate cyclase is necessary for biofilm development. It also performs as a messenger for bacterial virulence, motility, adhesion, secretion, and community behavior. 109
TOZ47572.1 was predicted as an alpha-2-macroglobulin (A2M) protein, which can structurally mimic proteins of eukaryotic innate immunity in invasive bacteria. Bacterial A2M are located in periplasm where they trap external proteases and provide cellular protection. 110 Both pathogenically invasive and saprophytically colonizing species possess A2M and mostly exploit higher eukaryotes as hosts. Therefore, bacterial A2M can be used as useful targets to increase vaccine efficacy in infections. 111
Membrane protein
A total of 38 proteins were characterized as integral component of the membrane and 1 protein as extrinsic component of the membrane. TOZ40775.1 was annotated as OmpA family protein. This family of proteins is surface-exposed porin proteins with anti-parallel β barrels in the outer membrane. 112 HMMTOP and TMHMM also predicted the presence of transmembrane helices for this protein (Supplementary Table 4). Several pathogenic roles including adhesion, invasion, intracellular survival, and host defenses have been assigned to OmpA. In various cases, OmpA proteins are being considered as potential vaccine candidates. 112
TOZ49620.1 was annotated as a TerC family protein. This type of protein is largely found in bacteria species and may influence host-pathogen interaction. 113 Moreover, TerC family proteins in Bacillus subtilis have been found to help prevent Manganese (Mn) intoxication. Mn is essential for virulence for many pathogens. 113 Mn detoxification helps in oxidative stress resistance and virulence in S aureus. 114
TOZ44410.1 and TOZ49766.1 were both characterized as EAL domain-containing proteins. EAL domain is a ubiquitous signal transduction protein domain involved in hydrolysis of second messenger cyclic dimeric GMP (c-di-GMP) as it is the exclusive substrate of EAL.114,115 The second messenger c-di-GMP regulates many lifestyle aspects and virulence of several gram-negative bacteria. 116 Moreover, EAL domain protein VieA from Vibrio cholerae inversely regulate biofilm-specific genes (vps) and virulence genes like ctxA by decreasing the amount of cellular c-di-GMP. This phenomenon is of particular interest as the shift in gene expression plays a major role in V. cholerae life cycle. 117 Upon entering to a host, V. cholerae tends to undergo a shift in gene expression, where vps expression ceases 118 and virulence genes are expressed.119 -123
Virulent protein prediction
MP3, DeepVF, VFDB, and PHI-base were used for virulence factor prediction with high confidence level. A total of 23 HPs were predicted by 2 or more tools to be virulent, and the remaining HPs were identified by either only one tool or not virulent at all (Supplementary Table 3). As virulence factors help bacteria to colonize and cause disease, the knowledge of biological function and mechanism of the virulence factors is necessary to understand their role in the pathogenesis of bacteria. 2 Moreover, virulent factors are potential therapeutic targets in case of bacterial infections. 124 Characterizing virulence factors include several secretion systems (Type I to Type VI secretory systems) 2-component signal transduction systems, quorum sensing, and biofilm formation.125,126 Virulent proteins are utilized by a large number of pathogenic bacteria, and therefore identifying inhibitors against essential factors for virulence factors is a new research interest, which is a different molecular approach than traditional drug discovery. 127 Annotated virulent HPs can obtain a better target-based approach and aid against bacterial infections as a subsidiary therapy to different antibiotics. 125
Virulent HPs with therapeutic potential
Antigenicity of the virulent HPs was studied, and it was observed that 7 of them have antigenic potential. All of these 7 proteins are likely to be non-allergenic and nontoxic. The subcellular localization of the protein was also explored, and we observed that the 7 antigenic proteins were either membrane bound or periplasmic proteins (Table 2). Our findings suggest that each of these 7 proteins could be a great candidate for vaccine development.128 -131
Prediction of antigenicity, allergenicity, toxicity, and subcellular localization of the virulent HPs.

Abbreviation: HPs, hypothetical proteins.
Subcellular localization and physiochemical prediction
In the study, amino acid sequences of 78 HPs were analyzed by using various tools, such as PSORTb v3.0, CELLO v.2.5, TMHMM 2.0, HMMTOP 2.0 and ProtParam for assessing their subcellular location along with physiochemical prediction (Supplementary Table 4). However, more attention was paid to the virulent HPs that were predicted to have roles in pathogenesis.
The cellular location along with secretion or signaling ability and transmembrane helices of the 23 HPs were predicted. Nine of them were found to have transmembrane helices predicted by both HMMTOP and TMHMM (TOZ49766.1, TOZ48809.1, TOZ40775.1, TOZ44410.1, TOZ43300.1, TOZ45909.1, TOZ49630.1, TOZ47361.1, and TOZ42186.1). About 19 proteins out of 23 were predicted by CELLO to be an inner or outer membrane and periplasmic proteins. However, pSORTdb predicted 9 proteins as cytoplasmic or cytoplasmic membrane proteins, and 7 proteins as outer membrane proteins. The SignalP 5.0 server predicted 10 proteins out of 23 to contain signal peptides for several secretion pathways. About five of them were predicted to be standard secretory signal peptides and cleaved by Signal Peptidase I. In addition, 5 more proteins were predicted to be lipoprotein signal peptides and cleaved by Signal Peptidase II. All ten proteins were predicted to be transported by the Sec translocon.
The pH at which no net electric charge of a molecule remains and does not move in an electric field of direct current is the theoretical pI.132,133 For the virulent proteins, the theoretical pI ranged from 4.58 to 9.47. Again, these 23 virulent HPs molecular weight ranged from 11390.68 to 179998.3. 2D gel electrophoresis visualization in laboratorial experiments can be accompanied by the combination of these 2 parameters. The extinction coefficient of the virulent HPs at 280 nm ranged from 8450 to 228165 M−1 cm−1 with respect to the Cys (cysteine), Trp (tryptophan), and Tyr (tyrosine) concentration. The extinction coefficient indicates the amount of light absorbent by a protein at a specific wavelength, which is useful for purifying and separating a protein in spectrophotometer. In addition, high extinction coefficient occurred in some HPs because of the presence of high concentration of Cys, Trp, and Tyr.132,134,135 The instability index estimates the stability of a protein in test tubes. Proteins with less than 40 instability index are predicted as stable proteins. 136 In the study, the 23 predicted virulent HPs instability index ranged from 20.3 to 59.09, and 16 out of 23 proteins were stable. Stable proteins have a longer half-life. The half-lives of several virulent effector proteins are integral to their function. For example, in Salmonella, virulent effector proteins’ half-life modulations are necessary for the pathogenic cellular functions. 137 Aliphatic index of a protein determines the relative volume obtained by the aliphatic side chains (alanine, valine, isoleucine, and leucine). Aliphatic index functions as a positive factor for the thermostability increase of globular proteins. 138 The aliphatic index of the virulent HPs in the study ranged from 60.98 to 124.62. Finally, the Grand Average of Hydropathy (GRAVY) value of a protein is the sum of hydropathy values of all amino acids divided by the number of residues in that sequence. 139 GRAVY values in the study ranged from -0.535 to 0.434, where 17 proteins had a score of < 0 and 6 proteins had a score of >0. Proteins with a GRAVY score <0 are considered to be relatively hydrophilic and proteins with GRAVY score >0 are relatively hydrophobic.139,140 This information can be helpful for localizing the proteins by identifying them as a globular protein or membranous protein. 132
PPI of virulent proteins
Interaction between proteins plays a fundamental role in the biological processes of an organism. 141 Through PPI, protein cellular functions can be analyzed since execution of a function depends on the contact or regulatory interactions with another protein.60,142 Furthermore, PPI can be useful to infer an unidentified or hypothetical protein function based on the evidence of their interaction with known proteome of a particular organism as it is rare for a protein to interact with different biomolecules. Therefore, the PPI network is required to understand protein function and complexity as well as biological networks and pathways.60,143,144
PPI network analysis was performed for the 23 predicted virulent proteins to identify their functions and roles in pathogenesis. Only 20 of them were identified by STRING (Supplementary Table 5) and interactions between them and other E. cloacae ATCC13047 proteins were evaluated as B13 strain was not present in String database (Fig S1).
TOZ48059.1 is an efflux transporter outer membrane subunit protein which interacts with 18 different proteins. This protein has strong interaction with 2 two-component system sensor kinase proteins, a multidrug efflux periplasmic linker protein and macrolide transporter ATP-binding/permease protein (ECL_A036, ECL_04898, ECL_00055, ECL_02770). These proteins help bacterial survival against antibiotics and in virulence.125,145 -147 This protein also interacts with at least 5 cus proteins (cusA, cusB cusF, cusR, cusS). Cus protein complex helps in maintaining copper homeostasis and mediates resistance to copper stress by cation efflux.148,149 Toxic properties of copper are often harnessed by the innate immune system, which helps the host to kill bacteria. Bacteria counter this defense by relying on genes for copper tolerance for virulence within the host. 150
The proteins TOZ41378.1 and TOZ40438.1 are Cu(+)/Ag (+) efflux RND transporter outer membrane proteins and demonstrated interactions with 18 and 16 proteins, respectively (Fig S1). They strongly interact with each other along with TOZ48059.1 and most of its interactive proteins. These 3 proteins remain in one cluster. The protein cluster appears to bear the function of 2 component regulatory system with high strength (Log10 observed/expected value is 1.43). Majority of the interacting proteins also contain Histidine kinase domain, and GAF domain, which are associated with osmoregulation, hyphal development and virulence in bacteria like Agrobacterium tumefaciens and Candida albicans.151 -154
TOZ48307.1 is an outer membrane lipoprotein carrier protein, which interacts with 15 other proteins. These proteins also form a cluster (Fig S1). TOZ48307.1 interacts with 4 acyl carrier proteins (ACP) (ECL_04843, ECL_04852, ECL_048550, and ECL_04854). In Pseudomonas aeruginosa, Acp3 has been found to be involved in oxidative stress response and Acp1 and Acp3 each contribute to the virulence. 155 Only Acp1 functions in the fatty acid synthesis in P aeruginosa 156 and fatty acid plays a multifactorial role in controlling bacterial viability and virulence in this organism.157 -159
Finally, TOZ43300.1, which was predicted as a diguanylate cyclase, interacts with 115 different proteins (Figure 3). Interacting proteins were mostly related to 2-component regulatory system, biofilm formation, diguanylate cyclase activity, and intracellular signal transduction. Environment factors helps to induce bacterial biofilm formation, which are microbial multicellular communities encased within extracellular matrix. Two-component signal transduction system (TCS) strategy is used by bacteria to connect input signals change in environment to changes in physiological output, and coordinate input signals to control biofilm formation. 160 In several E. cloacae outbreaks, biofilm formation has been suspected of contributing to its pathogenicity.4,161,162 This is alarming because biofilms can show resistance against antibiotics in nosocomial infections and almost 65% of the microbial and 80% of the chronic infections are related to biofilm formation. 163 In addition, fight against E. cloacae in NICU infection and outbreaks is still a major challenge. 164 Moreover, many interacting proteins strengthen the functional prediction of the protein.

Protein-protein interaction network of protein TOZ43300.1, which is a diguanylate cyclase.
Conclusions
Hypothetical proteins form a large portion of a bacterial proteome which play crucial biological roles. Identifying these proteins and their functional annotation will help us to understand about the organism in a better way. For this study, 78 HPs were from E. cloacae B13 strain was functionally annotated with high confidence. The pipeline used in the study obtained a great result and can be followed to assign function to HPs from different organisms. Most proteins were predicted as enzymes, binding proteins, transporter proteins, regulatory proteins or membrane proteins, and their subcellular localization and physiochemical parameters were crucial to the understanding of their characteristics. As E. cloacae is responsible for several outbreaks in hospitals and multidrug-resistant E. cloacae complex are an emerging global threat, the HPs were analyzed for their role in virulence. We identified several proteins which have potential role in virulence by incorporating antibiotic resistance activity, biofilm formation, quorum sensing, secretion pathway or others. The potential virulent proteins were further investigated for their interaction with other proteins. PPI helped to determine the relationship between these proteins and the known proteome of E. cloacae, which also strengthened our prediction of the virulence. Findings from this study will eventually help to fill the gaps in the proteome knowledge of E. cloacae and create the possibility to fight against nosocomial infections and NICU outbreaks caused by E. cloacae.
Supplemental Material
sj-jpg-1-bbi-10.1177_11779322221115535 – Supplemental material for Functional Annotation of Hypothetical Proteins From the Enterobacter cloacae B13 Strain and Its Association With Pathogenicity
Supplemental material, sj-jpg-1-bbi-10.1177_11779322221115535 for Functional Annotation of Hypothetical Proteins From the Enterobacter cloacae B13 Strain and Its Association With Pathogenicity by Supantha Dey, Sazzad Shahrear, Maliha Afroj Zinnia, Ahnaf Tajwar and Abul Bashar Mir Md. Khademul Islam in Bioinformatics and Biology Insights
Supplemental Material
sj-xlsx-2-bbi-10.1177_11779322221115535 – Supplemental material for Functional Annotation of Hypothetical Proteins From the Enterobacter cloacae B13 Strain and Its Association With Pathogenicity
Supplemental material, sj-xlsx-2-bbi-10.1177_11779322221115535 for Functional Annotation of Hypothetical Proteins From the Enterobacter cloacae B13 Strain and Its Association With Pathogenicity by Supantha Dey, Sazzad Shahrear, Maliha Afroj Zinnia, Ahnaf Tajwar and Abul Bashar Mir Md. Khademul Islam in Bioinformatics and Biology Insights
Supplemental Material
sj-xlsx-3-bbi-10.1177_11779322221115535 – Supplemental material for Functional Annotation of Hypothetical Proteins From the Enterobacter cloacae B13 Strain and Its Association With Pathogenicity
Supplemental material, sj-xlsx-3-bbi-10.1177_11779322221115535 for Functional Annotation of Hypothetical Proteins From the Enterobacter cloacae B13 Strain and Its Association With Pathogenicity by Supantha Dey, Sazzad Shahrear, Maliha Afroj Zinnia, Ahnaf Tajwar and Abul Bashar Mir Md. Khademul Islam in Bioinformatics and Biology Insights
Supplemental Material
sj-xlsx-4-bbi-10.1177_11779322221115535 – Supplemental material for Functional Annotation of Hypothetical Proteins From the Enterobacter cloacae B13 Strain and Its Association With Pathogenicity
Supplemental material, sj-xlsx-4-bbi-10.1177_11779322221115535 for Functional Annotation of Hypothetical Proteins From the Enterobacter cloacae B13 Strain and Its Association With Pathogenicity by Supantha Dey, Sazzad Shahrear, Maliha Afroj Zinnia, Ahnaf Tajwar and Abul Bashar Mir Md. Khademul Islam in Bioinformatics and Biology Insights
Supplemental Material
sj-xlsx-5-bbi-10.1177_11779322221115535 – Supplemental material for Functional Annotation of Hypothetical Proteins From the Enterobacter cloacae B13 Strain and Its Association With Pathogenicity
Supplemental material, sj-xlsx-5-bbi-10.1177_11779322221115535 for Functional Annotation of Hypothetical Proteins From the Enterobacter cloacae B13 Strain and Its Association With Pathogenicity by Supantha Dey, Sazzad Shahrear, Maliha Afroj Zinnia, Ahnaf Tajwar and Abul Bashar Mir Md. Khademul Islam in Bioinformatics and Biology Insights
Supplemental Material
sj-xlsx-6-bbi-10.1177_11779322221115535 – Supplemental material for Functional Annotation of Hypothetical Proteins From the Enterobacter cloacae B13 Strain and Its Association With Pathogenicity
Supplemental material, sj-xlsx-6-bbi-10.1177_11779322221115535 for Functional Annotation of Hypothetical Proteins From the Enterobacter cloacae B13 Strain and Its Association With Pathogenicity by Supantha Dey, Sazzad Shahrear, Maliha Afroj Zinnia, Ahnaf Tajwar and Abul Bashar Mir Md. Khademul Islam in Bioinformatics and Biology Insights
Footnotes
Acknowledgements
The authors acknowledge high performance computing facility support from Centre for Bioinformatics Learning Advancement and Systematics Training (cBLAST), University of Dhaka. The authors also acknowledge support of Biomolecular Research Foundation (BMRF), Dhaka, Bangladesh.
Author Contributions
ABMMKI conceived the project. SD, SS, and AT collected the data; SD, SS, and MAZ performed the analyses. SD, SS, MAZ, and ABMMKI wrote the manuscript. The manuscript was reviewed and approved by all authors.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data and Software Availability
All data added Table, figures and supplementary file and supplementary tables. In this research work publicly available free mostly online and few offline software/tools were used. Necessary link, reference of the software/tools provided in the method section.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.