
Abstract
Background:
Cronobacter sakazakii, a foodborne pathogen with a fatality rate of 33%, is a rod-shaped, Gram-negative, non-spore-forming bacterium responsible for causing meningitis, bacteremia, and necrotizing enterocolitis. Despite many unknown functions of hypothetical proteins in bacterial genomes, bioinformatic techniques have successfully annotated their roles in various pathogens.
Objectives:
The aim of this investigation is to identify and annotate the structural and functional properties of a hypothetical protein (HP) from Cronobacter sakazakii 7G strain (accession no. WP_004386962.1, 277 residues) using computational tools.
Methods:
Multiple bioinformatic tools were used to identify the homologous protein and to construct and validate its 3D structure. A 3D model was generated using SWISS-MODEL and validated using tools, developing a reliable 3D structure. The STRING and CASTp servers provided information on protein-protein interactions and active sites, identifying functional partners.
Results:
The putative protein was soluble, stable, and localized in the cytoplasmic membranes, indicating its biological activity. Functional annotation identified TagJ (HsiE1) within the protein, a member of the ImpE superfamily involved in the transport of toxins and a part of the bacterial type VI secretion system (T6SS). The 3-dimensional structure of this protein was validated through molecular docking involving 6 different compounds. Among these, ceforanide demonstrated the strongest binding scores, -7.5 kcal/mol for the hypothetical protein and −7.2 kcal/mol for its main template protein (PDB ID: 4UQX.1).
Conclusion:
Comparative genomics study suggests that the protein found in C. sakazakii may be a viable therapeutic target because it seems distinctive and different from human proteins. The results of multiple sequence alignment (MSA) and molecular docking supported HP’s potential involvement as a T6SS. These in silico results represent that the examined HP could be valuable for studying C. sakazakii infections and creating medicines to treat C. sakazakii-mediated disorders.
Introduction
Cronobacter sakazakii is a rod-shaped, motile, Gram-negative bacterium belonging to the Enterobacteriaceae family. While it infrequently causes illness in healthy adults, it has been linked with outbreaks in neonates (particularly premature infants), as well as isolated cases in highly immunocompromised individuals and the elderly. 1 Cronobacter sakazakii (C. sakazakii) has different kinds of strains, among all these strains Cronobacter sakazakii 7G is one of the strains. Its genome size is 4.3 Mb, having total gene number 4122, and its protein-coding gene number is 3899. 2 It can live in extremely dry conditions found in products like baby formula, protein shakes, powdered milk, and other dried foods.3,4 C. sakazakii infections are rare, they have been associated with meningitis and sepsis, particularly in newborns and young children.5,6 Due to its acid tolerance, C. sakazakii exhibits significant resistance to low pH environments. Additionally, this capacity of the bacterium to form biofilm enhances its resistance to antibiotics. 7 Premature babies may be particularly vulnerable because their stomach acid is not fully developed, even though adult gastric juice normally has a pH between 2 and 3. 5
Epidemiological studies have linked powdered infant formula (PIF) to over 90% of these Cronobacter illnesses. Contamination of PIF can occur during the manufacturing process, recovery, or storage. 8 Still, some Cronobacter species—C. sakazakii in particular—benefit from the growing conditions provided by dehydrated formulations. These bacteria are considered to be highly pathogenic, and their presence in baby formula raises serious concerns for public health since they can cause serious infections like meningitis and enterocolitis.5,9 Neonates comprise the majority of C. sakazakii infection cases. Reports indicate that case fatalities from necrotizing enterocolitis, meningitis, and sepsis can reach up to 33%. 10 C. sakazakii will therefore be the focus of this study. Despite the rarity of Cronobacter infections, the mortality rate can range from 33% to 80%.5,11,12 Newborns who survive Cronobacter infection often experience severe long-term effects, such as cerebral abscesses, developmental delays, and loss of hearing and vision. 13
The majority of disease-causing bacteria have become resistant to several medications. 14 Cronobacter species exhibit a higher degree of antibiotic resistance compared to other Enterobacteriaceae members. While limited studies have documented the presence of multi-drug resistance (MDR) in Cronobacter isolates from clinical and environmental sources, the underlying molecular mechanisms responsible for their antibiotic resistance remain largely unexplored. 15 It is recommended to treat neonatal infections caused by C. sakazakii with antibiotic therapy. 16 Most cases of Cronobacter spp. infections respond well to antibiotic therapy 17 and the conventional treatment typically involves a combination of ampicillin and gentamicin or chloramphenicol. 3 However, the proliferation of antibiotic-resistant strains has increased due to the overuse and improper administration of medications. 16 To address the issue of multidrug-resistant bacteria, it is crucial to identify new therapeutic targets given the proliferation of antibiotic-resistant strains. 14 Traditional drug development methods are characterized by substantial time and financial commitments. 18 Computer-based analysis utilizes various techniques to point out potential drugs and vaccines, design molecules based on structure, evaluate efficacy, study host-pathogen interactions, and conduct genome-based comparative research. This approach minimizes the need for extensive laboratory experiments.19,20
Next-generation sequencing (NGS) facilitates rapid data collection, yet determining gene functions remains a significant challenge.21,22 A significant proportion of bacterial genomes, ranging from 30% to 40%, consists of genes that are currently classified as hypothetical or of unknown function. 23 The biochemical and functional characterization of these hypothetical proteins is crucial for validating their roles. Bioinformatics plays a crucial role in annotating the functional characteristics of these hypothetical proteins, a process essential for drug discovery and development. Identifying these unknown proteins in-silico can improve the effectiveness of drugs and vaccines by uncovering biochemical pathways essential for bacterial survival and pathogenicity.24,25 This study utilized different bioinformatics tools to analyze the structural and functional aspects, as well as molecular docking, of a putative protein (accession no. WP_004386962.1) from C. sakazakii 7G. Figure 1 illustrates the overall procedure of this study.

An overview of putative protein annotation and the findings that followed.
Materials and Methods
Retrieval of FASTA (Fast alignment search Tool-All) sequence
About 1906 C. sakazakii genomes are accessible in the NCBI (http://www.ncbi.nlm.nih.gov/) 26 database. For this investigation, we filtered 257 hypothetical proteins from C. sakazakii and then we focused on a hypothetical protein composed of 277 amino acids, obtained from the C. sakazakii 7G strain (WP_004386962.1). The primary sequence of the protein was retrieved in FASTA format for subsequent analysis. Besides, we clarified that hypothetical proteins were filtered based on their annotation status in the NCBI database, with a focus on proteins of appropriate sequence length and completeness for downstream analysis. Additionally, we explicitly stated that proteins lacking sufficient annotation or with incomplete sequences were excluded in our study.
Exploration of physicochemical characteristics
The physicochemical properties of the protein were evaluated using the ProtParam tool (http://web.expasy.org/protparam) 27 available on the ExPASy server. The ProtParam tool calculates different physicochemical characteristics of proteins, which provide insights into their functional and structural characteristics. By analyzing a single protein sequence, ProtParam provided insights into the physicochemical properties of a protein, including molecular mass, stability, charge, amino acid composition, and hydrophobicity. This tool offers an accurate and efficient method for assessing the physical and chemical attributes of proteins. 28
Multiple sequence alignment (MSA) and phylogenetic tree evaluation
For the HP homologs (WP_004386962.1), a BLASTp (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins; Retrieved date: 5 Jan 2024) 29 search was performed against the NCBI non-redundant database using default parameters to identify homologous proteins. For determining homologous proteins, this method finds protein alignments locally. This study used MEGA 11 (version 11.0.13) software for Multiple sequence alignment and phylogenetic tree evaluation. 30 Using the FASTA sequence of our query protein (WP_004386962.1), along with 9 related proteins, we performed a multiple sequence alignment, WP_004386962.1, and homologous 9 proteins, WP_105653240.1, WP_063265155.1, WP_104671789.1, WP_076728080.1, WP_161584471.1, WP_105622741.1, ELY2789108.1, EGT4277351.1, and EJC1154322.1. Recent advancements in sequencing alignments have improved precision, scalability, and the ability to compare proteins with different domain designs, making them more efficient for inferring phylogenies and predicting protein structure. 31
Prognosis of protein solubility and subcellular localization
Understanding the subcellular distribution of proteins is crucial for identifying potential drug or vaccine targets. Membrane proteins can serve as therapeutic or immunological targets, whereas cytoplasmic proteins may be pharmacological targets. CELLO (http://cello.life.nctu.edu.tw/) 32 determined the subcellular location of a HP. To further validate the results, we utilized the PSORTb (https://www.psort.org/psortb) 33 and PSLpred (http://crdd.osdd.net/raghava/pslpred) 34 servers. These servers are commonly employed for forecasting the location of bacterial proteins within cells, providing additional confirmation for our findings. SOSUI (http://harrier.nagahama-i-bio.ac.jp/sosui/) 35 computes the solubility of a protein and average hydrophobicity.
Determining the motifs and domains of a protein
Domain evaluation was conducted using NCBI Conserved Domain (CD) Search Service (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) 36 and InterProScan (https://www.ebi.ac.uk/interpro/search/sequence/). 37 CD Search can be used to determine which domains in a protein sequence are conserved. RPS-BLAST was utilized to identify conserved domains within the query sequence by comparing it against itself using position-specific matrices from the Conserved Domain Database (CDD). 38 The Pfam (http://pfam.xfam.org/) 39 database, which provides protein family annotations and multiple sequence alignments generated from the Hidden Markov model (HMMs), served as the reference. Protein motifs were identified using the Motif tool on the Genome Net server (https://www.genome.jp/tools/motif/). 40 The coiled-coil structure of the protein was identified using the DeepCoil service. 41 In addition, protein folding patterns were found using the PFP-FunD SeqE server. 42
Structure determination and homology modeling of putative protein
The 2-dimensional structure of WP_004386962.1 protein was determined by PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred/) 43 and SOPMA (https://npsaprabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_sopma.html). 44 A correlation was found between the outcome of the PSIPRED study and the SOPMA result. The SWISS-MODEL program was employed to construct a 3D model of the target protein based on homology. 45 The service utilized BLASTp to identify suitable templates for the entire protein sequence. Based on the search results, the protein 4uqx.1.A was selected as the template for homology modeling. This X-ray diffraction model, with 38.28% sequence identity, represents a possible cytoplasmic protein from Pseudomonas aeruginosa and is a good base of reference for modeling. Further protein sequence was evaluated by the HHpred server (https://toolkit.tuebingen.mpg.de/tools/hhpred) using MODELLER (version 10.4) where we found 97% sequence identity. The 3D model structure was then displayed with BIOVIA Discovery Studio (version 20.1). A more accurate 3D model was generated using the highest-scoring template. This model was further refined and improved over time using the YASARA energy minimization server using steepest descent method. 46 It utilizes less energy to produce an accurate and reliable 3-dimensional structure of the target protein.
Quality evaluation of a three-dimensional model
During the last phase of homology modeling, the improved structure of a model protein underwent extensive testing to ensure internal consistency and reliability. Various criteria were utilized to determine the accuracy of this putative protein model. The psi/phi (ψ/φ) Ramachandran plot, which was generated by the PROCHECK examination, was used to analyze the backbone configuration. The quality of the model structure was evaluated using the ExPASy service of SWISS-MODEL Workspace’s PROCHECK (https://servicesn.mbi.ucla.edu/PROCHECK/), 47 Verify3D (http://nihserver.mbi.ucla.edu/Verify3D/), 48 QMEAN (https://swissmodel.expasy.org/qmean/), 49 and ERRAT (https://servicesn.mbi.ucla.edu/ERRAT/) 50 programs. Moreover, The UCSF Chimera software (version 1.16.0) was used to superimpose and visualize the model and template structure. 51 Also, the ProSA-web server (https://prosa.services.came.sbg.ac.at/prosa.php) computed Z scores for both proteins. 52
Protein-protein interaction network
Most biological activities are regulated and executed by protein-protein interactions (PPIs), which form a complex, continuous network of reactions. The STRING v11.0 (https://string-db.org/) 53 inquiry found a protein-protein functional interaction network.
Active site identification
To determine the active site of a protein, the web-based tool CASTp (http://sts.bioengr.uic.edu/castp) was used. CASTp is key in identifying, delineating, and quantifying geometric and topological characteristics essential for correcting the protein function. These include interior cavities, cross channels, and surface pockets. Furthermore, it aids in the mapping of functionally annotated residues onto protein 3D structures. 54
Molecular docking analysis
Preparation of receptor and ligand
The receptor for molecular docking was chosen to be the putative TagJ protein (accession no. WP_004386962.1). Following that, for comparing our binding affinities, the 3D structure of the primary template TagJ protein (PDB ID: 4uqx.1) 55 was examined using the RCSB PDB server (http://www.pdb.org). 56 After recognizing and removing co-crystallized compounds from the 4uqx.1 structure, within the 3-dimensional coordinate file crystallographic water molecules were retrieved.
In this in-silico docking experiment, we employed different anti-bacterial FDA-approved Therapeutics as ligands such as ceforanide (CID:43507), ceftriaxone (CID:5479530), latamoxef (CID: 47499), cefixime (CID: 5362065), pefloxacin (CID: 51081), and amikacin (CID: 37768 ). The choice of these ligands was guided by a literature review study, focusing on their proven antibacterial effectiveness in favor of a diverse range of Gram-negative bacteria, especially those from the Enterobacteriaceae family.57,58 Given that our selected hypothetical protein (WP_004386962.1) from C. sakazakii is a Gram-negative bacterium within the Enterobacteriaceae family, these compounds are promising candidates as inhibitors for treating this bacterial infection. The decision to use these inhibitors over other compounds was motivated by our goal to validate the 3D model of the TagJ protein from C.sakazakii and compare it with the template TagJ protein from Pseudomonas aeruginosa.
After retrieving the canonical SMILES ID of a drug from the PubChem database (https://pubchem.ncbi.nlm.nih.gov), 59 we converted the 3-dimensional SDF to the PDB structure of a drug using Pymol software. 60 Following this, we significantly simplified the research process by performing ligand optimization and converting it to the PDBQT files utilizing Autodock Vina Tools 1.5.7. 61
Molecular docking and binding interactions analysis
Autodock Vina facilitates the docking of small-molecule libraries to macromolecules, enabling the identification of lead compounds with specific biological functions. The Auto Grid engine generates the configuration file for grid specifications. 61 During the docking process, possible candidates for favored binding were identified using an average root-mean-square deviation (RMSD) of below 1 kcal/mol. The ligands with the highest binding affinity have the highest negative binding energy. For site-specific docking, the grid box was built to the following specifications: dimension x:y:z = 50:50:50, exhaustiveness = 8, and center_x:y:z = 9.175:−8.927: .75. Subsequently, hydrophobic and hydrogen bond interactions were analyzed using Biovia Discovery Studio. 62
Comparative genomics approach
To find human proteins that might be similar to the hypothetical protein WP_004386962.1, we used a BLASTp search of the human protein database. We employed stringent filtering criteria, setting a minimum bit score of 100 and an E-value cutoff of 0.005 to ensure the reliability of the identified hits. 63
Results
Physicochemical properties and subcellular localization
To understand the function and molecular evolution of a protein requires a comprehensive assessment of its physicochemical characteristics. With 277 amino acids, this protein exhibits a diverse composition. Among its constituents, Cys (3), Tyr (4), His (4), Lys (4), Met (5), Ile (7), Asn (7), Thr (9), Trp (9), Phe (9), Ser (11), Val (15), Arg (16), Asp (17), Pro (17), Gly (18), Gln (20), Glu (20), Leu (37), and Ala (41) are notably abundant, giving protein negative charge is evident, molecular weight of 30427.40 Da and a putative isoelectric point (pI) of 4.54. Table 1 presents a comprehensive overview of the physicochemical characteristics of the putative protein, encompassing its frequency and predominance with their constituent amino acids. According to the CELLO program, our target protein was predicted to localize to the “Cytoplasmic” region. Subsequent analysis using the PSORTb and PSLpred protein subcellular localization services also yielded strong localization scores, further confirming its cytoplasmic classification. Additionally, the SOSUI server projected the protein to be soluble.
The ProtParam tool was employed to analyze the physicochemical characteristics of the hypothetical protein (WP_004386962.1).

Phylogenetic tree construction and multiple sequences alignment
In our investigation, we employed BLASTp to survey nonredundant databases, revealing similarities with other bacterial type VI secretion system (T6SS) auxiliary proteins (Table 2). To delve deeper into these similarities, we aligned the FASTA sequence of the putative protein (WP_004386962.1) with the FASTA sequences of related annotated proteins (Supplemental Figure S1). Using several Type VI secretion system accessory protein TagJ protein sequences, we generated a phylogenetic tree using the MEGA11 program. Most of the TagJ proteins utilized in building the phylogenetic tree were sourced from Cronobacter sakazakii and other Cronobacter species, identified through BLASTp analysis. The resulting phylogenetic tree, with branch distances of 0.004, revealed that WP_104671789.1, WP_076728080.1, ELY2789108.1, and EGT4277351.1 were closely related to our query protein (Accession No. WP_004386962.1; Figure 2). Moreover, The E-value quantifies the statistical significance of an alignment, indicating how likely it is to be a random occurrence in a database of the specified size. The E-value varies with database size and query length. The alignment is best if the E-value is near zero. 64
Ten homologous proteins were chosen from the non-redundant BLASTp database due to their high similarity to our WP_004386962.1 query sequence.


A phylogenetic tree illustrating the ancestral connection of the target protein (boxed) to other TagJ proteins. The phylogenetic tree revealed that 4 proteins, WP_104671789.1, WP_076728080.1, ELY2789108.1, and EGT4277351.1, are closely related to our query (WP_004386962.1) with branch distances of 0.004, respectively.
Domains and motifs analysis
We used many annotation techniques to identify conserved domains of our target protein and potential functionalities. The target protein, TagJ (HsiE1), is a component of the bacterial T6SS and likely contains an ImpE-like domain (Table 3), as suggested by bioinformatics tools like NCBI-CD Search, Pfam, and InterProScan. The structure of this protein secretion system is similar to that of the bacteriophage puncturing mechanism. This protein is commonly found and contributes to the development of various opportunistic infections. T6SS enhances pathogen survival by injecting protein effectors into host cells and releasing toxins to nearby pathogens. 65 ClpV and the TssC/TssB sheath interact with it in a particular way. 55 Bacterial complexes that resemble phage tails are called type VI secretion systems, or T6SS for brief. When the TssBC sheath contracts, the toxin delivery mechanism releases poisons into the target cells. 66 T6SSs possess 13 essential proteins. These proteins form a puncturing complex similar to that found in bacteriophages. The complex includes a tube, a puncturing tip, and a contractile sheath. Other proteins help assemble the baseplate. TagA binds to a star-shaped protein to regulate the growth of the contractile sheath. 67 The NCBI-CDD server and Pfam both predicted that the ImpE superfamily domain contains 1 to 273 amino acid residues. Additionally, the NCBI-CDD server assigned an E-value of 1.38e-119 to this domain. Using a motif server like Pfam, 2 additional motifs were found: Tetratricopeptide repeat (PF14559) and ImpE protein (PF07024; Table 3). Tetratricopeptide repeat (TPR) motifs have been identified in a wide range of species from microbes to humans. Numerous biological activities, including nuclear and peroxisomal protein transport, transcriptional regulation, cell cycle regulation, neurogenesis, and protein folding, are mediated by proteins that contain TPRs. 68
List of the domains and motifs of the hypothetical protein (accession no: WP_004386962.1).

A “DNA-binding 3-helical bundle” fold was discovered in the protein sequence using the PFP-FunDSeqE method for protein fold pattern recognition. The helical contact of the protein is frequently less regular than a 3-stranded coiled. The formations known as 3-helix bundles are usually single-stranded and have loops connecting the helices. In the tertiary structures of many big proteins, this structural motif is frequently observed as a subdomain. Moreover, lysins, enzyme inhibitors, DNA-binding proteins, and enzymes are among the functionally diverse protein families represented by the 3-helix bundle. 69 The placements of amino acid numbers in the protein are shown on the X-axis of the coiled-coil graph in Figure 3, while the probability score of the coiled-coil is represented on the Y-axis.

WP_004386962.1 is a putative protein that has undergone functional analysis (coiled-coil interaction). The Y-axis shows the probability of a coiled-coil, while the X-axis shows the position in the sequence.
Protein structure prediction
The secondary structure of the protein is predominantly composed of turns, coils, helices, and sheets. According to the SOPMA server analysis, the protein contains a significant proportion of alpha helices (48.01%), followed by random coils (33.57%), beta-turns (5.78%), and extended strands (12.64%; Figure 4). A comparable outcome from the PSIPRED server (Supplemental Figure S2) confirmed the accuracy of the previous result. The 3-dimensional structure of this protein was predicted using the SWISS-MODEL server, with the template protein (4uqx.1.A) serving as a guide. The predicted structure shares 38.28% sequence similarity with the template. 70 The objective of the template protein is 2 Type VI Secretion Classes Differentiated by the Coevolution of the Accessory HsiE Protein, the TssB-TssC Sheath, and the ATPase ClpV. The bacteria known as Pseudomonas aeruginosa is the template protein. 55 The 3D structure that was produced using SWISS-MODEL is shown in Figure 4.

The putative protein WP_004386962.1 shows features reminiscent of ImpE through predictions of its (A) primary, (B) secondary, and (C) 3-dimensional structures. When its sequence of 277 amino acids was first obtained, it was examined to ascertain the makeup of its secondary structure. Significant amounts of alpha helices (48.01%) and extended beta strands (45.05%) were found in the secondary structure anticipating, but beta-turns (5.78%) and random coils (33.57%) were found in smaller proportions: (A) primary structure (NCBI), (B) 2-dimensional structure (SOPMA), and (C) 3-dimensional structure (SWISS-MODEL).
3D structural analysis and quality evaluation
The YASARA Energy Minimization Server improved the stability of the protein model by lowering its energy from −121848.9 to −158647.1 kJ/mol. Additionally, the final score increased from −1.33 to −0.50 kJ/mol, indicating a more robust protein structure. The quality of the 3D model was evaluated using the PROCHECK, Verify 3D, QMEAN, and ERRAT programs. PROCHECK analysis revealed that 94.0% of the amino acid residues were positioned in the most preferred region of the Ramachandran plot (see Table 4 and Figure 5A). The Verify 3D server confirmed that the model structure functioned as anticipated, with 83.33% of the residues having an average 3D to 1D score ⩾ 0.1. Using the QMEAN tool, the model was placed into the dark gray zone and had an outstanding QMEAN4 value of −0.53 (Figure 5B). ERRAT further forecasted that the protein structure has a good quality, with a quality factor of 95.7198. In Figure 5C, the model is superimposed on top of the template protein (PDB ID: 4uqx.1.A). The 3D model that was suggested by the RMSD value of 0.458 Å was discovered after superimposition in UCSF Chimera. The Z score of the model, which reflects its overall accuracy, can determine how the input structure compares to the typical scores reported for native proteins of a similar shape. Figure 5D and E illustrate that the model generated by ProSA exhibits homology with the template, having Z scores of −7.42 and −7.73, respectively.
Ramachandran plot analysis of the hypothetical protein (WP_004386962.1) provides insights into the conformational preferences of its amino acid residues.

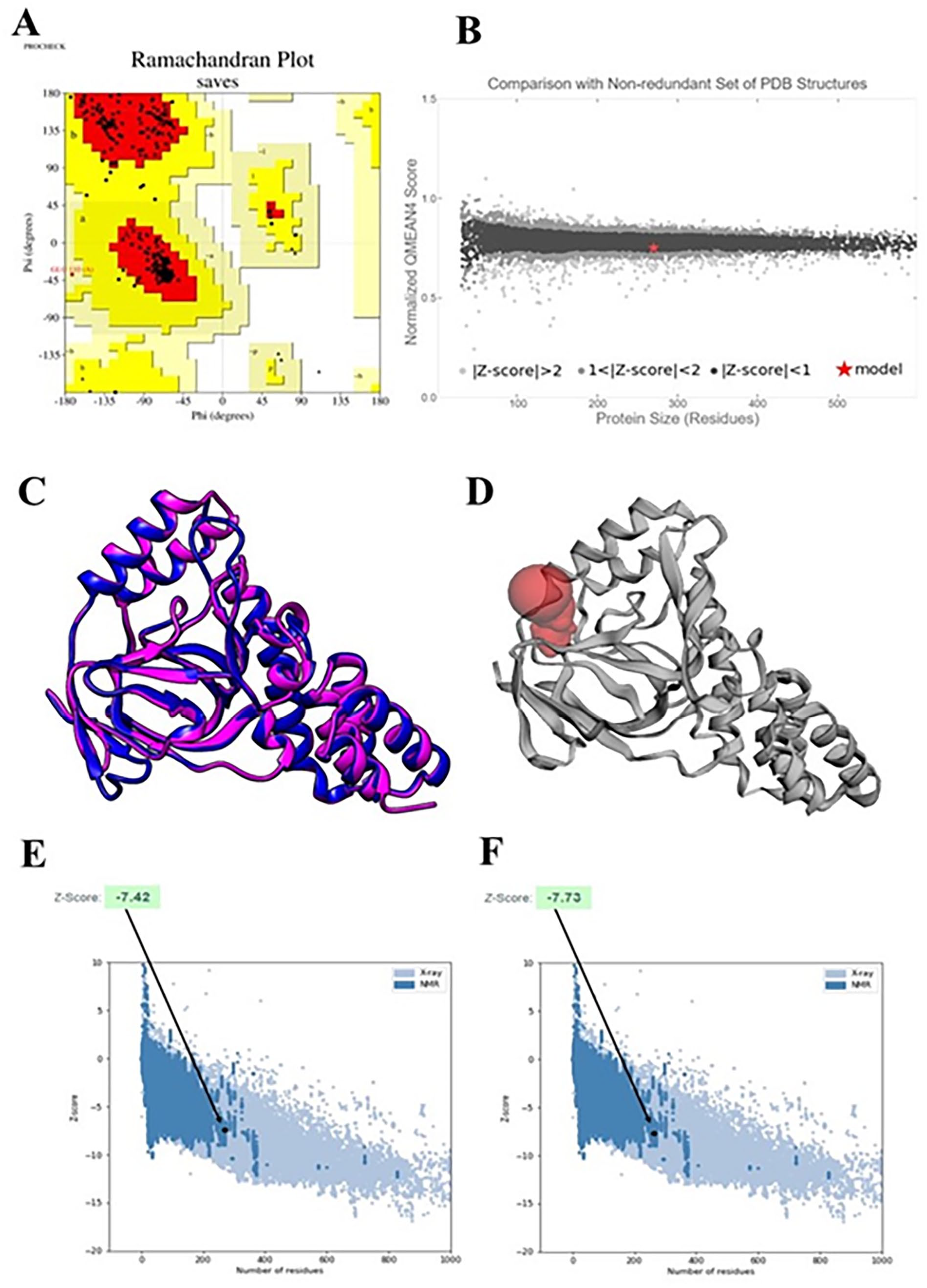
The quality of the model was assessed using 2 methods: (A) a Ramachandran plot generated by the PROCHECK tool to evaluate the structural accuracy; and (B) a QMEAN plot to compare the model’s structure with experimental structures of similar size. The QMEAN plot indicated a strong correlation between the model and experimental structures. (C) Using the UCSF Chimera software, (D) A CASTp server-based graphical depiction of an active site, superimpose the template and the model protein (model: royal blue, template: magenta). Z scores of the ProSA server for the target (E) and template (F) modeled protein. The 2 structures were within the usual range for similarly sized native proteins that have been experimentally characterized (NMR and X-ray).
Active site determination
The CASTp web server has shown to be a useful tool for several research projects, such as the evaluation of signaling receptors, the discovery of cancer therapies, the comprehension of drug action mechanisms, and the investigation of issues related to immunological disorders. 54 This CASTp server was used to analyze the active site and amino acid residues of a model structure (Figure 5F). Identifying and characterizing active site residues is crucial for drug or inhibitor design. Medication or antagonist design depends on the assessment and characterization of active site residues. CASTp forecast states that the model protein’s active residues (of 1 biggest active pocket with solvent accessible [SA] area of 120.790 and Volume [SA] area 64.195, respectively) were found to be Trp100, Trp100, Leu124, Leu124, Leu124, Leu124, Leu124, Glu125, Ala127, Ala127, Glu128, Glu128, Ala129, Asn130, Asn130, Asn130, Asn130, Phe148, Trp150, Trp150, Trp150, Trp150, Trp150, Trp150, Leu151, Leu151, Met152, Met152, Pro160, Pro160, Pro160, Pro160, Phe175, Phe175, Ser176, Ser 176 consist of the target protein’s ImpE superfamily domain in line with the predictions of Pfam, InterProScan, and NCBI-CD Search (described in the “Protein family and phylogeny analysis” section).
Protein-protein interactions analysis
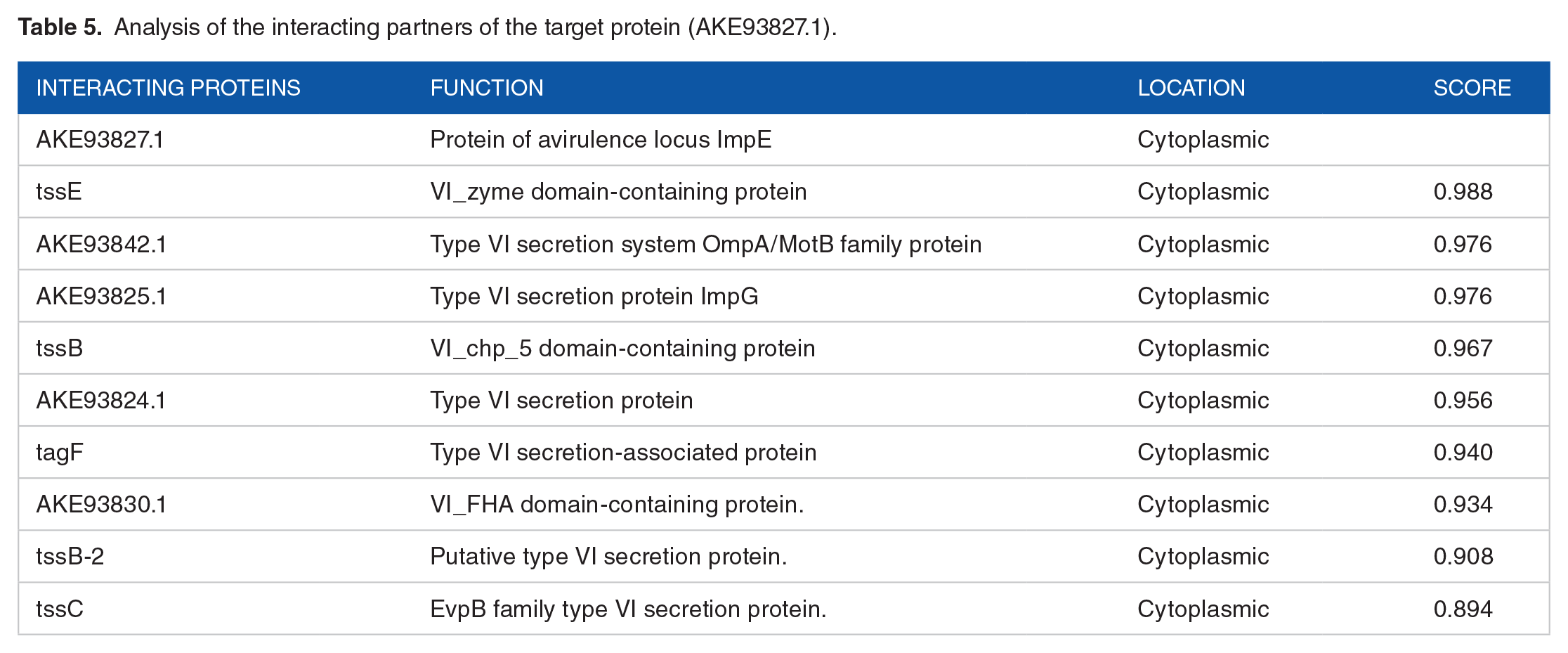
Many predicted interaction partners of the desired protein were found using the STRING database (Figure 6). TssB-2, tssC, AKE93833.1, tssE, AKE93842.1, AKE93825.1, tssB, and AKE93824.1 were the proteins that interacted with each other. These cellular locations and molecular functions of a protein were found through literature mining and are listed in Table 5. Located in the cytoplasm, the majority of the proteins interact with our hypothetical target protein. The proteins are mostly associated with the supramolecular bacterial complex known as the type VI secretion system (T6SS), which looks like phage tails. When the TssBC sheath transcriptional regulatory system contracts, the toxin delivery system discharges poisons into target cells. 67 This is crucial to our understanding of the putative protein’s identity as the avirulence locus ImpE protein. The lowest (0.15), medium (0.40), high (0.70), and highest (0.90) default thresholds are the ones that STRING advises choosing. Only the edges with a score equal to or higher than the designated threshold are included in the construction of an unweighted network for each of these criteria, creating a network of interconnected proteins. 71 Every protein that interacts has a score of more than 0.70, indicating a strong interaction.

Network of protein-protein interactions for the hypothetical protein from the STRING server. The query proteins are depicted by colored nodes, while the second shell of interactors is represented by white nodes. Proteins with unknown 3D structures are indicated by empty nodes, and those with known or predicted 3D structures are represented by filled nodes.
Analysis of the interacting partners of the target protein (AKE93827.1).
Molecular docking and binding affinity analysis
A docking investigation between the ligands and the model was carried out using the Auto Dock Vina. Hence, there were 6 ligands docked with both the C. sakazakii model protein (WP_004386962.1) and the template Pseudomonas aeruginosa PAO1 (Q9I746.1) protein. Each ligand had a high binding affinity with both proteins. The binding affinities of the ligands for the model and template proteins ranged from −6.6 to −8.0 kcal/mol. Several interacting residues in the active site were discovered to be the same in both proteins. The results are also in line with the active site assumption of CASTp. Figures 7 and 8, and Table 6 illustrate the binding interactions of 6 compounds within a specific protein pocket. CID: 43507 engages in hydrogen bonding with Ala107, Ala109, Glu110, Asp112, Asp237, and Ala238, while forming hydrophobic bonds with Pro113, Ala116, Pro235, and His242. CID: 5479530 is capable of forming hydrogen bonds with Arg88, Leu108, Thr111, Pro216, Thr219, and Asp237, and engages in hydrophobic interactions with Ala82, Ala86, Ala116, Arg120, Leu217, and His242. CID: 47499 primarily binds via hydrogen bonds to Ala86, Leu108, and Pro235, while also forming hydrophobic interactions with Arg120, Asp237, and Ala116. CID: 5362065 forms hydrogen bonds with Ala82, Leu108, Arg120, and Arg157, and hydrophobic interactions with Ala82, Ala86, and Ala116. CID: 51081 generates hydrogen bonds with Leu85, Leu108, Glu110, and Thr111, and a hydrophobic bond with Ala86. Lastly, CID: 37768 establishes hydrogen bonds with Ala82, Ala86, Leu108, Thr111, Arg120, Asp237, Ala238, and Glu239, while interacting hydrophobically with Ala107.

(Left side) 3D ligand-bound structure of hypothetical protein WP_004386962.1. (Right side) 3D ligand-bound structure of Pseudomonas aeruginosa PAO1 (Q9I746.1).
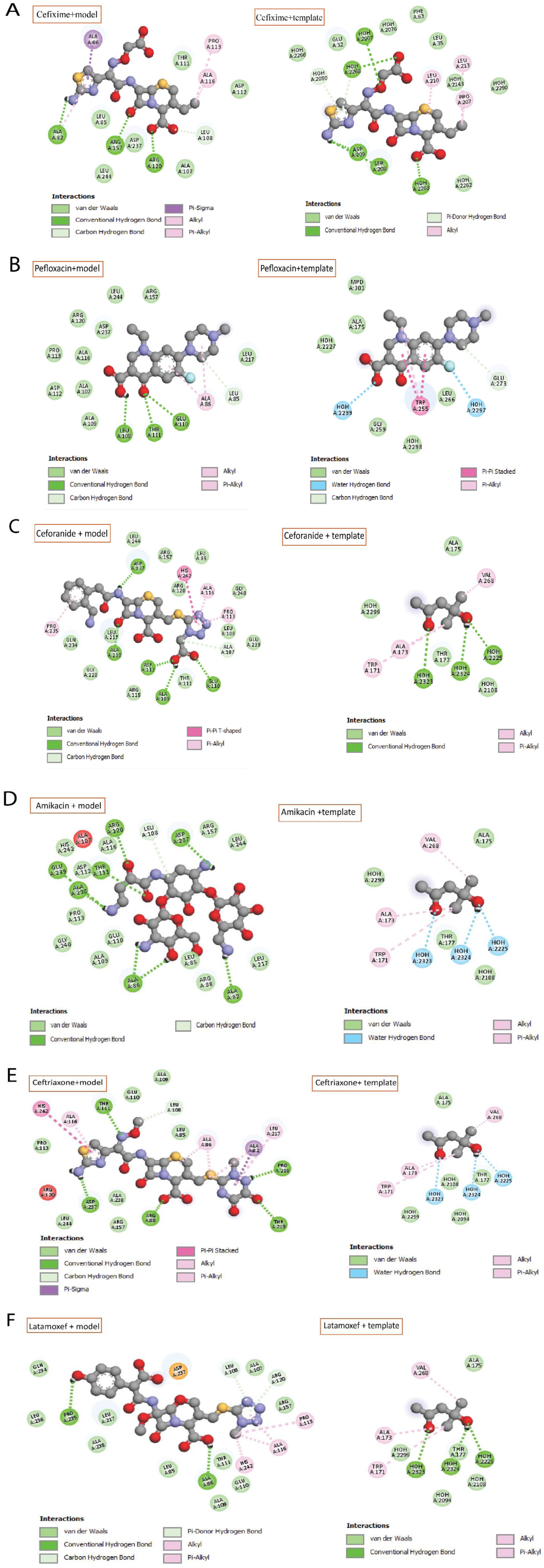
Key interacting residues of hypothetical protein (WP_004386962.1) model and also a template (Q9I746.1) with: (A) Cefixime, (B) Pefloxacin, (C) Ceforanide, (D) Amikacin, (E) Ceftriaxone, and (F) Latamoxef antibiotics.
Interacting residues of target protein and model using Auto Dock Vina.

After the structural and functional annotation of the hypothetical protein was completed successfully, the comparative genomics approach was used to better define our target protein. A BLASTp search was conducted to identify human proteins similar to the target protein. The target protein was identified as a novel C. sakazakii protein after no resemblance was found to any known human protein. To reduce side effects, a good therapy option would target microbial proteins that are not comparable to human proteins.
Discussion
The increasing affordability of sequencing technology has led to a surge in genomics and proteomics data. However, research on hypothetical proteins has lagged. Studying hypothetical proteins could provide valuable insights into bacterial metabolism, disease progression, drug discovery, and disease prevention strategies. 72 Using several bioinformatics resources, this study structurally and functionally described the putative protein WP_004386962.1 from the Cronobacter sakazakii 7G strain. The protein analyzed had 277 amino acids, a molecular weight of 30427.40 Da, a theoretical pI of 4.54, and a GRAVY of −0.140 (Table 1). These characteristics suggest a soluble protein. The CELLO server predicted that this protein is localized within the cytoplasm. The secondary structure of the protein consists of an alpha helix, a beta-turn, a random coil, and an extended strand, with the random coil being a significant prevalent. Based on domain and motif analysis, we predicted that the desired putative protein would be an ImpE superfamily protein, which is categorized with high confidence by all annotation methods as TagJ (also known as HsiE1) of T6SS (Table 3). The BLASTp analysis, which compared the sequence against a nonredundant database, revealed a high degree of sequence similarity (up to 99%) with various T6SS TagJ proteins. This strong similarity further confirms the earlier prediction. TagJ (HsiE1) is an auxiliary protein in the type VI secretion system (T6SS) of bacteria. It contributes to TssC/TssB and ClpV the sheath’s interaction. 55 The type VI secretion system (T6SS), like phage tails, is a bacterial supramolecular group. It is a toxin delivery system that injects poisons into target cells when the TssBC sheath contracts. 66 The Type VI secretion system (T6SS) increases the pathogenicity of dangerous bacteria like Pseudomonas aeruginosa, Escherichia coli, and Vibrio cholerae. This increases the formation of biofilm, protects the host immune system, and increases antibiotic resistance. Many strains of E. coli are known for their significant drug resistance. T6SS consists of 2 tubular structures, TssB and TssC, which are disassembled by ClpV. TagJ/HsiE is commonly associated with one of these components.55,73 The 3D structure of a protein, obtained via the SWISS-MODEL service, completed various model quality assurance tests, including PROCHECK, QMEAN, Verify 3D, and ERRAT. The YASARA energy minimization method enhanced the reliability of the 3D structure. When the hypothetical protein was compared to the Pseudomonas aeruginosa potential cytoplasmic protein (PDB ID: 4UQX) using UCSF Chimera, the 3D structures aligned well with a root-mean-square deviation (RMSD) of 0.458 Å (explained in this “Structure analysis and model quality assessment” section). The RMSD value indicates high sequence identity, suggesting minimal variability in protein structures. Our result, approaching zero, indicates a strong similarity between the template structure and the hypothetical model. 74 The CASTp server calculated active site amino acid residues in the ImpE superfamily domain area, which aligned with functional annotation tools’ predictions. Using the Autodock Vina, molecular docking was used to determine how the 6 ligands interacted with target proteins. According to the docking result, hydrogen and hydrophobic bonds were identified with interacting residues. These interactions are essential for protein folding, structure maintenance, and molecular recognition, thereby stabilizing ligands and enhancing therapeutic efficacy.75,76 Our findings were further verified by the observation of a high binding affinity among these ligands and both the target protein and the Pseudomonas aeruginosa TagJ protein (Table 6). The active sites of the proteins were shown to have a large number of identical interaction residues. As a second-generation cephalosporin, ceforanide has antibacterial activity against many Enterobacteriaceae. 77 Ceftriaxone is a popular third-generation cephalosporin antibiotic renowned for its broad-spectrum activity. It is particularly valued for its effectiveness against multidrug-resistant Enterobacteriaceae. 58 Latamoxef is highly effective against Gram-negative bacteria, including Enterobacteriaceae and Bacteroides fragilis. Its potent activity makes it a promising option for treating intra-abdominal infections in immunocompromised patients, as well as neonatal Gram-negative bacillary meningitis, due to its strong efficacy against Gram-negative bacilli and user-friendly administration. 57 Cefixime is a third-generation, semisynthetic cephalosporin, and an oral broad-spectrum antibiotic. 78 Cephalosporins, which belong to the beta-lactam antibiotic class, can be used to treat meningitis, resistant bacterial strains, skin infections, and inflammation induced by Gram-positive and Gram-negative bacteria. Pefloxacin effectively targets a wide range of bacteria, including both Gram-positive and Gram-negative types, making it a versatile antibiotic. 79 Amikacin is particularly efficient against more resistant Gram-negative bacteria and inhibits a lot of aerobic Gram-negative bacteria in the Enterobacteriaceae family. The 6 ligands used in this study have antibacterial activity against a wide range of Gram-negative bacteria, from those of the Enterobacteriaceae family. Given that our chosen hypothetical protein from C. sakazakii is also a Gram-negative bacterium within the Enterobacteriaceae family, these inhibitors represent promising options for treating this bacterial infection. We have clarified that, due to the lack of experimental validation, the in silico characterization of the hypothetical protein may not fully reflect its biological complexity. TagJ (HsiE1), an auxiliary component of the type VI secretion system (T6SS), is a known virulence factor, but its role in Cronobacter species is unknown. 80 The emphasis on a single putative protein may overshadow other critical proteins and the significance of the TagJ-associated type VI secretion system (T6SS), limiting our understanding of the metabolic flexibility of C. sakazakii. The limited nature of this study emphasizes the crucial importance of further investigating the metabolic pathways of the organism. Developing possible therapeutics for this bacterium infection will require additional molecular study utilizing both in-vivo and in-vitro models.
Conclusions
This study focuses on identifying a hypothetical protein (WP_004386962.1) associated with the type VI secretion system (T6SS) in C.sakazakii utilizing in-silico methods and molecular docking. The research highlights the protein’s sub-cellular localization, functional domains, motifs, and docking interactions with various ligands, all of which are crucial for understanding the pathogenesis of C. sakazakii. The characteristics of a protein, including its role in drug resistance, are investigated to improve our understanding of C. sakazakii pathophysiology and to contribute in medication discovery. Six FDA-approved inhibitor compounds were tested against this hypothetical protein, with Ceforanide showing (−7.5 kcal/mol) the most promising binding interaction score. Further research is required to validate these findings. Furthermore, a comparative genomics investigation identified a unique protein in C. sakazakii as a possible therapeutic target. The T6SS is instrumental in biofilm formation, which increases bacterial antibiotic resistance and complicates infection treatment. Further studies and experimental validations are necessary to fully comprehend T6SS and its effectors, supporting the development of effective treatment strategies for this pathogenic bacterium.
Supplemental Material
sj-docx-1-evb-10.1177_11769343251327660 – Supplemental material for Structural and Functional Characterization of a Putative Type VI Secretion System Protein in Cronobacter sakazakii as a Potential Therapeutic Target: A Computational Study
Supplemental material, sj-docx-1-evb-10.1177_11769343251327660 for Structural and Functional Characterization of a Putative Type VI Secretion System Protein in Cronobacter sakazakii as a Potential Therapeutic Target: A Computational Study by Nurun Nahar Akter, Md. Moin Uddin, Nesar Uddin, Israt Jahan Asha, Md Soyeb Uddin, Md. Arju Hossain, Fahadul Alam, Siratul Kubra Shifat, Md. Abu Zihad and Md Habibur Rahman in Evolutionary Bioinformatics
Footnotes
Acknowledgements
We want to express our gratefulness to the members of the Center for Advanced Bioinformatics and Artificial Intelligence Research Lab, led by Dr. Md Habibur Rahman, for their assistance and insightful contributions to the study.
Author Contribution Statement
Nurun Nahar Akter: Provided concept, experimenting, and built the paper; Md. Moin Uddin: Data analysis, edited and reviewed the manuscript; Israt Jahan Asha, Md Soyeb Uddin, and Fahadul Alam: Analyzed and interpreted the data; Md. Arju Hossain: Edited and reviewed the manuscript; Nesar Uddin, Siratul Kubra Shifat, and Md. Abu Zihad: Data analysis and formal analysis; Md Habibur Rahman: Designed the experiments and supervised the whole project.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability
The corresponding author can provide the data that was utilized to support the study upon request.
Ethical Statement
No ethical approval was required for this manuscript.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.