
Abstract
Leishmania infantum chagasi is an intracellular protozoan parasite responsible for visceral leishmaniasis, a fatal disease in humans. Heparin-binding proteins (HBPs) are proteins that bind to carbohydrates present in glycoproteins or glycolipids. Evidence suggests that HBPs present on Leishmania surface participate in the adhesion and invasion of parasites to tissues of both invertebrate and vertebrate hosts. In this study, we identified the product with an HSP90 (heat shock protein 90) domain encoded by lipophosphoglycan (LPG3) gene as a L infantum chagasi HBP (HBPLc). Structural analysis using the LPG3 recombinant protein suggests that it is organized as a tetramer. Binding analysis confirms that it is capable of binding heparin with micromolar affinity. Inhibition of adenosine triphosphatase activity in the presence of heparin, molecular modeling, and in silico docking analysis suggests that heparin-binding site superimposes with the adenosine triphosphate–binding site. Together, these results show new properties of LPG3 and suggest an important role in leishmaniasis.
Introduction
Components from the extracellular environment can exert their functions in cells by binding specifically to the cell surface receptors, which when activated may be internalized or may transmit intracellular signals to the nucleus to regulate different biological functions. These receptors sometimes use glycosaminoglycans (GAGs) from the cell surface to recognize their ligands or to regulate their activation. 1 Heparin, a polysaccharide belonging to the family of sulfated GAGs, possesses numerous biological functions related to interactions with different proteins 2 that play a major role in cancer, such as in curing wounds, inflammation, and infectious diseases. 3
Visceral leishmaniasis is a potentially fatal disease that represents a serious public health problem, with the main focus in Central and South America and in Mediterranean countries. This disease is responsible for approximately 20 to 40 000 deaths per year worldwide, including casualties caused by Leishmania donovani and Leishmania infantum chagasi. 4 Specific receptors present on the surface of Leishmania can bind to GAGs using a structure similar to heparin present in the host tissue. 5
Heparin receptors (also known as heparin-binding proteins [HBPs]) can influence host-parasite interactions during the infection process.5,6 The HBPs belong to a group of ubiquitous proteins, whose main characteristic is the ability to bind carbohydrates present in glycoproteins or glycolipids. Evidences suggest that HBPs present on Leishmania surface participate in the regulation of biological activities of the host cell and participate directly in the adhesion and internalization of parasites in cells from both vertebrate and invertebrate hosts: experiments with L donovani promastigote forms showed that the parasite HBP induces the inhibition of protein kinase C activity.7–9 Other studies have revealed the presence of these proteins in Leishmania braziliensis promastigote forms and their influence on the adhesion of parasites in the insect vectors by recognizing molecules present in the Lutzomyia intermedia and Lutzomyia whitmani midgut.5,7,10 Our research group purified and characterized HBP from L infantum chagasi promastigotes (HBPLc), showing its distribution on the surface as well as inside the parasite. In addition, incubation with heparin promoted a decrease in the frequency of internalization of parasites by macrophages, suggesting the participation of HBP in the parasite infection process. 6
In the present work, an HBPLc was sequenced and identified as product of lipophosphoglycan 3 (LPG3) gene. The protein was expressed in heterologous system as a recombinant HBPLc (rHBPLc) and their structural and functional properties were analyzed. In silico analyses were conducted to predict the 3-dimensional (3D) structure and the heparin-binding site of the protein. Our results demonstrated that rHBPLc is a tetrameric complex with an estimated molecular mass of 348 kDa. Each monomer is constituted by structures in α-helices interspersed by β-sheets. The recombinant protein can bind to heparin and displays hydrolytic activity over adenosine triphosphate (ATP). Interestingly, ATP and heparin-binding sites overlap in the protein, indicating that the sugar may be used as an inhibitor of its adenosine triphosphatase (ATPase) activity. Noteworthy, HBPLc coded by LPG3 gene has 2 regions described in the literature: heat shock protein 90 (HSP90—folding-function domain) and H+-ATPase (ATP-binding domain). Together, these findings corroborate our results about the ATPase activity of the rHBPLc and allow us to establish LPG3 as a new HBP.
Methods
Protein sequencing
Native HBPLc was purified and submitted to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The band corresponding to HBPLc (molecular mass = 105 kDa) 6 was extracted from the polyacrylamide gel and treated as follows: the gel was destained in acetonitrile solution (ACN; 50%/NH4HCO3 25 mmol L−1, pH 8.0) and dehydrated with 200 µL of pure ACN. The gel pieces were dried on SpeedVac (miVac; GeneVac, Ipswich, UK), reduced in dithiothreitol (65 mmol L−1/NH4HCO3 100 mmol L−1) at 56°C for 30 minutes and alkylated with 100 µL iodoacetamide (200 mmol L−1/NH4HCO3 100 mmol L−1) at room temperature in the dark. The gel was washed with 200 µL of 100 mmol L−1 NH4HCO3 by stirring for 10 minutes, dehydrated with 200 µL ACN, rehydrated with 200 µL of 100 mmol L−1 NH4HCO3, dehydrated again with ACN, and then dried on SpeedVac for 15 minutes. For protein digestion, the gel pieces were treated with 30 µL of the solution of 25 mmol L−1 NH4HCO3/2.5 mmol L−1 CaCl2/25 ng/µL trypsin at 4°C/45 minutes. In the sequence, 50 µL of 25 mmol L−1 NH4HCO3/2.5 mmol L−1 CaCl2 were added and the reaction occurred overnight at 37°C. Peptides were extracted, purified in a Pierce C18 Spin Column ZipTip (Thermo Scientific, Rockford, IL, USA) and mass spectrometry was performed in the MALDI-TOF-TOF mass spectrometer (Ultraflex III; Bruker Daltonics, Fremont, CA, USA). The peptides were prepared in 10 mg/mL matrix α-cyano-4-hydroxycinnamic 1:1 (v/v) prepared in 50% ACN/0.1% trifluoroacetic acid and, in the sequence, the sample was submitted to the sequencing. The peptides were detected in duplicate. The processing of MS1 and MS2 data was performed on FlexAnalysis 3.3 software (Bruker Daltonics, Bremen, Germany) and the identification of the amino acid sequence was performed using MASCOT 2.3.2 software (Matrix Science, London, UK).
Bioinformatics and sequence analysis
The sequence obtained in MASCOT was submitted to analysis in pBLAST-NCBI (National Center for Biotechnology Information) implemented in UNIPROT KB (European Bioinformatics Institute, Hinxton, UK; version August 28, 2015), parameterized with the values of E-threshold 0.0001, matrix blosum-62, filtering in low-complexity regions, gapped and hits 50. Genome analysis was performed in pBLAST implemented in TritrypDB. 11 The signal peptide prediction was performed using SignalP 4.1 server,12,13 and the presence of glycosylphosphatidylinositol (GPI)-anchor regions was assessed using GPI-SOM 1.5, 14 big-PI Predictor, 15 and PredGPI-GPI. 16 To predict regions with N- and O-linked glycosylation, we used the NetNGlyc 1.0 17 and NetOGlyc 4.0 servers, 18 respectively. The search for domains was performed using Pfam 30.0 database. 19 The multiple alignment of the LPG3 amino acid sequence for the species L infantum chagasi (LinJ.29.0790), Trypanosoma cruzi (TcCLB.506989.190), L braziliensis (LbrM.29.0780), Leishmania tarentolae (LtaP29.0800), Leishmania major (LmjF.29.0760), L donovani (LdBPK_290790.1), and Leishmania mexicana (LmxM.08_29.0760) was performed using the program Clustal Omega. The phylogenetic tree was constructed using the MEGA 6 with probabilities calculated by Clustal W, parameterized with BLOSUM matrix, and phylogenetic analysis by the maximum likelihood method, reliability by bootstrap method with 1000 resampling and pattern remaining of maximum likelihood. 20
Recombinant protein expression and purification
The DNA sequence encoding LPG3 was synthesized in vitro and cloned by GenOne (GenOne, Rio de Janeiro, RJ, Brazil). It was inserted into the cloning vector pUC19 in the NdeI and XhoI restriction site. The gene was subcloned into pET 28a(+) (Novagen, San Diego, CA, USA), which codes for His-Tag in the N-terminal portion of the polypeptide. Escherichia coli DH5α strain cells were transformed with the modified plasmid for the propagation of the vectors containing the insert of interest. The recombinant protein was expressed in E coli Rosetta (DE3) strain cells transformed with pET 28 constructs and grown at 37°C in the Luria-Bertani (LB) medium supplemented with kanamycin (50 μg/mL) and chloramphenicol (34 μg/mL) to achieve an optical density of 0.6 to 0.8, followed by expression induced by isopropyl β-
The cells were lysed by sonication in 10 mmol L−1 sodium phosphate/150 mmol L−1 sodium chloride, pH 7.0 (buffer A). The supernatant of soluble proteins was recovered and submitted to affinity chromatography using Ni-NTA resin (Qiagen, Hilden, Germany) equilibrated in buffer A. Immobilized proteins were eluted in buffer A containing 500 mmol L−1 imidazole (buffer B). The proteins were analyzed by 10% SDS-PAGE. Subsequently, the eluates were separated by affinity chromatography on heparin-agarose assembled in a liquid chromatography system (AKTA purifier; GE Healthcare Life Sciences, Freiburg, Germany) equilibrated with buffer A. The adsorbed fraction was eluted with buffer A plus NaCl (2 mol L−1). Later, the eluate was separated on a Superdex 200 10/300 GL column (GE Healthcare Life Sciences) assembled in the same equipment. The purity of the eluate was assessed by Coomassie-stained 12% SDS-PAGE.
Isothermal titration calorimetry analysis
The binding affinity of recombinant LPG3 (rLPG3) for heparin (16 000 Da), as well as affinity for mannose at a concentration of 1.5 mg/mL, were determined at 20°C using a VP-ITC calorimeter (MicroCal, Northampton, MA, USA). The protein diluted in buffer A at a concentration of 0.54 mg/mL was added to the VP-ITC cell unit, and the sugars loaded into the injection syringe were titrated in independent trials. Titrations were performed in a series of 28 injections of 5 µL each to achieve a complete isotherm binding. Titration of heparin or mannose in buffer was performed to obtain the heats of dilution, which were subtracted from the binding curve. The dissociation constant (Kd), number of sites (N), and enthalpy were obtained using the Microcal ITC Origin software using one site binding model.
Circular dichroism
The circular dichroism (CD) spectra of the protein were performed using spectropolarimeter Jasco Model J-815 CD Spectrometer (Jasco, Tokyo, Japan) connected to the temperature control system (Peltier). The spectra were collected in the range of 190 to 280 nm using a quartz bucket of 0.1 cm optical path. Spectra were determined as the average of 6 scans at a rate of 100 nm min−1. The structural analysis was performed at 25°C and the thermal stability was assessed from 25°C to 90°C in 5°C intervals with 5 minutes equilibration time in each temperature. Heparin (1.5 mg/mL) was added to protein at a proportion of 2:1 protein/sugar. The spectra were converted to molar ellipticity average per residue (MRW—mean residue weight), θMRW, λ defined as follows:

where

M is the molecular mass of the protein (Da) and N is the number of amino acids. θλ is the ellipticity in degrees at wavelength λ, d corresponds to the optical path (cm), and c is the protein concentration (g mL−1). 21
Molecular exclusion chromatography
The quaternary structure of rLPG3 was assessed by molecular exclusion chromatography on a Superdex 200 10/300 GL column (GE Healthcare Life Sciences). The apparent molecular mass of the protein was calculated based on its elution volume plotted in a calibration curve, designed using the following protein standards: ribonuclease (13.7 kDa), carbonic anhydrase (29 kDa), ovalbumin (43 kDa), conalbumin (75 kDa), alcohol dehydrogenase (150 kDa), and ferritin (440 kDa). The calibration curve was constructed according to the constant Kav, calculated considering the elution volume of each protein (Ve, absorbance maximum volume), the column void volume (Vo, corresponding to the blue dextran elution volume), and the column volume (Vc), where:

Cross-linking assay
The cross-linking reaction was initiated by adding 1 mmol L−1 disuccinimidyl suberate (DSS) or 1 mmol L−1 ethylene glycol-bis succinimidyl succinate (EGS) in 0.4 mg/mL recombinant protein solution. After 25 minutes at room temperature, the reaction was stopped by adding the sample buffer containing β-mercaptoethanol, followed by heating to 95°C. Proteins were separated by SDS-PAGE on 10% polyacrylamide gel and stained by Coomassie blue.
Native gel electrophoresis
Electrophoresis in a native gel (polyacrylamide gel electrophoresis [PAGE]) was performed on 8% polyacrylamide gel submitted to 90 V/4 h on an ice bath.
ATP hydrolysis assay
The hydrolytic activity of rLPG3 over ATP was measured by high-performance liquid chromatography (HPLC) as described elsewhere 22 with minor modifications. Overall, 14 µmol L−1 recombinant protein was incubated with 45 µmol L−1 ATP or 1.5 mg/mL heparin plus 45 µmol L−1 ATP in the presence of 5 mmol L−1 MgCl2. Aliquots were removed at different time intervals and immediately frozen in liquid nitrogen. Nucleotides were extracted from the samples as follows: ice-cold HClO4 (0.5 mol L−1 final concentration) was added to the samples and incubated at 4°C/10 min. The protein pellet was separated by centrifugation at 16 000 g at 4°C/10 min. KOH and K2HPO4 were added to the supernatant (one-sixth volume of 3 mol L−1 and 1 mol L−1 stock solutions, respectively) followed by the addition of 0.5 mol L−1 acetic acid (final concentration), and centrifugation at 20 000 g at 4°C/10 min. The ATP was separated from adenosine diphosphate (ADP) by anion exchange chromatography on a Protein Pack DEAE 5 PW 7.5 mm × 7.5 cm column (Waters, Milford, MA, USA) attached to a Waters 2695 chromatography system. The column was equilibrated with 25 mmol L−1 Tris at pH 8.0, and 200 µL of each sample were loaded into the system and eluted with a NaCl gradient of 0.1 to 0.45 mol L−1 in 10 minutes at 1 mL/min flow rate at room temperature. The absorbance was monitored at 253 nm. The retention times of each guanine nucleotide were compared with a mixture of 5 µmol L−1 ADP and 5 µmol L−1 ATP measured in the same sample buffer after treatment with HClO4.
Molecular modeling
Protein secondary structure predictions were performed using Phyre2 and Jpred4 software.23,24 The 3D structure of LPG3 protein was predicted by homology modeling using Phyre2 software: multiple protein templates were recovered from RCSB PDB 25 with at least 40% of identity, and a confidence score of 100% was used to build 1000 models. The best scored model was selected and optimized by molecular dynamics simulation using YAMBER 3 package 26 for model refinement, reducing the steric clashes between residues. The high quality of model was evaluated by PROCHECK 27 and ProSA 28 softwares. The protein-protein docking to tetrameric complex modeling was performed using M-ZDOCK29,30 Web server. The top 10 models of this program were compared and the most repetitive structure with smaller root-mean-square deviation against other models was selected as the consensus model. The heparin- and ATP-binding sites were predicted using 2 independent methodologies. For heparin, we used the specific optimized package for this molecule implemented by ClusPro Web server31,32 and the algorithm AutoDock Vina 33 to calculate the predicted affinity energy. The ATP-binding site was also performed with AutoDock Vina and SwissDock Web servers33,34 using flexible ligand and rigid protein. PyMOL 1.3 was used to analyze the polar contacts between proteins in the tetramer and ligand-protein in the heparin-LPG3 complex as well as to design figures. 35
Results
HBPLc is coded by LPG3 gene
HBPLc was obtained from the total extract of L infantum chagasi promastigotes, submitted to SDS-PAGE, and the 105-kDa band was extracted from the gel, as described elsewhere. 6 The protein was subjected to sequencing by MALDI TOF-TOF MS positive ion reflective mode, revealing the peaks analyzed in MS2 (Supplementary Figure 1A).
Tryptic peptides analysis performed in MASCOT database using the method of peptide mass fingerprinting was compared with the NCBI database for investigating the specific Leishmania species and allowed to detect matches with X peptides, producing a highly significant overall score of 556 (scores >33 are considered significant—data not shown). Alignment of identified peptides using pBLAST implemented on Uniprot KB resulted in a 15% sequence coverage of the protein A4I4C9_LEIIN (Supplementary Figure 1B), a L infantum protein identified as a putative LPG3. This protein has 2 domains described in the Pfam database: ATP-binding domain (H+-ATPase 3 domain) and a domain related to protein folding (HSP90 domain—Supplementary Figure 1C). Using pBLAST implemented on TritrypDB, 11 the gene sequence was identified as LinJ.29.0790—coding a heat shock protein 90/putative LPG3. HBPLc may hence be identified as the LPG3 protein. Analysis of LPG3 protein indicates the presence of a signal peptide, 3 possible N-linked glycosylation regions, and 2 O-linked glycosylation regions (Supplementary Figure 1B), but it was not possible to find the GPI-anchoring regions.
rLPG3 can bind heparin
rLPG3 was detected in the soluble fraction by SDS-PAGE, submitted to nickel affinity chromatography, followed by heparin affinity chromatography and later submitted to size-exclusion chromatography (Supplementary Figure 2).
Isothermal titration calorimetry (ITC) analysis was performed to evaluate the sugar affinity of rLPG3. The binding isotherms obtained from rLPG3 titrated with heparin (Figure 1, left) revealed that the reaction with heparin is exothermic, with an enthalpy value (ΔH) of −4.368 × 104 ± 4265 kcal/mol and a Kd of 2.33 × 10−6 mol L−1. The number of heparin-binding sites (N) was 0.507 ± 0.0402, indicating that not all sites on rLPG3 are occupied simultaneously or that 1 heparin binds to 2 molecules of protein and the binding can occur at the dimerization interface (see section “Discussion”). As a negative control, titration with mannose was performed and indicated no binding with the protein (Figure 1, right).

rLPG3 sugar-binding analysis. Graphics represent raw isothermal titration calorimetry data (top) and binding isotherms (bottom) from the sugar-binding assays using rLPG3 (6.5 µmol L−1 or 0.54 mg/mL) in standard buffer titrated with 1.5 mg/mL heparin or 1.5 mg/mL mannose. Each titration was performed with 28 injections of 5 µL.
LPG3 is a tetrameric complex
Purified rLPG3 was subjected to size-exclusion chromatography, which resulted in a chromatogram with a single peak, indicative of a pure and homogeneous sample (Supplementary Figure 2A). Comparison of the elution volume of the peak from rLPG3 sample with the calibration curve allowed the determination of an apparent molecular weight of 380 kDa (Figure 2A). To confirm this result, rLPG3 was submitted to PAGE. The electrophoresis allowed the detection of 2 bands compatible with expected masses of a monomer (~86 kDa) and a tetramer (~348 kDa) (Figure 2B—asterisk and arrow, respectively). A third band slightly smaller than 669 kDa was detected, probably indicating protein polymerization. Cross-linking assay of rLPG3 with 2 different bonding agents (EGS or DSS) allowed the detection of 2 bands with sizes compatible with a monomer and a tetramer (Figure 2C—asterisk and arrow, respectively).
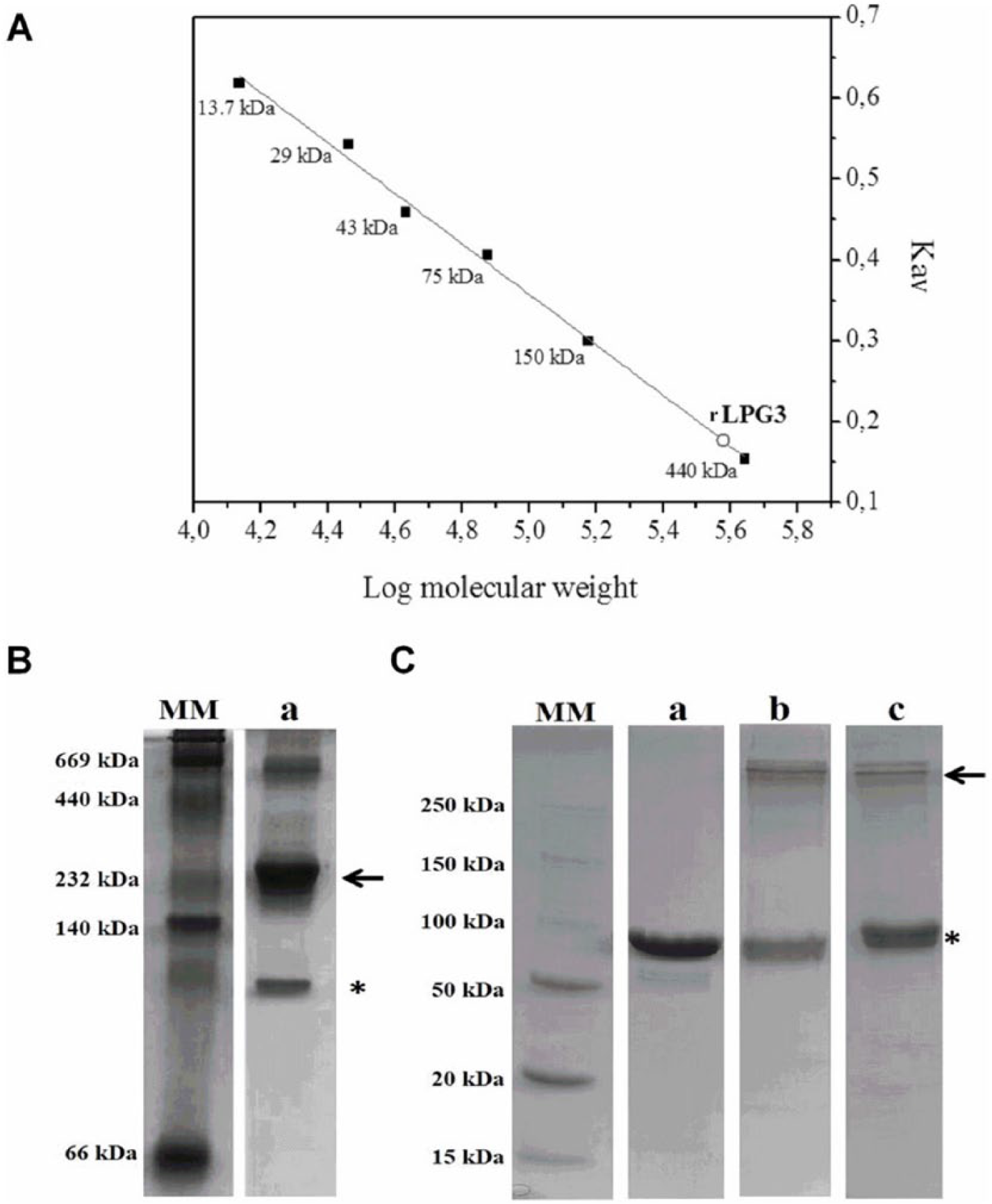
rLPG3 oligomerization. (A) Molecular mass of rLPG3 was estimated by size-exclusion chromatography using Superdex 200 10/300 GL column, as described in the methodology. Calibration curve was designed using the standards: ribonuclease (13.7 kDa), carbonic anhydrase (29 kDa), ovalbumin (43 kDa), conalbumin (75 kDa), alcohol dehydrogenase (150 kDa), and ferritin (440 kDa). Estimated rLPG3 molecular mass: 380 kDa. (B) Native gel electrophoresis: 20 µg of rHBPLc (a) was submitted to polyacrylamide gel electrophoresis and stained with Coomassie blue, as described in the methodology. (C) Cross-linking assay: rLPG3 was incubated in the presence of EGS or DSS by 25 minutes. The reaction was stopped and the samples were subjected to 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis, as described in the methodology. a—20 µg rLPG3 without bonding agent; b—20 µg rLPG3 + 1 mmol L−1 EGS; c—20 µg rLPG3 + 1 mmol L−1 DSS. DSS indicates disuccinimidyl suberate; EGS, ethylene glycol-bis succinimidyl succinate; MM, molecular mass standard.
Modeling of LPG3 structure
The modeling of LPG3 protein was performed using the Phyre2 program. 23 The structural modeling was performed by homology against multiple templates, and the model was refined by molecular dynamics. After undergoing strict quality control (Supplementary Figure 3), the distribution of secondary structures was compared with the predictor Jpred, 24 resulting in high compatibility among them (Figure 3A). Structures of α-helices interspersed by β-sheets were detected (Figure 3A and B), as confirmed by CD analysis (data not shown).

Secondary structure and 3D model predictions to LPG3 protein. (A) Secondary structure of LPG3 protein predicted by Phyre2 and Jpred softwares, (B) secondary structure of LPG3 protein in 3D model, and (C) surface structure prediction of LPG3 protein. Red ribbons, yellow arrows, and green lines represent α-helices, β-sheets, and unstructured regions, respectively. 3D indicates 3-dimensional.
The structure of the tetramer was predicted by modeling of a protein-protein docking using M-ZDOCK.29,30 Of the top 10 models, 8 models with the highest score created by the program showed similar conformation (Supplementary Table 1), suggesting that the model is suitable to represent the tetramer (Figure 4A). The heparin-binding site was identified using a specific and highly validated algorithmic function implemented in ClusPro,31,32 predicting a binding interface between 2 monomers (Figure 4B). This site was also confirmed using the Autodock Vina program, 33 and the predicted binding energy was higher to the tetramer than to the monomeric protein. Furthermore, this binding model suggests that heparin can influence the binding between 2 monomers. In fact, analysis of the secondary structure by CD under different temperatures revealed that the rLPG3 is stable against temperature and that heparin provides greater stability for the protein at higher temperatures (Figure 5).
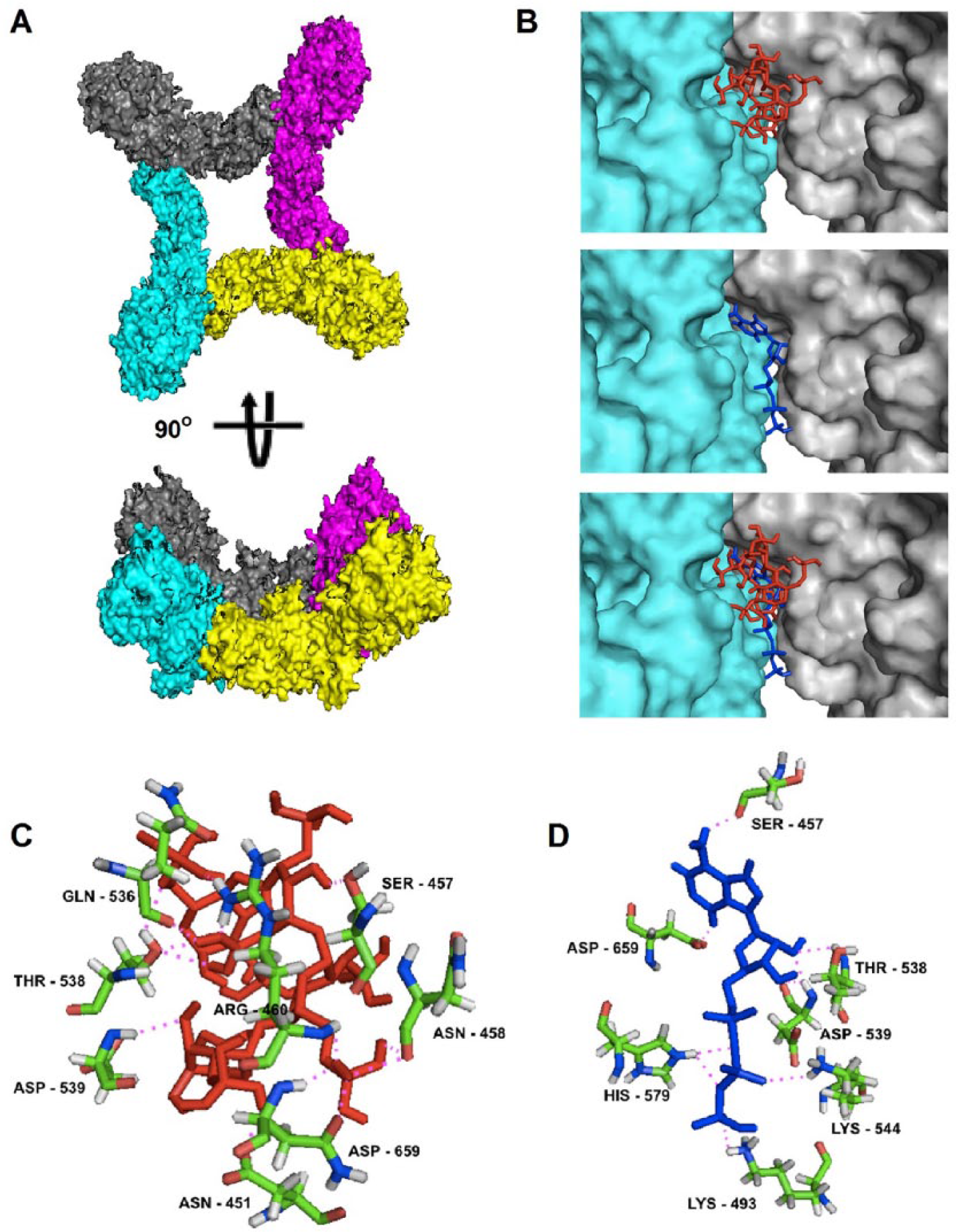
The structural modeling of the LPG3 complex and ligand-binding sites. (A) Complex surface structure created by M-ZDOCK software, (B) predicted heparin- and ATP-binding sites over intermolecular interface of contact between 2 LPG3 protein monomers and colocalization of heparin- and ATP-predicted sites, (C) predicted interactions between amino acids from heparin-binding site and heparin molecule, and (D) predicted interactions between amino acids from ATP-binding site and ATP molecule. The surface structure of each LPG3 protein is represented by gray, cyan, purple, and yellow colors in (A) and (B). Blue and red lines represent ATP and heparin molecules, respectively. Magenta dotted lines represent predicted polar contacts among atoms from LPG3 amino acids and ligand molecules. ATP indicates adenosine triphosphate.

Thermal stability analysis of the rLPG3 secondary structure in the presence of heparin. (A) rLPG3 and (B) rLPG3 associated with heparin were analyzed by circular dichroism at different temperatures, as described in the methodology.
In silico experiments of ATP-binding site predict an overlap with the site of heparin, suggesting that heparin could acts as a competitive inhibitor of ATP hydrolytic activity (Figure 4B). Four key amino acid residues from LPG3 predicted to allow ATP binding also interact with heparin, including SER-457, THR-538, ASP-539, and ASP-659 (Figure 4C and D).
rLPG3 displays hydrolytic activity over ATP
The ATPase activity assay was performed by incubating ATP with rLPG3 in the presence or absence of heparin for different intervals. Both ATP and ADP contents were determined by HPLC (Figure 6A and B, respectively). From 0.5 hours, onward, the amount of ATP was reduced gradually, being practically depleted within 2.5 hours (Figure 6C). When the protein was previously incubated with heparin, depletion of ATP took more than 3.5 hours (Figure 6C), being suggestive of a catalytic inhibition promoted by heparin.

ATPase activity of rLPG3. The profile of adenosine nucleotides amounts after elution in DEAE-5PW anion exchange column: ATP and ADP were measured at 253 nm at different time intervals after incubation of ATP in the presence of rLPG3 or rLPG3 + heparin, as described in the methodology. (A) rLPG3 activity, (B) rLPG3 + heparin activity, (C) representative graphic of the AUC for each ATPase activity at different time points. ADP indicates adenosine diphosphate; ATP, adenosine triphosphate; ATPase, adenosine triphosphatase; AU, absorbance units; AUC, area under the curve.
Evolutionary analysis
Knowing that LPG3 can interfere in the parasite infectivity, 6 we used its amino acid sequence and functional heparin-binding domain to compare L infantum chagasi with other trypanosomatids of medical importance. It was observed that the protein is highly conserved between the evaluated species, with 68% similarity to T cruzi, 87% to L braziliensis, 90% to L tarentolae, 98% to L major and L mexicana, and 100% to L donovani LPG3 (data not shown). The percentage of similarity of heparin-binding site from L infantum chagasi was >89% for T cruzi, 95% for L tarentolae and L braziliensis, and 100% for the other evaluated species, revealing the evolutionary pressure for the conservation of this site (Figure 7A). We also observed heparin- and ATP-binding sites overlapping after amino acids alignment using the program Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo/) (black highlights in Figure 7A). Reconstruction of the phylogenetic tree suggests that the LPG3 followed an evolutionary history similar to parasite speciation, allowing complete separation between Trypanosoma and Leishmania genus based on protein sequences (Figure 7B). This bias is strengthened by the presence of a clade associated with visceral Leishmania species (L infantum chagasi and L donovani) in the evolutionary model. The residues from the proposed heparin- binding site are highly conserved, which is suggestive of a critical role in protein function.

Conservation of predicted heparin- and ATP-binding sites and evolutionary relationship among LPG3 from trypanosomatides. (A) Amino acids from heparin-binding site (8 Å) are highlighted. Gray and black highlighted residues: heparin-binding site (identity >89% for the species analyzed). Black highlights correspond to the overlap of heparin- and ATP-binding sites. Amino acid positions are numbered. (B) Phylogenetic tree generated from multiple alignment of LPG3 protein comparing Leishmania infantum chagasi and others trypanosomatides. The numbers on the tree nodes are posterior probabilities calculated by Clustal W in maximum likelihood method. LPG3 amino acids sequence for the species Trypanosoma cruzi (TcCLB.506989.190), Leishmania braziliensis (LbrM.29.0780), Leishmania tarentolae (LtaP29.0800), Leishmania major (LmjF.29.0760), L infantum chagasi (LinJ.29.0790), Leishmania donovani (LdBPK_290790.1), and Leishmania mexicana (LmxM.08_29.0760) were aligned using the program Clustal Omega, as described in the methodology. ATP indicates adenosine triphosphate.
Discussion
Previously, we demonstrated that binding of HBPLc with heparin resulted in decreased infection rates, thus suggesting that this protein participates in the infection by L infantum chagasi promastigote forms in macrophages. 6 In this study, we provided further molecular evidence of the interaction between HBPLc and heparin and explore possible consequences to protein structure and function.
Identification of HBPLc was possible through proteomic analysis. Data from the analysis using MALDI TOF-TOF mass spectrometer were compared with MASCOT database, revealing that HBPLc is encoded by LPG3 gene. Since the end of the past century, studies using L donovani LPG3 genes had described the presence of LPG class I (LPG1 and LPG4A) and LPG class II (LPG2 and LPG3) genes. LPG1 and 4A genes encode glycosyltransferases and enzymes important to the biosynthesis of LPG linkages, which make it an important target for leishmaniasis chemotherapy, because they are not found in mammalians. LPG 2 and 3 genes encode proteins involved in the guide of LPG to the secretory pathway in the parasite. 36 In addition, our data obtained from peptide analysis in pBLAST implemented on Uniprot KB indicate that LPG3 gene codes for the sequence of a “putative lipophosphoglycan biosynthetic protein,” which has 2 functions already described: ATP binding and protein folding, derived from 2 functional domains (H+-ATPase_C and HSP90, respectively). Therefore, the ability to bind heparin is a novel function of the protein coded by the LPG3 gene.
The presence of HSP90 domain in LPG3 gene suggests the importance of LPG3 protein for the maintenance of the parasite life cycle. HSP90 proteins have been described to contribute to the adaptation to stress caused during transmission from the insect vector to the mammalian host, where there is a change in the ambient temperature of over 10°C, which would be a key to trigger the promastigote to amastigote differentiation.37,38 However, HSPs can stimulate the innate immune response of the host, improving its adaptive immune response. In addition, they induce dendritic cells to produce pro-inflammatory cytokines such as IL-1, IL-6, TNF-α (tumor necrosis factor α), and IL-12,39,40 which consequently induce strong TH1 immune response and strong cellular immunity. 41 These observations lead us to consider LPG3 protein as a strong candidate for anti-Leishmania vaccine, as also suggested by other authors.42–44
In silico analysis of LPG3 sequence allowed the detection of a signal sequence, which suggests that previously reported presence of the protein in the parasite vesicles and plasma membrane 6 probably occurs through the canonical secretion pathway. The observed presence of HBPLc in the plasma membrane suggests the presence of an uncleaved signal peptide or an interaction with protein and carbohydrate components of the parasite membrane.
rLPG3 was produced in the E coli heterologous system and enables us to overcome limitations from previous studies, where limited amount of native protein was extracted and purified from the parasites. 6 The recombinant protein is highly soluble and produced in satisfactory purity grade, which was evident by the presence of a single peak after purification on Superdex 200 and the presence of a single band of approximately 86 kDa on SDS-PAGE.
Gel filtration chromatography, nondenaturing electrophoresis, and cross-linking experiments suggest that LPG3 is organized mostly as a tetrameric complex. This is not surprising because previous studies have shown that porcine HSP90 proteins are able to polymerize into tetrameric, hexameric, and dodecameric complexes. 45 The low abundance of tetramers in cross-linking experiments suggests reduced efficiency of these reactions and therefore this experiment must be analyzed with caution. The model of the tetramer produced in silico suggests that it might be further stabilized by heparin bond in the interface between 2 monomers. Its secondary structure is composed predominantly of α-helices, as predicted by in silico analysis and confirmed by CD, which revealed 2 negative peaks at around 208 and 222 nm. Circular dichroism spectra of the protein in varying temperatures indicate rLPG3 spectra transition between 50°C and 60°C, with the increase in a negative peak at ~215 nm suggestive of increase in the content of β-sheets. The CD spectra also indicate that the protein remains structured in temperatures up to 80°C. It has been previously described that porcine HSP90 proteins undergo a structural change between 53°C and 63°C and that such change is important for the observed oligomerization of the protein in high temperatures.46,47 Therefore, it is possible that the observed changes in structure in rLPG3 generated by temperature increase are analogous to that described for porcine HSP90. Further studies are necessary to evaluate whether the observed structural changes in rLPG3 are also associated with the changes in the oligomerization states. This change is noticeable in samples with and without heparin, thus suggesting that the presence of this ligand does not influence such possible changes in oligomerization states.
The ITC analysis indicates that rLPG3 is able to bind to heparin with micromolar affinity. Moreover, the fact that heparin stabilizes rLPG3 structure at high temperatures provides further evidence of this interaction. It has been previously suggested that human HSP90 proteins are anchored in the human cell surfaces through interactions with heparan sulfate proteoglycans. This hypothesis is based on observations that treatment of cells with sodium chlorate, heparinase, and heparin leads to detachment of HSP90 proteins from cell surface and inhibits binding of exogenous HSP90 proteins. 48 Therefore, it is tempting to propose that rLPG3 could also interact with human cells using a similar mechanism.
Heparin-binding sites are commonly observed on the protein surface corresponding to shallow pockets of positive charge. A minimum requirement for binding that appears to be common among HBPs is the concentration of basic residues in a particular region of the protein surface (not necessarily near each other in a sequence), oriented in a geometry that matches the pattern of sulfate groups along heparin. Moreover, the presence of a geometry that can accommodate an elongated molecule seems to be necessary for the binding.2,3,49 The molecular interactions that promote binding between heparin and protein are ionic interactions, wherein positively charged clusters of basic amino acids form ion pairs with sulfo- or carboxyl groups of negative charge in the heparin chain. However, in some cases, there is contribution of nonionic interactions such as hydrogen bond and hydrophobic forces, which may play a minor role in heparin-protein interaction. 2 The pattern of expected binding site was also observed in the binding model of heparin and rLPG3. Most of the amino acids near the binding site are composed of basic side chains such as lysine, histidine, and arginine. The enrichment of these amino acids suggests a positive formal charge of the binding site. However, the model predicts that polar intermolecular interactions are also important to stabilize heparin binding, including hydrogen interactions with aspartate residues.
The ATP hydrolysis measurements allowed inferring that heparin displays an inhibitory activity toward ATP hydrolysis. In silico prediction suggests that such effect would be the consequence of a superposition between the ATPase and heparin-binding domain. Interestingly, it has been shown that heparin blocks the binding of ATP in the protein fibroblast growth factor 2 (FGF2) and that mutations in the heparin-binding site of this protein reduced the affinity for ATP. 50 Therefore, it is possible that certain general characteristics in the architecture of ATP-binding sites favor the binding of heparin, thus explaining the coincident effect of inhibition of ATP hydrolysis by heparin in both FGF2 and HSP90 proteins. Further studies using single or multiple mutants in amino acids involved in the heparin and ATP binding, such as the critical residues Ser 457, Thr 538 and Asp 539, could explain mechanistically this inhibitory activity of rLPG3 over ATP hydrolysis.
The ATP hydrolytic activity of rLPG3 is consistent with the presence of H+-ATPase domain detected using the HMM search against Pfam database. The parasites of the genus Leishmania are devoid of de novo synthesis of purine nucleotides and need to use preformed purine from the host environment to satisfy their demand for nucleotides, translocating these molecules through their plasma membrane. 51 The ATP has previously been shown to be a “danger” extracellular signal induced by infection of pathogens or injury that can trigger different cellular events such as proliferation, differentiation and chemotaxis, release of cytokines and lysosomal constituents, and the generation of oxygen or nitrogen reactive species. 52 The ecto-ATPase activity of pathogens is an adaptive parasitic behavior that makes these organisms more virulent because of their interference in the extracellular pro-inflammatory content, as a result of a decrease in the extracellular ATP and a subsequent increase in ATP metabolites with anti-inflammatory properties, which, additionally, in the case of trypanosomatides, may be internalized and used to form purine nucleotides.53–55 As the LPG3 is well distributed in the parasite plasma membrane, 6 its involvement in the purine salvage process and parasite virulence mechanism is possible. It should be noted, however, that LPG3 catalytic activity occurs only on ATP and cannot provide adenosine monophosphate (AMP) to further hydrolysis aiming to nucleotide internalization. Ecto-NTPDases can be considered more suited for such purpose, as they are able to hydrolyze both ATP and ADP to AMP. 56 Interestingly, we also observed overlapping of the putative heparin- and ATP-binding sites, which could explain the observed changes in the hydrolytic activity from LPG3 over ATP in the presence of heparin. These results suggest that heparin possesses regulatory properties over the ATP metabolism and, consequently, over the acquisition of extracellular metabolites used to form purine nucleotides. The conservation of the heparin-binding domain in LPG3 from different parasite species reinforces the idea that these residues belong to an active binding site and is suggestive of the importance of this protein to trypanosomatid biology. The fact that LPG3 might be involved in several processes critical for parasite survival reinforces the idea of LPG3 as a target for drug research and anti-Leishmania vaccine development.
Conclusions
In this work, we demonstrated a novel function for LPG3 protein: the ability to bind heparin. Moreover, in silico protein modeling followed by docking analysis suggests that heparin interacts with LPG3 by contacting several residues, including some that belong to an ATP-binding domain. The fact that incubation with heparin reduces the ATPase activity of LPG3 provides support for this model and suggests that heparin interferes in the ability of the parasite to acquire extracellular nucleotides by delaying ATP hydrolysis. Considering these results and the previously demonstrated importance of HBPs for adhesion and internalization of Leishmania into the host cell and the conservation of the heparin-binding domain of LPG3 among different trypanosomatid species of medical importance, the characterization of LPG3 as an HBP reinforces the potential of this protein as target for new drugs and vaccine development against leishmaniasis.
Supplemental Material
Supplementary_Material – Supplemental material for Lipophosphoglycan 3 From Leishmania infantum chagasi Binds Heparin With Micromolar Affinity
Supplemental material, Supplementary_Material for Lipophosphoglycan 3 From Leishmania infantum chagasi Binds Heparin With Micromolar Affinity by Thaís Viana Fialho Martins, Ana Eliza Zeraik, Natália Oliveira Alves, Leandro Licursi de Oliveira, Tiago Antônio de Oliveira Mendes, Ricardo DeMarco and Eduardo de Almeida Marques-da-Silva in Bioinformatics and Biology Insights
Footnotes
Acknowledgements
The authors are grateful to the Núcleo de Análise de Biomoléculas of the Universidade Federal de Viçosa for providing the facilities for the conduction of the experiments.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors acknowledge the financial support by the following brazilian agencies: Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG), Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), Coordenacão de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Financiadora de Estudos e Projetos (FINEP) and Sistema Nacional de Laboratórios em Nanotecnologias (SisNANO)/Ministério da Ciência, Tecnologia e Informação (MCTI).
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
EdAM-d-S conceived the idea for the project. TVFM conducted most of the experiments, analyzed the results, and wrote most of the paper. AEZ and TVFM conducted ITC, cross-linking, circular dichroism, molecular exclusion chromatography, and ATP hydrolysis assays. NOA and TVFM conducted experiments of rHBPLc expression and purification. TAdOM conducted in silico analyses. EdAM-d-S, TAdOM, LLdO, RDM, and TVFM analyzed the results and wrote the paper. All authors reviewed and approved the final manuscript.
Disclosures and Ethics
As a requirement of publication, authors have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the authorship and contributor ship, conflicts of interest, privacy and confidentiality. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.