
Abstract
α-Amylase is one of the important enzymes in the starch-processing industry. However, starch processing requires high temperature, thus resulting in high cost. The high adsorptivity of α-amylase to the substrate allows this enzyme to digest the starch at a lower temperature. α-Amylase from Saccharomycopsis fibuligera R64 (Sfamy R64), a locally sourced enzyme from Indonesia, has a high amylolytic activity but low starch adsorptivity. The objective of this study was to design a computational model of Sfamy R64 with increased starch adsorptivity using bioinformatics method. The model structure of Sfamy R64 was compared with the positive control, ie, Aspergillus niger α-amylase. The structural comparison showed that Sfamy R64 lacks the surface-binding site (SBS). An SBS was introduced to the structure of Sfamy R64 by S383Y/S386W mutations. The dynamics and binding affinity of the SBS of mutant to the substrate were also improved and comparable with that of the positive control.
Introduction
α-Amylase, or 1,4-α-
The raw starch–digesting amylases are mostly produced by fungi, such as Aspergillus sp., Rhizopus sp., and Corticium rolfsii.7,8 In Indonesia, the best identified amylolytic enzyme–producing microorganism was a strain of Saccharomycopsis fibuligera R64. 10 However, at the enzymatic level, the isolated α-amylase from S fibuligera R64 (Sfamy R64) showed no adsorption to the raw starch. 11 Thus, unlike the other raw starch–digesting α-amylase, Sfamy R64, is predicted without the SBD and/or SBS. Unfortunately, the structure of Sfamy R64 is still not available.
Computer-Aided Molecular Design (CAMD) is one of the promising methods to develop a modified enzyme with desired properties. Fungamyl, a thermostable amylase-like enzyme at acidic pH, is one of the successful products which was engineered using CAMD technique. 12 Therefore, to improve the substrate adsorption of Sfamy R64 without compromising its excellent amylolytic activity is expected to be achieved by CAMD approach. In this study, we used the crystal structure of Aspergillus niger α-amylase, which shares 71% homology with Sfamy R64, as a positive control. It has one SBS in the C-domain which is bound to maltose. The complex structure was resolved at 1.8 Å resolution. 13
Therefore, this study aims to investigate the effect of new SBS on the model structure of Sfamy R64 toward the substrate adsorption using computational methods. The model was developed using homology modeling method. The structural dynamics behavior of Sfamy R64 as compared with the positive control was explored using molecular dynamics (MD) simulation. Moreover, the substrate binding was calculated using molecular mechanics-generalized Born surface area (MM/GBSA) method. It is hoped that results would be useful to the development of Sfamy R64 as a locally sourced enzyme for industrial purposes.
Methods
Modeling of Sfamy R64
The structure of Sfamy R64 was constructed by homology modeling method using MODELLER 9.15.14,15 The sequence of Sfamy R64 was retrieved from NCBI (https://www.ncbi.nlm.nih.gov/) with GI (GenInfo Identifier) number 315451018. 16 The functional regions within Sfamy R64 sequence were predicted using Pfam analysis (http://pfam.xfam.org/). 17 The templates for protein modeling were selected based on the sequence similarity, folding pattern, and structure quality (PDB ID 2GUY 13 and 3VM718). The discrete-optimized protein energy (DOPE) value, a statistical potential energy to assess the model, was calculated for the structures of model and template. The quality of model structure was evaluated by the Ramachandran plot using PROCHECK. 19 The mutant of Sfamy R64 was modeled by adding an SBS to the structure of Sfamy R64 wild type using Biovia Discovery Studio Visualizer. 20 The serine at positions 383 and 386 (382 and 385 in PDB 2GVY numbering scheme) was replaced by tyrosine and tryptophan, respectively.
MD simulation
A crystal structure of A niger α-amylase in complex with maltose (PDB ID 2GVY) 13 was used as positive control. The residue type of cysteine and histidine were adjusted manually, based on their specific chemical environment. A box of TIP3P water model was added to the system, where the shortest distance between protein and the edge of the box was 10 Å. The system was neutralized by the adding sodium ions.
All the minimization and MD simulations were performed using AMBER14. 21 Initial minimization of 1000 steps using the steepest descent algorithm was conducted. Then, 2000 steps of conjugate gradient minimization with 500 kcal/molÅ2 of harmonic restraints were applied to the backbone atoms. A final 1000 steps of unrestrained conjugate gradient minimization were performed to remove any sterical clashes among the atoms.
The system was gradually heated to 50°C ≈ 323 K, its optimum temperature, 11 for 60 ps in NVT ensemble using harmonic restraints of 5 kcal/molÅ2 on the backbone atoms. Furthermore, 1000 ps of NPT equilibration was performed, where harmonic restraints on the backbone were slowly decreased by 1 kcal/molÅ2 until it reaches 0. Then, 20 ns of the production run in NPT ensemble was performed with all hydrogen atoms constrained using the SHAKE algorithm. The temperature was controlled with Langevin thermostat with a collision frequency of 1 ps−1, whereas the pressure was controlled using Berendsen barostat with the coupling constant of 1 ps and the target pressure of 1 bar. The time step value during the production stage was 2 fs. The nonbonded cutoff value of 9 Å was used, and the long-range electrostatics was treated using particle mesh Ewald. The MD trajectories were analyzed using the cpptraj module in AmberTools15.
Calculation of binding affinities
All binding energy calculations were done using MMPBSA.py 22 in AmberTools15. According to the MM/GBSA theory, binding free energy (ΔGbind) between an enzyme (E) and a substrate (S) to form a complex ES is calculated as follows:



where ΔH is the enthalpy, T is the temperature in Kelvin. ΔEMM is the MM energy change in the vacuum, which is composed of ΔEint as the internal energy, ΔEelec as the Coulomb electrostatic term, and ΔEvdw as the van der Waals interaction term. ΔGsolv is the solvation free energy which is composed of ΔGGB as the electrostatic solvation energy (polar contribution) calculated by GB model and ΔGSA as the nonelectrostatic solvation component (nonpolar contribution). The interval step of 10 ps for MM/GBSA calculation and the salt concentration of 150 mM were applied.
Results
Modeling of Sfamy R64
Two functional regions were revealed when the Sfamy R64 sequence was submitted to the Pfam analysis (http://pfam.xfam.org/), 17 ie, a catalytic domain of α-amylase (A/B-domain) and an unknown domain at the C-terminal. Further analysis by Phyre2 server showed that 79 residues at its C-end had a high folding similarity to a glycosyl hydrolase domain, namely, C-domain, not a CBM (Supplementary data).
The model of Sfamy R64 was constructed using multitemplate approach. The selected templates were PDB ID 2GUY 13 and 3VM7. 18 The first template was a structure of A niger α-amylase which has the best sequence similarity to that of Sfamy R64 (54% identity and 71% homology), whereas the second template has the best similar folding pattern to Sfamy R64 detected by Phyre server (http://www.sbg.bio.ic.ac.uk/), 23 an α-amylase structure from Malbranchea cinnamomea (50% identity and 66% homology). These crystal structures were resolved at good resolutions, ie, 1.59 and 2.25 Å, respectively. The multiple sequence alignment of Sfamy R64 and the templates is presented in Figure 1. A gap in Sfamy R64 sequence was found at the positions 401 to 404 when compared with the A niger α-amylase.

Multiple sequence alignment of Sfamy R64 with the templates (PDB ID 2GUY and 3VM7).
Furthermore, the quality of Sfamy R64 model was assessed by the Ramachandran plot. It is shown that 92.3% residues were located in the most favored regions, whereas 7.2% and 0.5% residues fell into the additional allowed and generously allowed regions, respectively (Figure 2). None of the residues were located in the disallowed region. In general, a protein structure with more than 90% residues in the allowed region is categorized as a good model. 19 The superimposition of the model with the templates is presented in Figure 3. Moreover, the successful effort in modeling the structure of Sfamy R64 using multitemplates was indicated when comparing its quality with the single-template–based models. The model which was constructed based on PDB 2GUY and 3VM7 individually had only 90.9% residues in the most favored region (Supplementary data). Moreover, their DOPE profiles were higher than that of the multitemplate-based model (Supplementary data). Also, the multitemplate-based model showed close structural similarity to both the templates, which is indicated by small root-mean-square deviation (RMSD) values, ie, 0.67 and 0.74 Å for PDB 2GUY and 3VM7, respectively.

Ramachandran plot of the model structure of Sfamy R64.

The superimposition of the Sfamy R64 model and its templates: PDB ID 2GUY, and 3VM7, which are visualized in red, blue, and gray colors, respectively.
Structural comparison between SBS in A niger α-amylase and Sfamy R64
There are 3 categories of SBS (A, B, and C). The type A is a flat surface that is composed of 1 or 2 aromatic residues. 24 Aspergillus niger α-amylase falls into type A because of 2 aromatic residues which are exposed on its surface as an SBS, namely, Y382 and W385. Although Sfamy R64 was predicted to lack the CBM, interestingly, the total number of aromatic residues (tyrosine, tryptophan, and phenylalanine) in A niger α-amylase (58) and Sfamy R64 (57) were similar (Figure 4A). The A/B-domains between these 2 structures share 75% similarity. However, a visible difference between C-domain of A niger α-amylase and Sfamy R64 was observed when their structures were visualized using the solvent surface model. A patch of aromatic residues was exposed on the C-domain of one of the A niger α-amylases, but not Sfamy R64 (Figure 4B). Further analysis showed that at the position of aromatic residues in the SBS of A niger, that of Sfamy R64 is occupied by 2 serines (Figure 4C). Therefore, it can be suggested that Sfamy R64 lacks the SBS too, despite having a similar number of aromatic residues with the positive control. The intermolecular interaction between the substrate and SBS in A niger α-amylase was studied using the cocrystallized maltose (represents the shortest chain of the substrate of amylase) in PDB ID 2GVY. 13 It is shown that the maltose formed hydrogen bond with K383 and stabilized by Y382 and W385 through the CH-π stacking interaction (Figure 5). These interactions were considered to be important in many biological systems. 25 For this reason, the low substrate adsorptivity of Sfamy R64 might be due to the absence of SBS which is composed of aromatic residues in this region. To test the hypothesis, a new model of Sfamy R64 with mutations of S383Y/S386W, namely, Sfamy R64MT, was constructed and further analyzed by MD simulation.

(A) The presence of aromatic residues in Sfamy R64 (red cartoon) and Aspergillus niger α-amylase (blue cartoon). (B) The aromaticity of Sfamy R64 (left) and A niger α-amylase (right) which are visualized by solvent surface. The edge and face sides of aromatic residues are colored in blue and brown, respectively. Nonaromatic residues are colored in white. (C) The residual structural alignment of Sfamy R64 (yellow stick) and A niger α-amylase (purple stick) at the SBS region of A niger.
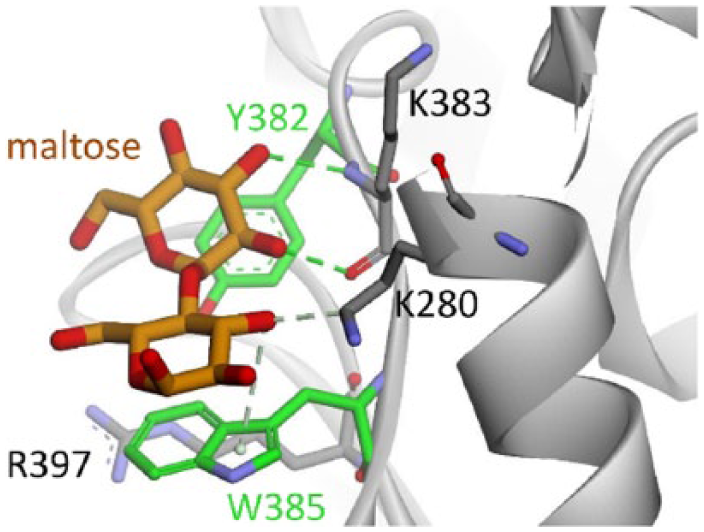
The intermolecular interaction between maltose (brown stick) and SBS residues of Aspergillus niger α-amylase. The aromatic and nonaromatic residues are visualized in green and gray sticks, respectively. Hydrogen bond is depicted in green dashed line.
Substrate binding in A niger α-amylase, Sfamy R64, and Sfamy R64MT
MD simulations of 20 ns were performed to investigate the substrate binding in the positive control (A niger α-amylase), Sfamy R64, and Sfamy R64MT. The coordinate of maltose in Sfamy R64 and Sfamy R64MT was determined by superimposition with the positive control system.
The overall structural stability of all systems was evaluated by RMSD (Figure 6A). It is showed that the Sfamy R64MT was generally more stable than its wild type (average value of 1.39 and 1.43 Å, respectively). Furthermore, the residual fluctuation of all systems was computed by the root-mean-square fluctuation calculation (Figure 6B). The highest fluctuations were observed in the N-terminal and C-terminal because these regions were not restrained, whereas the fluctuation of the catalytic site (around residue numbers 25, 125, and 150) in both wild and mutant types of Sfamy R64 was also high. On visual inspection, the catalytic site in all systems was transformed from the open to closed conformation due to the absence of a substrate in the A/B domain. The other high fluctuations which are observed around residue numbers 300, 375, and 425 were due to the natural flexibility of loop structure. Nevertheless, it is noted that these regions were distant from the SBS region. Thus, it might not reflect the harmful influence of the new SBS addition to the structure of Sfamy R64.

(A) RMSD of protein backbone and (B) residual RMSF profile of all molecular dynamics systems throughout 20 ns of simulation. RMSD indicates root-mean-square deviation; RMSF, root-mean-square fluctuation.
The RMSD of maltose in all systems was also computed to observe its mobility throughout the MD simulations (Figure 7). In the positive control, maltose was remained stable in its initial position, ie, at the SBS of A niger α-amylase. As expected, the absence of SBS in Sfamy R64 would result in the unstable binding of maltose in C-domain. In this system, maltose was moving out from its initial position after 6 ns. Interestingly, the fruitful effort of introducing the SBS in Sfamy R64 was reflected by the stable binding of maltose until 17 ns of simulation. The time evolution snapshots of MD trajectories that were taken every 2 ns confirmed the better stability of substrate in Sfamy R64MT (Figure 8A) than in its wild type (Figure 8B). However, it is worth noting that the maltose binding in positive control was completely persisted more than 20 ns of simulation (Figure 8C). Furthermore, the evidence of SBS on the surface of C-domain in Sfamy R64MT was visually confirmed using the solvent surface which is colored by the aromaticity of residues (Figure 9). It is shown that SBS was already present in the Sfamy R64MT, similar to the positive control, but not in the Sfamy R64.

The movement of maltose during 20 ns of molecular dynamics simulation, which is represented by its RMSD in Aspergillus niger α-amylase (blue), Sfamy R64 (red), and Sfamy R64MT (orange). The initial position of maltose was determined by its coordinate in the surface-binding site of A niger α-amylase (PDB ID 2GVY). RMSD indicates root-mean-square deviation.

The visual representation of maltose mobility at the surface-binding site region of (A) Sfamy R64MT, (B) Sfamy R64, and (C) Aspergillus niger α-amylase. The snapshots were taken every 2 ns from 20 ns of molecular dynamics simulation, which are colored from the initial (red) to the end position (blue).
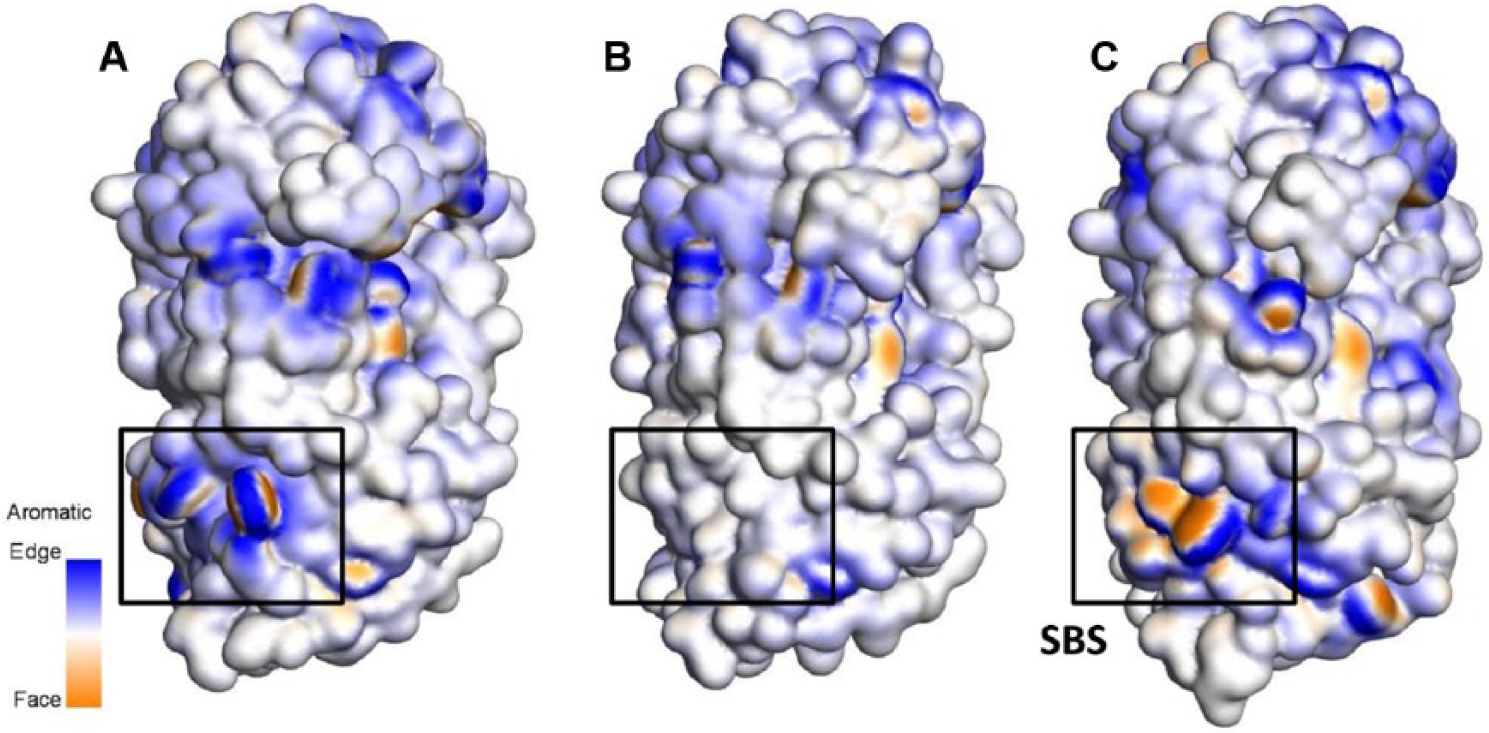
The differences of the aromaticity of (A) Sfamy R64MT, (B) Sfamy R64, and (C) Aspergillus niger α-amylase, which are visualized by solvent surface. The edge and face sides of aromatic residues are colored in blue and brown, respectively. Nonaromatic residues are colored in white.
The strength of maltose binding in all systems was evaluated too. In agreement with the observed behavior of maltose throughout the simulations, its intermolecular energy with the C-domain was found the weakest in Sfamy R64. Interestingly, after the mutation of S383Y/S386W, the binding energy of maltose was increased from −3.0 to −22.3 kcal/mol (Table 1). Besides the CH-π interaction between aromatic SBS and maltose, hydrogen bond had an important role to the overall strength of binding. Hydrogen bond analysis on the SBS region showed that the occupancy of hydrogen bond formation with N384 in the mutant was increased from 4% to 62% (Table 2). Nevertheless, the major hydrogen bond in the positive control was formed with K383, instead of N384. This observation was in accordance with the pairwise decomposition energy computed by MM/GBSA method. It is shown that interactions with SBS residues (Y383 and W386) in Sfamy R64MT were comparable with that of A niger α-amylase (Figure 10).
Interaction energies between maltose and 3 α-amylase systems calculated by molecular mechanics-generalized Born surface area method.

Hydrogen bond analysis of maltose (GLC) binding in all systems throughout 20 ns of molecular dynamics simulation.

GLC refers to substrate.
Residue number according to PDB ID 2GVY.
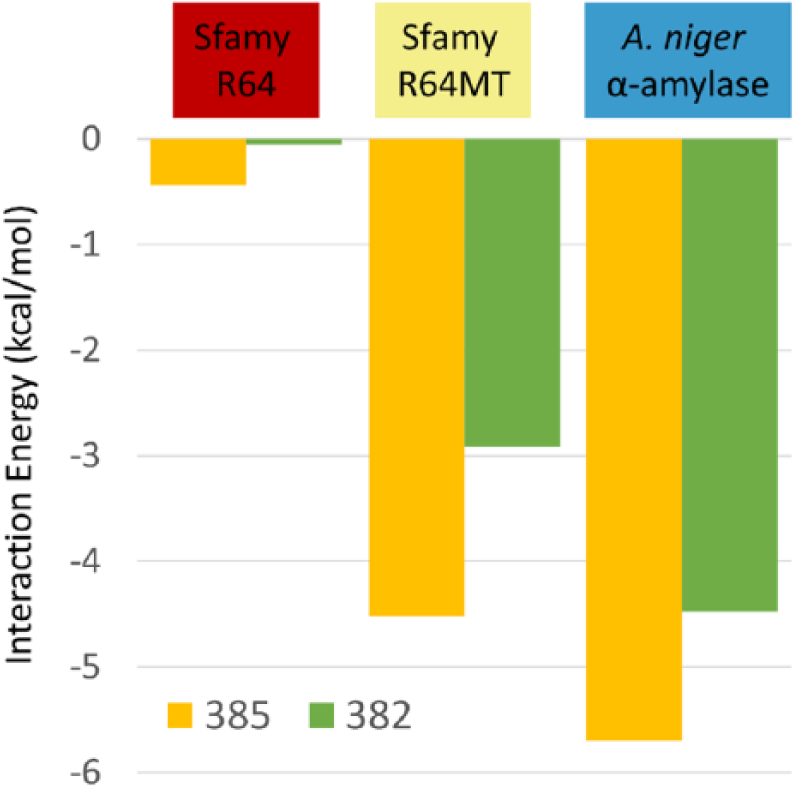
Pairwise decomposition of interaction energy between maltose and surface-binding site (the side chain of residues 382 and 385 in 2GUY numbering) in Sfamy R64, Sfamy R64MT, and Aspergillus niger α-amylase.
Discussion
Cockburn et al 26 suggested that the CBM or SBD and the SBS are responsible for the raw starch digestion ability of α-amylase. Surface-binding site has a physiological role in the binding of raw starch (and the other polysaccharide chains) through the stacking interactions with aromatic residues, eg, tyrosine, tryptophan, and phenylalanine. 13 The mutations of W278A/Y380A, W279A/Y380A, and W278A/W279A/Y380A in SBS region eliminated the ability of a barley α-amylase in binding the starch granules. 26 Moreover, the catalytic efficiencies of the mutants to the complex substrate were decreased by 60-fold to 400-fold as compared with the wild type. 26 Not only SBS but also the removal of aromatic residues on the CBM had reduced its affinity to the polysaccharide. 27 Moreover, the removal of CBM58 and SBS in SusG α-amylase decreased its activity on the insoluble cornstarch by 71% and 56%, respectively. 28
However, it is worth noting that the location of SBS should not be necessarily close to the catalytic site of α-amylase. For example, SBS in A niger α-amylase is situated far from the catalytic site, ie, ~20 Å. Therefore, although Sfamy R64 has a good amylolytic activity on the soluble substrate, it does not always have a good raw starch adsorptivity. We had performed an additional computational experiment to calculate the binding energy of acarbose, a soluble substrate, in the catalytic site of A. oryzae α-amylase (PDB ID 7TAA) and Sfamy R64. The result showed a comparable strength of acarbose binding in the catalytic site of both the enzymes (−66 and −77 kcal/mol, respectively), indicating their good amylolytic activity. Thus, the amylolytic capacity of α-amylase, especially on the soluble starch, is predicted to be independent of the presence of CBM/SBD and SBS.
Many efforts have been done to improve the substrate affinity by introducing the SBS to the structure of the enzyme. Cuyver et al 29 studied the effect of aromatic residues on the SBS of xylanases from Bacillus subtilis and Aspergillus niger. It was expected that the increase in hydrophobic stacking and hydrogen bond interactions would be achieved by mutating several amino acids with the aromatic one. Nevertheless, the binding affinity of these enzymes for the substrate was not enhanced. Also, the strength of hydrophobic and hydrogen bond interactions of the mutant was still similar to that of the wild type. In addition, some of the mutations by aromatic residues even resulted in the changes of substrate binding to a lower degree. 29 Mutagenesis study to increase the aromaticity of C-domain of Sfamy R64 has also been done. A tyrosine at position 375 was replaced with a bulkier tryptophan to provide more hydrophobic stacking interaction with the substrate. 30 However, this mutation was not sufficient to change the substrate binding of Sfamy R64. In comparison with our study, it is shown that W375 is rather located at the other side of C-domain, not at the same position with SBS in A niger α-amylase. Moreover, Figure 6B shows that the fluctuation of residues around position 375 was relatively high, indicating its low potency to stabilize the substrate. From these 2 studies, introducing SBS could be very challenging due to the specific position and environment around the aromatic residues. Cuyver and colleagues proposed that the mutation, although the crystal structure guided it, might result in the wrong orientation of the substrate, thus not delivered correctly to the active site. Besides, too many aromatic residues might cause a too strong binding, leading to the inefficient catalytic activity. 29 For this reason, an MD-guided mutation might be useful to evaluate the new formation of SBS regarding structural behavior and energetics of substrate binding. In addition, MD can be used to design a new SBS at a specific location and orientation rationally.
Based on the sequence alignment between Sfamy R64 and the A niger α-amylase, there is a putative N-glycosylation site on the N218 (numbering of A niger). At the same position, Sfamy R64 has N198. On inspection, N198 is located far away (~40 Å) from the SBS. For this reason, glycan at the N198 of Sfamy R64 and its mutant was not modeled, also to simplify the protein system in our MD simulation. It is noted that the deglycosylation did not affect the stability of Aspergillus oryzae α-amylase. 31
Besides the effort to enhance the substrate binding, some studies were done to improve the physicochemical properties of Sfamy R64.32,33 Ismaya et al 32 modified the Sfamy R64 chemically using nonpolar group. Interestingly, the stabilization factor of the enzyme was increased by 18-fold, despite the decrease in its specific activity. Moreover, Natalia et al 33 introduced a new disulfide bond between the A and C domains, based on the model structure of Sfamy R64. It is shown that activity and optimum pH were still similar to the wild type.
Although the increase in maltose affinity in Sfamy R64MT was observed, the maltose was deviated after 17 ns of MD simulations (Figure 7). Therefore, there might be another structural feature of Sfamy R64MT which is different from the positive control. The sequence alignment (Figure 1) showed that Sfamy R64MT still lacks a short loop after S383. Figure 11 indicates that this loop is required to support the conformation of Y383 to be in a similar manner to that of A niger. Moreover, the bulkier N421 had caused a different conformation of tryptophan, as compared with that in the positive control (Figure 11). The further computational experiment is needed to investigate the role of surrounding residues of mutation points to the stability of SBS. Also, the more expensive simulation involving complex substrate mimics the behavior of granular starch. In silico study should provide valuable insight to rationalize the design of new Sfamy R64 before the recombinant protein experiment in the lab.

The superimposition of surface-binding site in Sfamy R64MT and Aspergillus niger α-amylase, represented in blue and yellow colors, respectively. The differences between them are highlighted in black circle, ie, a short loop and N421.
Conclusions
A possible reason of the poor raw starch adsorptivity of Sfamy R64 was due to the absence of SBS. Based on the structural comparison between A niger α-amylase (positive control) and Sfamy R64, the latter lacks the aromatic residues at the SBS region. Molecular dynamics simulations showed that the maltose mobility in Sfamy R64 was high, whereas it remained stable at the SBS of A niger. For this reason, double mutations of S383Y/S386W were introduced to the structure of Sfamy R64. It is shown that the stability of maltose on the mutant system was better than that of the wild type. As a conclusion, mutations of S383Y/S386W are proposed to improve the raw starch adsorptivity of Sfamy R64, thus highlighting the importance of SBS in the substrate binding of α-amylase.
Footnotes
Peer review:
Three peer reviewers contributed to the peer review report. Reviewers’ reports totaled 1338 words, excluding any confidential comments to the academic editor.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Internal Grant RKDU from Universitas Padjadjaran.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
MY, UB, SDR, and TS conceived and designed the experiments. MY, UB, and KH analyzed the data. MY, UB, and TS wrote the first draft of the manuscript and contributed to the writing of the manuscript. MY, UB, SDR, KH, SI, and TS agree with manuscript results and conclusions and jointly developed the structure and arguments for the paper. MY, SI, and TS made critical revisions and approved final version. All authors reviewed and approved the final manuscript. MY and UB equally contributed to the article.
Disclosures and Ethics
As a requirement of publication, author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality, and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.