
Abstract
Vibriosis is regarded as an important disease of penaeid shrimps affecting larvae in hatcheries. Among the Vibrio species, Vibrio parahaemolyticus, Vibrio vulnificus, Vibrio furnissii, Vibrio campbellii, Vibrio harveyi, Vibrio alginolyticus, and Vibrio anguillarum are often associated with diseases in finfish and shellfish of brackishwater ecosystem. Accurate species differentiating methods for the organisms present in an ecosystem are required for precise classification of the species and to take steps for their management. Conventional methods like 16s rRNA phylogeny and multilocus sequence typing (MLST) have often failed to correctly identify Vibrio species. This has necessitated a comprehensive investigation on methodologies available to distinguish Vibrio species associated with brackishwater aquaculture system. To achieve this, 35 whole genomes belonging to 7 Vibrio species were subjected to phylogenetic analysis based on 16s rRNA gene, MLST genes, single-copy orthologous genes, and single-nucleotide polymorphisms. In addition, genome-based similarity indices like average nucleotide identity (ANI) and in silico DNA-DNA hybridization (DDH) were computed as confirmatory tests to verify the phylogenetic relations. There were some misclassifications occurred regarding phylogenetic relations based on 16s rRNA genes and MLST genes, while phylogeny with single-copy orthologous genes produced accurate species-level clustering. Study reveals that the species identification based on whole genome-based estimates or genome-wide variants are more precise than the ones done with single or subset of genes.
Introduction
Vibriosis is one of the major diseases of concern to the aquaculture. 1 Several Vibrio spp. of harveyi clade such as Vibrio parahaemolyticus, Vibrio alginolyticus, Vibrio harveyi, Vibrio owensii, and Vibrio campbellii infect farmed aquatic animals. 2 They affect both fish and shrimp in marine environment and brackishwater aquaculture. Vibrio harveyi infections result in 80% to 100% mortality during the early larval stages and inflict huge economic losses to shrimp hatcheries. 3 Vibrio parahaemolyticus is another opportunistic pathogen, which devastated the shrimp aquaculture sector in Southeast Asia in recent years by causing acute hepatopancreatic necrosis disease (AHPND), also called as early mortality syndrome (EMS). 4 Due to the outbreak of AHPND, Thailand lost about 7% of production in 2012, Vietnam experienced USD7.2 million losses and Mexico lost USD118 million. 5 Diagnosis of vibrios is done by phenotypic or polymerase chain reaction (PCR) tests for identification of these bacteria at species level. Accurate identification of species is difficult with the conventional phenotypic methods. Often, the 16s rRNA sequencing also fail to correctly identify Vibrio species. In such a scenario, application of bioinformatics tools can aid in differenti-ating these pathogenic strains. 6 Vibrios are Gram-negative bacteria in the family Vibrionaceae under the phylum Gammaproteobacteria and are found abundantly in marine environments. 7 Genome of vibrios contains 2 chromosomes, of which chromosome 1 is longer than chromosome 2 and is about two-thirds of total genome regarding its length. The longer chromosome carries housekeeping genes, while the smaller one has accessory genes.8,9 The GenBank (https://www.ncbi.nlm.nih.gov/) consists of 88 complete genomes of vibrio species as on April 2019.
Recent advances in next-generation sequencing made it possible to study the phylogenetic relations of targeted organisms more accurately at genome level using in silico approaches. Several whole genomes of Vibrio spp. sequenced from different laboratories located around the globe were deposited at GenBank and made readily available for reanalysis studies. Present study aims at finding appropriate methods for differentiating and finding evolutionary distances of Vibrio spp. that are more commonly found in brackishwater ecosystem with the available complete genomes and modern bioinformatics tools.
Materials and Methods
Genomic data
In all, 35 genome sequences were downloaded from GenBank including LB102 strain of V campbellii, a brackishwater isolate sequenced in-house. 10 Complete genomes representing 7 Vibrio spp., V parahaemolyticus, Vibrio vulnificus, Vibrio furnissii, V campbellii, V harveyi, V alginolyticus, and Vibrio anguillarum were downloaded from GenBank (https://www.ncbi.nlm.nih.gov/). One genome of Vibrio cholerae was also included to serve as an out group for phylogenetic comparisons.
Phylogenetic analysis
Data sets with different genomic features representing single 16s rRNA genes and different gene clusters were prepared for phylogenetic analysis. RNAmer v1.2 was used to extract 16s rRNA sequences from genome datasets. 11 Fetched sequences were trimmed using Bioedit v7.0.5.3, aligned with MEGA version 7, and subjected to phylogenetic analysis using maximum likelihood (ML) method of RaxML v8.2.12.12-14 Similarly, housekeeping genes listed at http://pubmlst.org for Vibrio spp. were used for phylogenetic analysis using RaxML (Randomized Axelerated ML). 15
For phylogeny of orthologous genes, at first coding sequences from the genomes were predicted using Prodigal v2.60. 16 Prodigal output was subjected for gene clustering using OrthoMCL v2.0, which has resulted 2085 single-copy orthologous genes. 17 Each gene present across 36 genomes was aligned using Molecular Evolutionary Genetic Analysis (MEGA) and trimmed using trimAl v1.4 with strictplus option. 18 All the 2085 genes from each genome were concatenated and subjected to phylogenetic analysis using RAxML. Figtree v1.4.2 (http://evomics.org/resources/software/molecular-evolution-software/figtree/) was used for visualizing all the consensus trees generated by RAxML. To construct whole genome–based phylogenetic trees, variant detection and phylogenetic analysis pipeline kSNP3.0 were used. kSNP3.0 accepts genomes as input and does not require genome alignments and reference genomes. It estimates phylogenetic trees by parsimony, neighbor-joining and ML methods. 19 The number of replicates for bootstrapping was set at 500 for all phylogenetic trees built in this study.
Genome similarity indices
Two confirmatory metrices of closeness between the genomes, namely average nucleotide identity (ANI) and Genome-to-Genome Distance, were computed using pyANI v0.20 and GGDC server (http://ggdc.dsmz.de/), respectively. 20 Heatmaps for visualizing the genome similarity based on ANI and isDDH were generated using R stats library ggplot2.
Results
Genome statistics of all the 35 strains along with the out-group entry, that is, V cholerae are given in Table 1. The average number of genes present in each genome is around 4700 genes. Smallest genome among the genomes studied is V anguillarum which contains 3686 genes and the largest being 5818 genes containing V campbellii. Vibrio campbellii BB120 has the lowest gene translated to protein with 93% and V parahaemolyticus O3:K6 substr. RIMD 2210633 has highest gene translated percentage of 97%. The guanine-cytosine (GC) content of genomes ranges from 44.37% to 47.49% with an average of 45.68% except for NCTC 11218 isolate of V furnissii whose GC content is 50.63%. Draft assembly of V campbellii LB102 which was isolated from tiger shrimp hatchery located at south east coast of India contains 90 scaffolds and 5145 genes.
The genomes of Vibrio species used in the study.

Abbreviations: GC, guanine-cytosine.
The phylogenetic tree with ML method was generated based on 16s rRNA genes from 36 Vibrio genomes (Figure 1) along with branch lengths and bootstrap values. There was no clear distinction observed between the Vibrio sp. in this phylogeny. Misclassifications were observed regarding V alginolyticus strain ATCC33787 and V vulnificus YJ016. Even though the members of the harveyi clade, namely V harveyi and V campbellii were found to be clustered together, they were also found in close proximity to other members, such as V alginolyticus and V parahaemolyticus. Lower bootstrap support for many of the nodes is a notable observation in this tree. It is has been observed that bootstrap value as low as 13% for one of the branches signifies poor clustering pattern.
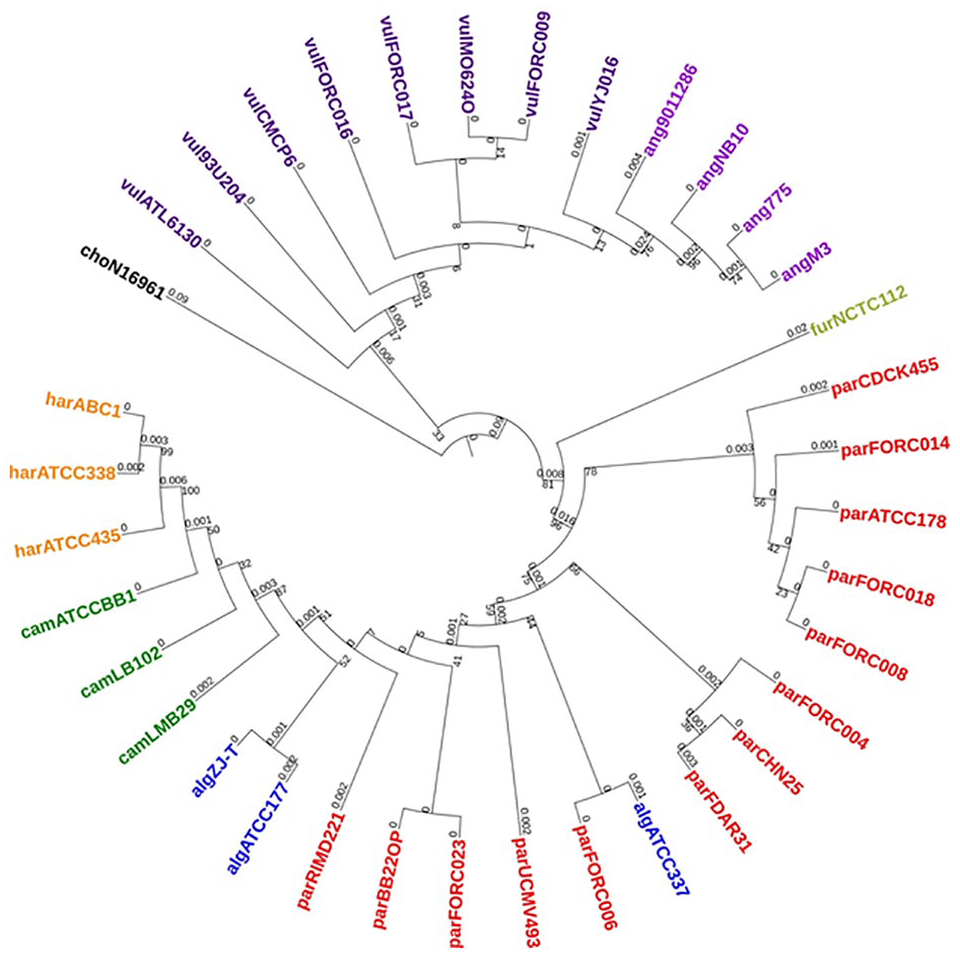
Phylogenetic tree based on 16s rRNA genes.
The phylogenetic tree built using housekeeping genes has been depicted in Figure 2. Relatively a better clustering pattern observed in this tree compared to the one built using 16s rRNA genes. Distinct clades between the species were observed except for harveyi and campbellii. Bootstrap support for branching between species is ranged from 64% to 100%.

Phylogenetic tree based on MLST sequences. MLST indicates multilocus sequence typing.
The ML tree constructed for 36 Vibrio spp. based on orthologous genes has been depicted in Figure 3. The tree indicated distinct monophyletic clades for each of the species considered in this study. Bootstrap support is 100% for between species indicate significance of this feature set. Wrong clustering patterns noticed regarding entries of harveyi clade have been corrected with this method of phylogenetic analysis. Tree generated using kSNP3 (Figure 4) pipeline is observed to be comparable with the tree generated using single-copy orthologous genes.
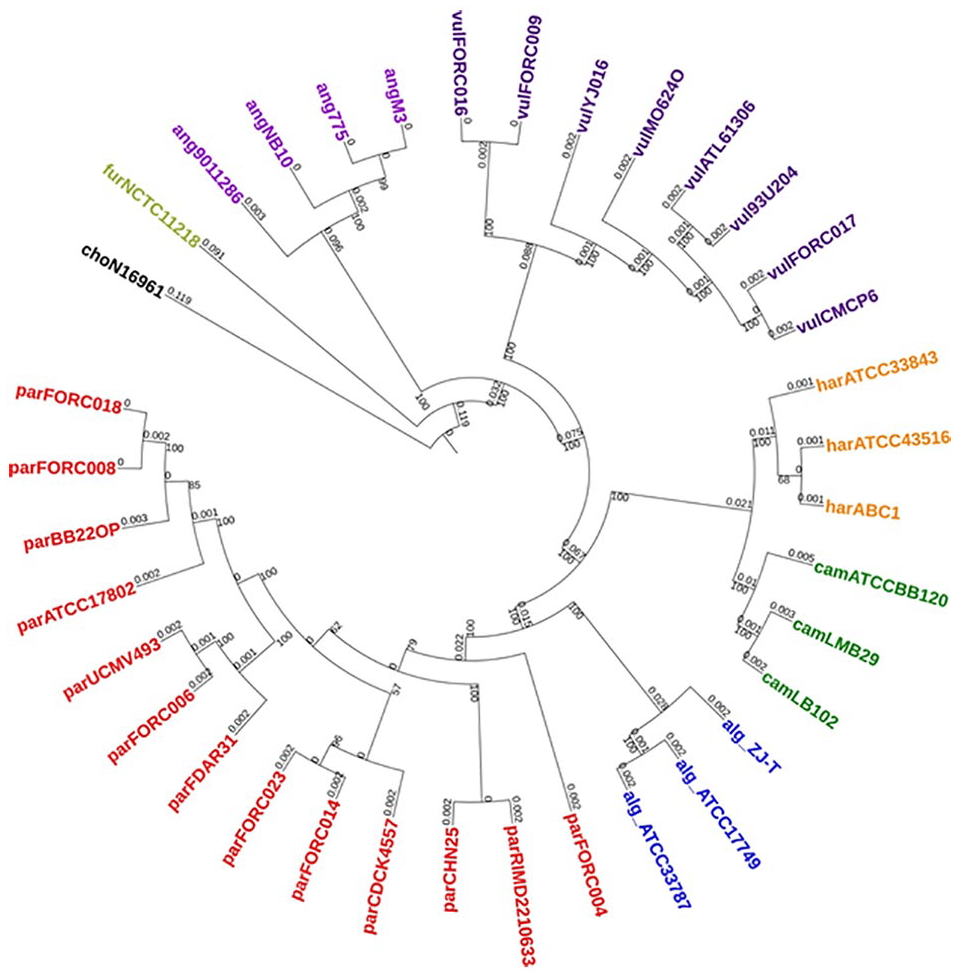
Phylogenetic tree constructed with single-copy orthologous genes.

Phylogenetic tree generated with kSNP3.0.
The ANI values between all the genomes of the study ranged from 0.8323 to 0.9998. The lowest ANI value observed is between V anguillarum 90-11-286 and V furnissii NCTC11218 strains, while the highest is between V parahaemolyticus strains FORC 008 and FORC 018. The heat map generated with ANI values is depicted in Figure 5. Pair-wise computed DDH values for the 36 genomes including out-group have been plotted in Figure 6 to display similarities between the Vibrio species. The values are in ranges from 14.8% to 100%. DDH values of more than 70% are considered to be an important factor in classifying species. 21

Heatmap generated based on average nucleotide identities for selected Vibrio species.

Similarity matrix based on DDH values for selected Vibrio species.
Discussion
The most commonly used bacterial signature sequences are 16s rRNA genes due to their presence in all the bacteria, limited evolutionary changes over time, and sufficiency of their length for analytical purposes. 22 These gene sequences are widely applied in the studies related to species identification and to calculate evolutionary distances between or within the species. Misclassifications or ambiguities in identification of the species can arise due to poor quality of the sequences available at public domain databases. But in recent times, due to advent of accurate and high throughput sequencing technologies, more accurate and complete prokaryotic genomes are available at public domain databases for mining and identifying true signatures of a species required for identification and clustering of it with related ones. Here in, we used 35 genomes of Vibrios having relevance with brackishwater aquaculture for finding the phylogenetic relations between the species and precise methods for clustering of the genomes. Correct species-level assignment, formation of monophyletic clades for each species, and high degree of bootstrap support are the metrics chosen for deciding the accuracy of species classification based on phylogenetic analysis. Misclassifications observed in the phylogenetic tree with 16s rRNA genes regarding alginolyticus ATCC33787 and vulnificus YJ016 strains imply that the variation present in these genes is not sufficient for proper delineation. With no species-level monophyletic clades and lesser bootstrap support necessitated other criteria which can take more variation present between the genomes for accurate classification.
Multilocus sequence typing is a sequence-based approach to unambiguous characterization of bacterial strains with sequences of internal fragments of housekeeping genes. 23 The multilocus sequence typing (MLST) databases like the one at www.pubmlst.org houses MLST allelic profiles and sequences for different bacterial species. Figure 2, plotted based on MLST sequences, has overcome misclassification errors unlike the one made with 16s rRNA genes. But issues of distinct monophyletic clades and bootstrap support were not found to be addressed with MLST also.
Orthologs are genes that diverged through a speciation event unlike paralogous genes, which diverged after a duplication event. 24 Size of these genes over a set of genomes depends on their evolutionary relationship and the quality of genome assemblies. With 4700 average number of genes among the selected Vibrio spp., 2085 single-copy orthologs were found to be present, which accounts nearly 44% of genes. Phylogenetic analysis based on these single-copy orthologs met all the criteria set for best classification (Figure 3). Monophyletic clades with 100% bootstrap support for between species indicate superiority of this feature set compared to previous ones. The utility of orthologous genes to address species ambiguities was demonstrated by Ke et al 6 in an attempt to correct the misclassification of V campbellii with the strains present in harveyi clade. But the clustering, extraction, and curation of the sequences and tree building with single-copy orthologous genes were observed to be computationally intensive and time consuming.
kSNP 3.0 pipeline identifies single-nucleotide polymorphisms (SNPs) from the input genomes and does phylogeny based on the core SNP data matrices, which include only SNPs detected at loci that were present in all genomes. The program runs faster and requires less memory compared to previous approach. Phylogenetic tree generated from kSNP 3.0 is comparable to the one built with single-copy orthologous genes (Figure 4) in terms of accuracy in classification.
The complete genome is used as reference standard to determine phylogeny which in turn determines taxonomy of the species in genome-based similarity indices. 25 Average nucleotide identity is one such method which depends on large number of genes unlike the ones depends on 16s rRNA, and it was considered to be better measure of relatedness. When the large number of genes considered for estimating relatedness of genomes, the indices are unaffected by varied evolutionary rates of the genes as fast-evolving genes are compensated by slow-evolving genes. 26 Dendrogram based on ANI values in Figure 5 accurately classified each species and made monophyletic clades. Genome-genome distance measure isDDH is another metric of relatedness computed using whole genomes. Average nucleotide identity values of 95% is equal to DDH values of 70%, which signifies highly related species. 27 In Figure 6, clusters having more than 70% similarity belonged to the same species. Both the measures have confirmed the clustering patterns established through single-copy orthologous genes. These similarity metrics have become gold standard for in silico species identification in recent times.
Conclusions
Information on bacterial species present in an ecosystem along with their phylogenetic distances and right methods for identification or classification has got significance in evolutionary biology. Here, we used different subsets of whole genome data, namely, 16s rRNA gene, MLST genes, and single-copy orthologous genes pertaining to 35 Vibrio species which are native to brackishwater ecosystem. Phylogenetic trees based on 16s rRNA and MLST sequences resulted the wrong classification patterns. To clear the ambiguities in classification, it was further tested with single-copy orthologous genes dataset as well as kSNP 3.0 pipeline. Clearly distinguished clades were observed in both of these methods among which kSNP3.0 requires less computational resources. Genome similarity indices like ANI and in silico DDH methods supported the validity of trees built with single-copy orthologous genes and kSNP3.0. The work needs to be continued from time to time by including newly sequenced genomes with proposed methods to clear ambiguities in classification of a new species.
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The Indian Council of Agricultural Research (ICAR), New Delhi, provided financial support under the “Network Project on Agricultural Bioinformatics and Computational Biology.”
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
JAK, KVK conceived the study. JAK, VA, and SAs conducted the analysis. JAK drafted the manuscript with inputs from SAv, KVK, SK, and BS, MG, AR, SVA, and KKV reviewed the manuscript and gave critical comments.