
Abstract
Cholera is a bacterial disease featured by dehydration and severe diarrhea. It is mainly caused by alimentary infection with Vibrio cholerae. Due to the wide applicability of quinazolin-2,4-dione compounds in medicinal and pharmaceutical chemistry, a new series of N-containing heterocyclic compounds was synthesized. We used the in silico docking method to test the efficacy of quinazolin-2,4-dione compounds in the prevention of cholera in humans. The newly synthesized compounds showed strong interactions and good binding affinity to outer membrane protein OmpU. Moreover, the pharmacokinetic properties of the newly synthesized compounds, such as absorption, distribution, metabolic, excretion, and toxicity (ADMET), were predicted through in silico methods. Compounds with acceptable pharmacokinetic properties were tested as novel ligand molecules. The synthesized compounds were evaluated in vitro for their antibacterial activity properties against Gram-negative Escherichia coli O78 strain using the minimum inhibition concentration (MIC) method. Compounds
Background
In the last years, the incidence of cholera disease has dramatically increased around the world. Due to its impact, it is considered the world’s most infectious disease, being responsible for 21 000 to 143 000 deaths worldwide. Vibrio cholerae is a Gram-negative bacterium that on colonization of the small intestine and induction of virulence factors causes severe diarrhea. 1 The regulation and expression of its virulence genes have been studied earlier.2,3 Like most of the Gram-negative bacteria, V. cholerae during their growth produce outer membrane vesicles (OMVs), which entrap periplasm for adherence promotion and transferring bacterial DNA. 4 A wide variety of V. cholerae virulence genes are regulated by ToxR and/or ToxS that regulates and expresses genes, which encode a porin such as OmpO and OmpT.5,6 About 60% of Vibrio cholera outer membrane proteins are OmpU if it has grown in salt-free medium. 7 This same porin forms nonspecific-barrel channels that allow free diffusion of hydrophilic molecules throughout the OM. Apart from its porin function, OmpU has also been shown to confer the pathogen with resistance to bile and antibacterial peptides. 8 Lembke et al 9 demonstrated that ToxR is blocked independently of ToxS; hence, sodium deoxycholate was used for inactivation of ToxR by linking bile to ToxRS complex formation and further activation of its transcription factor activity. Li et al 10 reported a high-resolution crystal structure of the outer membrane protein OmpU. Interestingly, the impact of ZnO2 nanoparticles on the outer membrane of Vibrio cholera affects only the OmpT porins, while OmpU was not affected as recorded by Salem et al. 11 Consequently, there is a need to identify novel compounds acting as drug-like molecules against cholerae.
In recent years, considerable evidence has been found on the importance of quinolone and tetracycline as having high medicinal value in the treatment of cholera.12,13 The nitrogen-containing heterocyclics, such as quinazolindione, are crucial structural motifs of different pharmaceutical compounds and are found in some commercial drugs like ketanserine and pelanseine. 14 Due to the significant importance of quinazolindione derivatives as anticonvulsant, 14 antileishmanil, 14 and antimicrobial agents, 15 a series of quinazolin-2,4-dione derivatives have been synthesized and in silico evaluated to predict the inhibitory activity against the target protein OmpU. 16 In this context, we have selected OmpU protein as a target for predicting the action of the newly synthesized quinazolin-2,4-dione compounds. However, this study focused on the screening of the newly synthesized compounds against the target protein. PyRx tool is one of such programs which is used to predict the strength of intermolecular docking interactions between the ligand molecules and the binding site of the target protein. 17 In addition, pharmacokinetics properties of the compounds were also in silico predicted.
To extend the applicability of newly quinazolindione derivatives
Results and Discussion
Herein, we reported the synthesis of some new quinazolindione derivatives, and in silico molecular docking study was carried out to identify their activity toward cholera disease.
Chemistry
Eight new quinazolin-2,4-dione derivatives were synthesized by (4-methylene-2-oxo-3-phenyl-3,4-dihydro-2H-quinazolin-1-yl)-acetic acid hydrazide

Synthesis of quinazolindione compounds

Synthetic strategy of compound
In silico study
The outer membrane protein OmpU is selected as a target protein for the identification of novel leads against cholera disease. Recently, the crystal structure of the outer membrane protein OmpU (PDBID: 5ONU) of 341 amino acid residues, with a high resolution, was reported by Li et al, which is a significant virulence factor in V cholerae and delivers a vigorous mechanical basis for the colonization of host cells surfaces and the phage-recognition mechanism. The crystallographic structure of OmpU was downloaded from protein data bank website (rcsb.org) as shown in Figure 1.

The three-dimensional (3D) structure of OmpU protein. The 3D model of OmpU consists of 6 α-helices and 21 β-strands as predicted from PDBsum. N-terminal indicates the starting residue and C-terminal indicates the end residue.
Furthermore, the physicochemical properties of the target protein were predicted using the ProtParam tool.
23
The molecular weight was 36 645 Da and the isoelectric point (pI) was 4.46. The results show that the amino acid residues ALA, GLY, ASP, ASN, TYR, and VAL were present in high percentages in OmpU protein. In molecular docking method, all the synthesized compounds were docked into the binding site of OmpU. The binding site prediction tools such as MetaPocket and ProBiS declare that the amino acid residues ALA14, ASP29, ARG107-TYR110, ASN125-ASP126, and ARG155-LYS172 are crucial for OmpU to bind with quinazolindione derivatives. An in-house database of 8 quinazolindione compounds was created in standard file format (SDF). All the compounds were energy minimized using Universal Force Field (UFF),
24
to obtain structures with proper bond length between different atoms, and then used in the virtual screening study. In addition, doxycycline was chosen as a reference compound to compare the docking score of synthesized compounds. Herein, PyRx software was employed to simulate the ligand into the active site of the protein to calculate the binding energy of the ligand-receptor complexes. Computer-based docking predicted 9 conformers for each ligand-protein complex. The preferable binding orientation between protein and compound, with more negative binding affinity score, was considered for further study.
25
Docking interactions between compounds and protein are shown in Figure 2. The docked ligand molecules scored good binding affinity values of −10.6 to −8.0 kcal/mol. Compound

Molecular interactions of compounds
The binding affinity (kcal/mol) of various compounds

The 8 molecules (
Drug-likeness scores of the screened quinazolindione compounds were filtered by in silico prediction of ADMET, using admetSAR.
28
The compounds with acceptable ADMET properties in the permissible range are considered as novel molecules.
29
The pharmacological properties of 8 compounds
List of ADMET properties of the newly synthesized molecules (
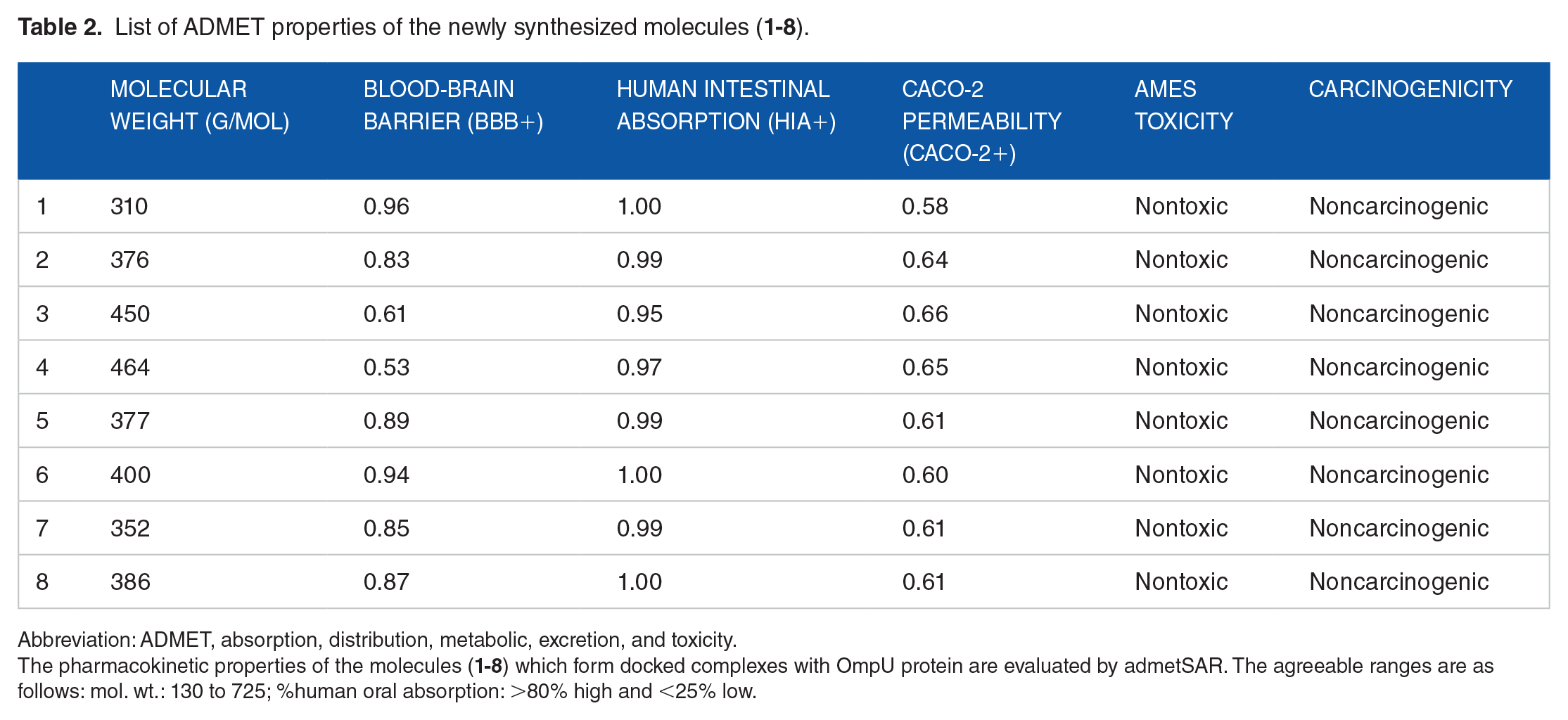
Abbreviation: ADMET, absorption, distribution, metabolic, excretion, and toxicity.
The pharmacokinetic properties of the molecules (
Our study concluded that the newly synthesized quinazolin-2,4-dione compounds such as (4-methylene-2-oxo-3-phenyl-3,4-dihydro-2H-quinazolin-1-yl)-acetic acid hydrazide
Antibacterial activity of quinazolindione compounds against E. coli O78 strain
Eight newly synthesized compounds

The relationship between the antibacterial activity and binding affinity of all synthesized compounds
Minimum inhibitory concentration (MIC) values of the quinazolindione derivatives against Gram-negative Escherichia coli O78 strain.

The compounds
The MICs
The colorimetric iodonitrotetrazolium (INT)-formazon assay of compounds
Methods
Chemistry
Materials and apparatus
All melting points of the compounds were determined on Griffin apparatus and are uncorrected. The IR spectra of samples were recorded in KBr via a Shimadzu FT-IR 8101 PC infrared spectrophotometer. The NMR spectra were run at 400 MHz using tetramethylsilane (TMS) as the internal standard. Chemical shifts were measured in ppm (δ) related to TMS (0.00 ppm). Mass spectra were measured on a GCMS-QP1000 EX spectrometer at 70 eV. Elemental analyses were carried out at the Micro Analytical Center of Cairo University, Giza, Egypt. Thin-layer chromatography (TLC) was conducted on 0.25 mm precoated silica gel plates (60F-254). Purities of these compounds were established by TLC.
Spectral analysis of quinazoline compounds
(4-Methylene-2-oxo-3-phenyl-3,4-dihydro-2H-quinazolin-1-yl)-acetic acid hydrazide 1
A mixture of (2,4-dioxo-3-phenyl-3,4-dihydro-2H-quinazolin-1-yl)-acetic acid ethyl ester (1 mmol) and hydrazine hydrate (1 mmol) in absolute ethanol was refluxed for 6 hours; after cooling, the solid formed was filtered off and recrystallized from ethanol to give compound
1-[2-(3,5-Dimethyl-4,5-dihydro-pyrazol-1-yl)-2-oxo-ethyl]-3-phenyl-1H-quinazolin-2,4-dione 2
To a solution of compound
Arylsulfonyl N′-[2-(4-methylene-2-oxo-3-phenyl-3,4-dihydro-2H-quinazolin-1-yl)-acetyl]-hydrazide 3-4
A stirred solution of 4-methylene-2-oxo-3-phenyl-3,4-dihydro-2H-quinazolin-1-yl-acetic acid hydrazide
Benzenesulphonyl N′-[2-(4-Methylene-2-oxo-3-phenyl-3,4-dihydro-2H-quinazolin-1-yl)-acetyl]-hydrazide 3
Yield 74%; m.p. 240 to 241. FT-IR (KBr, υ, cm−1) = (NH), 3380 (C=Os), 1663, and 1685. 1H-NMR (DMSO d6, 400 MHz): δ (ppm) = 11.5 (s, 1H, NH), 7.2 to 8.0 (m, 14H, Ar-H), and 4.3 (s, 2H, CH2). MS (EI): m/z (%) = 450 [M]+. Anal. calcd for C22H18N4O5S: C, 58.66%; H, 4.03%; N, 12.44%. Found C, 58.90%; H, 4.32%; N, 12.71%.
Toluenesulphonyl N′-[2-(4-Methylene-2-oxo-3-phenyl-3,4-dihydro-2H-quinazolin-1-yl)-acetyl]-hydrazide 4
Yield 78%; m.p. 270 to 272. FT-IR (KBr, υ, cm−1) = (NH), 3365 (C=Os), 1660, and 1690. 1H-NMR (DMSO d6, 400 MHz): δ (ppm) = 11.5 (s, 2H, 2NH), 7.2 to 8.0 (m, 13H, Ar-H), 4.3 (s, 2H, CH2), and 1.3 (s, 3H, CH3). MS (EI): m/z (%) = 464 [M]+. Anal. calcd for C23H20N4O5S: C, 9.47%; H, 4.34%; N, 12.06%. Found C, 59.50%; H, 4.65%; N, 12.15%.
Cyano-acetic acid N′-[2-(2,4-dioxo-3-phenyl-3,4-dihydro-2H-quinazolin-1-yl)-acetyl]-hydrazide 5
Heating of hydrazide
(2,4-Dioxo-3-phenyl-3,4-dihydro-2H-quinazolin-1-yl)-acetic acid N′-benzyl-hydrazide 6
A mixture of (4-methylene-2-oxo-3-phenyl-3,4-dihydro-2H-quinazolin-1-yl)-acetic acid hydrazide
Acetic acid N′-[2-(2,4-dioxo-3-phenyl-3,4-dihydro-2H-quinazolin-1-yl)-acetyl]-hydrazide 7
(4-Methylene-2-oxo-3-phenyl-3,4-dihydro-2H-quinazolin-1-yl)-acetic acid hydrazide
Chloro-acetic acid N′-[2-(2,4-dioxo-3-phenyl-3,4-dihydro-2H-quinazolin-1-yl)-acetyl]-hydrazide 8
To a solution of (4-methylene-2-oxo-3-phenyl-3,4-dihydro-2H-quinazolin-1-yl)-acetic acid hydrazide
In Silico Study
Binding site identification
Various computational methods were used for the prediction of drug-like molecules against cholera. The binding site region was identified using computational prediction tools like MetaPocket2.0 and ProBiS.32,33 A grid with dimensions 25A° × 25A° × 25A° is created around the binding site of OmpU to perform the screening studies.
In silico docking and ADME prediction
Next, in silico molecular docking of 8 quinazolindione derivatives and reference compound onto the target has been carried out using PyRx-virtual screening tool. The intermolecular interactions between the docked compounds-protein complexes such as H-bonds, π-π, π-cation, and π-sigma interactions add more stability to the target protein. 34 Because of these interactions, compounds are arranged in a certain position within a protein. Discovery Studio® Visualizer 2016 software (Accelrys Software Inc., San Diego, CA, USA) was used to visualize the interactions. In addition, the prediction of absorption, distribution, metabolic, excretion, and toxicity (ADMET) properties plays a key role in drug design. Blood-brain barrier, human intestinal absorption (HIA), Caco-2 Permeability (Caco2+), Ames toxicity and carcinogenicity were calculated using admetSAR (http://lmmd.ecust.edu.cn/admetsar1/). The promising results for the tested compounds indicate that they can be used as drug candidates.
Biological screening (antibacterial activity)
The antibacterial activity of compounds
Conclusions
Cholera is common in places with famine and poor sanitation that can cause acute diarrhea. This study provides an insight into the efficiency of quinazolindione derivatives as new chemical entities (NCEs) against cholera. The resulted data from in silico study are helpful to identify new potential lead molecules for the treatment of cholera throughout the blocking mechanisms of OmpU binding sites. The antibacterial activities of the newly synthesized compounds were evaluated and revealed that the compounds
Supplemental Material
Supplementary__File – Supplemental material for Synthesis, Characterization, Antibacterial Activity, and Computer-Aided Design of Novel Quinazolin-2,4-dione Derivatives as Potential Inhibitors Against Vibrio cholerae
Supplemental material, Supplementary__File for Synthesis, Characterization, Antibacterial Activity, and Computer-Aided Design of Novel Quinazolin-2,4-dione Derivatives as Potential Inhibitors Against Vibrio cholerae by Mohamed El-Naggar, Mahmoud Eldeeb Mohamed, Ahmed Mohamed Mosallam, Wesam Salem, Huda RM Rashdan and Aboubakr Haredi Abdelmonsef in Evolutionary Bioinformatics
Footnotes
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
AHA, MEM, and AMM designed the study and were responsible for synthesis of compounds. AHA did the virtual screening approach and he wrote the preliminary draft of the manuscript. ME and HRMR were responsible for the spectral analysis section. WS did the antibacterial evaluation of the compounds. All the authors contributed to the revision of the drafts and agreed on the final version to be submitted.
Availability of Data and Materials
All data generated or analyzed during this study are included in this manuscript.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.