
Abstract
Defensins as 1 of major classes of host defense peptides play a significant role in the innate immunity, which are extremely evolved in almost all living organisms. Developing high-throughput computational methods can accurately help in designing drugs or medical means to defense against pathogens. To take up such a challenge, an up-to-date server based on rigorous benchmark dataset, referred to as iDEF-PseRAAC, was designed for predicting the defensin family in this study. By extracting primary sequence compositions based on different types of reduced amino acid alphabet, it was calculated that the best overall accuracy of the selected feature subset was achieved to 92.38%. Therefore, we can conclude that the information provided by abundant types of amino acid reduction will provide efficient and rational methodology for defensin identification. And, a free online server is freely available for academic users at http://bioinfor.imu.edu.cn/idpf. We hold expectations that iDEF-PseRAAC may be a promising weapon for the function annotation about the defensins protein.
Introduction
Defensins, a kind of small cysteine-rich antimicrobial proteins, are considered as a part of the non-specific immune response. Researches have indicated that defensins were widely distributed in compartments of body in almost all living species, and these peptides increased in some pathogenic body cells apparently. Cells containing these host defense peptides render assistance in combating against bacterial, 1 viral, 2 and fungal infections.3–5 Evidence is also increasing that an imbalance or a reduction 6 of defensins in organisms may predispose the occurrence of many diseases. 7 As for its action mechanism, defensin peptides are mainly through the destruction of the structure of bacterial cell membranes.8–10 Specifically, most defensins form a pore-like membrane defect by binding to a microbial cell membrane, and then allow necessary ions and nutrients to flow out through permeabilization. 11
As defensins have a wide range of importance in application of various industries, 12 it is extraordinarily important to design and distinguish defensins which can be used for the special needs. Nevertheless, the traditional experimental methods, such as the nuclear magnetic resonance, 13 own a higher cost and demand more stringent requirements. In addition, there are also even some limitations that make it impossible to analyze the precise functional characteristic of proteins. As we all know, the specific codes endow sequence motifs with unique structures or functions, and the differential combination and arrangement of the motifs with specific codes determine the isoforms that possess multiple functions. 14 In this case, the bioinformatics methods appear as more efficient approaches for providing some unique perspectives of protein research.15,16 Over the past few years, a vast number of antimicrobial peptide predictors have been reported,17,18 which inspired us to develop prediction methods for defensin.
The first computational study of defensin family was given by our team based on diversity measure in 2009 19 and the results were further optimized by support vector machines (SVMs) method and a free online web server was developed. 20 Subsequently, another group designed a new classifier, DEFENSINPRED, which classified human defensin proteins and their types based on pseudo amino acid compositions. 21 In 2015, we proposed a novel iDPF-PseRAAAC servers concentrated on distinguishing defensin peptide family and subfamily with the help of Protein Blocks. 20 As the performances in these existing predictors are still not perfect, further work is valuable and we have further built a more effective web server for defensins.
In this article, the measures we proposed to improve predictive power was based on a new reduced amino acid resource, PseRAAC_Book (http://bioinfor.imu.edu.cn/pseraacbook), which contains more than 600 types of reduced amino acid clusters (RAACs). We expect that such an idea will reduce the complexity inherent of the protein to a greater extent and will be widely used in machine classifiers to establish better web servers for the defensin family. Compared with the standard amino acid composition, the reduced amino acid alphabet can accurately extract higher quality predictive result of protein sequences and improve the level of research. Our better results can also demonstrate and prove this. At last, a useful online server iDEF-PseRAAC was freely established for the convenience of basic academic use.
Materials and Methods
Dataset
Most machine-learning classification algorithms work properly if they are trained by a skewed benchmark dataset. In the study, the benchmark dataset used was selected from Zuo et al. 20 First, the raw peptides were downloaded from the Defensins Knowledgebase dataset, 22 and this defensin sequence is strictly verified by experiments. Redundant cut-off is enforced using the program CD-HIT 23 to remove sequences with pairwise sequence identity ⩾80% with a ultrahigh speed then. In addition, with the latest annotation, we checked the new family annotations of database. And 5 misclassified defensins were deleted from the subsets. There is no big or main defensins included in the datasets. 24 The final benchmark data sets, which were used in this analysis, are formulated by the following equation:

where set S composes of 328 defensin proteins, it can be classified into 5 families: while the subset S1 consists of insect defensins, S2 contains invertebrate defensins, S3 contains plant defensins, S4 contains unclassified defensins, and S5 contains vertebrate defensins, and
The sequence profile used in this study.

Reduced amino acid alphabet
As machine-learning algorithms are unable to directly process sequence samples, such as SVMs, neural networks, random forests, we need to encode defensin peptides through a mathematical expression that conveys sequential properties. Suppose a protein sequence P with L amino acid residues can be represented as follows:

where R1 represents the amino acid residue at the sequence position 1, R2 represents the amino acid residue at position 2, and so on, and L is between 22 and 183.
For the second category, interaction of individual amino acids and more detailed sequential information were effectively excavated using typical N-peptides composition (N = 1, 2, 3). We do not choose an N value greater than 3 to extract features because too high a dimension may lead to overfitting problems and may reduce the generalization ability of the model. For example, the dipeptide composition of 20 natural amino acids can be expressed by equation (3). 25

where
For the last category, the amino acid can be clustered by the similarity of hydrophobic and polar characteristics to achieve reduction of the amino acid composition vector. 26 The amino acid reduction method was first proposed in 1976 27 and it was gradually used in various subcellular localization as well as protein prediction and activity prediction. 20 Compared with the general amino acid composition, the RAACs performed sufficient ability for decreasing protein complexity and withdrawing the conservative feature hidden in the noise signals that affect protein sequence researches. After reducing amino acid composition, the protein sequence will be significantly simplified, which could improve computational efficiency, decrease information redundancy, and reduce chance of overfitting.19,28–30 Therefore, it is reasonable to formulate an amino acids sequence by using reduced amino acid composition. Here, based on RAAC book, more than 70 types of reduced amino acid alphabet were applied to preprocess the data. According to the method of reduced amino acid described above, the feature extraction method we obtained can be expressed by the following mathematical formula:

where

where N is the N-peptide, T is the type of different amino acid alphabets, and C is the cluster of reduced amino acid alphabet. The current method also has some limitations, for example, all of the alphabets from the RAAC book were manually curated from published literatures, and there is no uniform method for different protein dataset, which are our further efforts.
F-score method of feature selection
The F-score method is the feature extraction method adopted in this article. In general, there is a trade-off between the accuracy of the results and the recall rate, while F-score has a unique balance of accuracy and recall. By calculating the F-score for each feature, we can effectively extract appropriate and valid data dimensions. F-score can be represented by the following mathematical expression31–33:
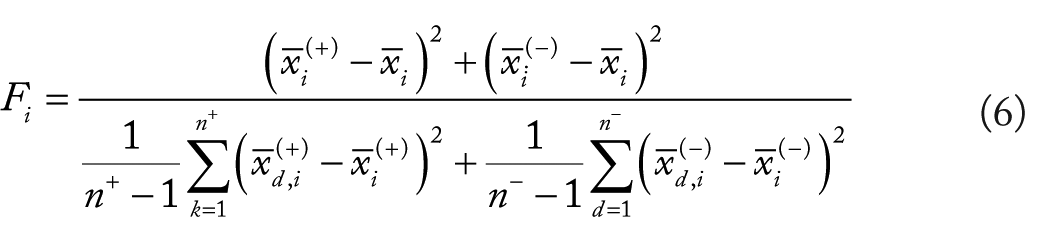
where
SVM
The SVM model have better predictive power because they can reduce noise on the dataset. The basic idea of SVM is to construct an N-dimensional space hyperplane based on finite sequence sample information, and then map the feature vector X~ in the input space to the high-dimensional Hilbert space through the kernel function,34,35 so as to infer which category belongs to the same species. 36 The trained SVM can be speculated which category they belong to. And 4 types of kernel functions can be chosen for prediction in the software, including linear functions, polynomial functions, S-shaped functions, and radial basis functions (RBFs). 20 The best classification hyperplane was obtained by RBF, 36 which was shown as follows 34 :

Furthermore, the user-defined parameters in SVM are adjusted by a grid search method.
The regularization parameter C and kernel parameter

From equation (8), it can be rigorously concluded that the optimal values are C and γ. For the concrete description of the SVM and how it works, see the guide of SVM. 37 In this article, all calculations are performed using the Scikit-learn Python package.20,35,38
Performance evaluation
The Sensitivity (Sn), Specificity (Sp), overall accuracy (OA or ACC), and Matthews correlation coefficient (MCC) were computed to measure the performance of models across the prediction process. According to the definition of these evaluation quantities, it can be expressed as follows17,20:

where N is the whole number of the samples, M = 5 refers to the number of subsets, TP(i) refers to the value of the positive samples correctly predicted for the ith subset, FP(i) stands for the value for positive sample that predicted incorrectly to the negative sample. TN(i) refers to the value of the negative samples correctly predicted for the ith subset, FN(i) stands for the value for negative sample that predicted incorrectly to the positive sample.
Jackknife test
Among the 3 cross-validation methods, the jackknife test is deemed the most objective that can always yield a unique result for a given benchmark dataset, and hence has been increasingly used by investigators to examine the accuracy of various predictors.33,39–41 In this study, the jackknife test was used to test the predictive performance of our proposed model.
Results and Discussion
The predictive performance of different reduced amino acid alphabets
To investigate the best alphabets for predicting defensin proteins, we calculated accuracies of all the descriptors from the RAAC book using SVM based on the stringent jackknife test. Figure 1A shows prediction density profile of different N-peptides composition (N = 1, 2, 3). We can observe that the 2-peptides and 3-peptides achieve the better performance than the 1-peptides composition, and the best prediction result occurred in the 2-peptides. So we gave the detailed accuracy (ACC) of 2-peptides compositions (Figure 1B). To improve the efficiency of the calculation and display convenience, the numbers of clusters less than 8 were not given in Figure 1B.

(A) Binary precision density maps illustrate the distribution of different descriptors based on different N-peptide composition. (B) The predictive accuracy of defensin families based on 2-peptide composition using different types of reduced amino acid alphabet. A bluer box indicates a higher accuracy, while a lighter box has the opposite.
In the heat map of Figure 1B, a more reddish box indicates a higher accuracy and a more greenish one is the opposite. It can be observed that more than half of the methods have a predictive accuracy greater than 70%. And when the accuracy is greater than 80%, the general size is relatively high. Performance is generally degraded due to the lack of critical order information in highly simplified alphabets.
The optimal cluster of reduced amino acid alphabet
According to observations, the dipeptide and tripeptide feature is better for the predictive method. The dipeptide composition of Type 5, Cluster 19 (T = 5, C = 19) gave the best discriminative ability, the OA achieves to 91.16%. Here we found the C (cluster) and T (type) is the optimal reduction method for identifying defensins (Figure 2). In this reduction method, named secondary-structure method, the stepwise reduction is by joining the highest-scoring pair, and reestimating the new set of pairs is shown going down to 2 groups. 42

(A) Cluster size of reduced amino acid alphabet based on secondary-structure method. (B) The prediction results of different cluster sizes for alphabet type 5. (C) The comparison of different alphabet types with the same cluster number (C = 19).
In addition, a comparison was also made to replicate different types of reduced amino acid process by the jackknife test because other amino acid descriptors required a further and fair comparison. Figure 2B is the comparison of different clusters in the specific alphabet type, which achieves the highest precision, and Figure 2C is the comparison of different alphabet types with the same cluster number. In dipeptides, when calculating all the sizes based on type = 5, we can clearly see that the highest OA is about 91.16% of cluster 19 of reduced amino acids (Figure 2B). After evaluating cluster 19 with different alphabet types, we still got the best result in type = 5 (Figure 2C). Such result can prove the accuracy of the best reduction method. It is important to note that although there are several methods for obtaining the highest precision in the tripeptide, we have chosen the smallest cluster to reduce the complexity of the protein as much as possible.
Feature selection can further improve the performance of prediction
Appropriate dimension is very important for the result of the prediction, hence there is a need to examine the accuracies of different dimensions of the N-peptide composition. It can be clearly reflected, in Figure 3, that the prediction ability does not always possess a linear increase with the feature dimensions augmenting. To check which is the most suitable value of dimension that helps in achieving better performance, we used the F-score of Incremental Feature Selection (IFS) process with jackknife test. And the results optimized by F-score are described below. In case of basic amino acid composition (N = 1), it achieved the highest accuracy of 78.35% using 19 features from the type 19. As for the tripeptide’s composition (N = 3), more than 1000 dimensions was selected as the input parameter, the maximum accuracy for predicting 5 defensin families was no more than 90.00%. In case of the dipeptide (N = 2, T = 5, C = 19), it shows most optimal accuracy 92.38% by selecting 329 features based on F-score of Feature selection, which performed better than other models conspicuously.
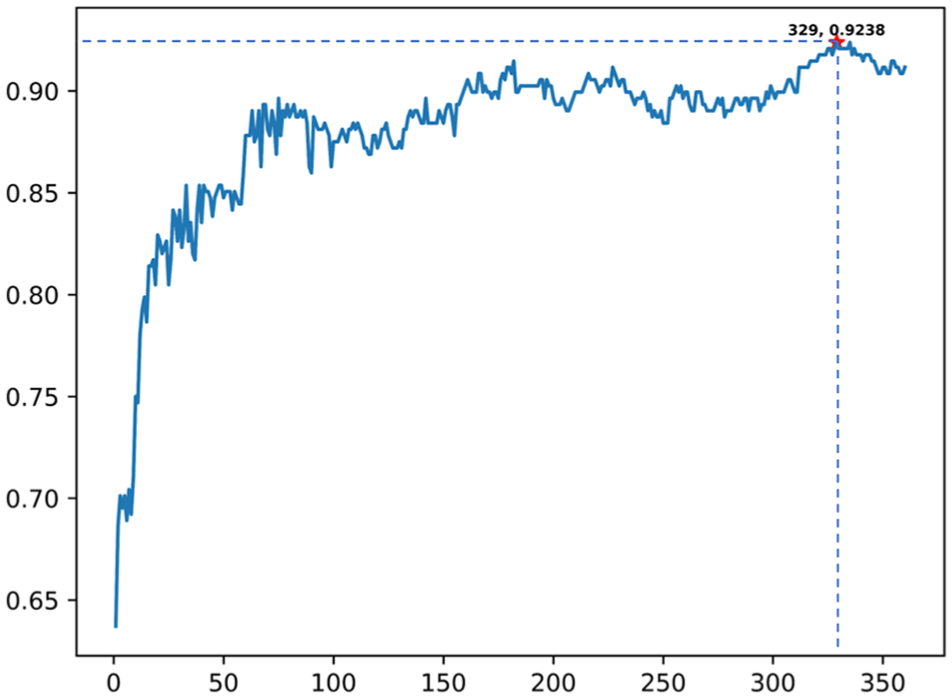
The IFS curve shows feature extraction process using F-score of different features in dipeptide composition (T = 5, C = 19). An IFS peak of 92.38% was obtained when using the 329 optimal features. IFS indicates incremental feature selection.
Defensin family prediction
The predictor engine achieved from the predictive procedure is called iDEF-PseRAAC, where “i” means “identify,” “DEF” means “defensin,” “Pse” means “Pseudo,” and “RAAC” means “reduced amino acid cluster.” Better performance was obtained by the iDEF-PseRAAC when the different types (T) of reduction methods are added to peptide sequence based on N-peptide compositions (N). From the prediction performance based on different vector dimensions depicted in Figure 1B and Figure 3, we observed that the OA reached a maximum 92.38% based on selected 329 features from 2-peptide composition for type 5 with 19 clusters (N = 2, C = 19, T = 5). Although the best results are in the dipeptide properties, the results obtained by the tripeptide properties are still better in general. See Figure 4 for the overall trend of the accuracies in the process of defensin family prediction.

Univariate density map of ACC for the defensins prediction based on RAAC. ACC indicates accuracy; RAAC, reduced amino acid cluster.
Vertebrate defensin subfamily prediction
To determine the feasibility of our method for predicting the 3 subfamilies of vertebrates (including alpha-, beta-, and theta-defensins),24,43 we repeated the previous process and performed a comprehensive analysis of the results. Based on the tri-peptide composition of type 7, cluster 3, [W, C, GPHNDERQKASTFYVMIL], the best OA we achieved was 98.79%, and the Sn, Sp, and MCC are 0.99, 0.99, and 0.91, respectively. This high result is more indicative of the accuracy of the method we use and its effectiveness for the vertebrate subfamily.
Comparison with previous methods
To prove the superiority of our proposed method, it is necessary to compare it with other existing methods. As we use the same benchmark dataset as the iDPF-PseRAAAC model, we compare it directly with our approach. To achieve a more accurate acquisition of sequence information, we used a more mature and simpler N-peptide sequence representation in bioinformatics. On this basis, to achieve a better sequence representation, we have found a sequence simplification method that is more accurate or suitable for the prediction model than the method iDPF-PseRAAAC. This method, called the secondary-structure method, processes amino acid redundancy information strictly by joining the highest-scoring pair and re-estimating the new set of pairs is shown going down to 2 groups. Such a process has apparently led to an increase in the reduction performance.
According to Table 2, it is apparent that the method proposed in this article produces higher precision than the previous method. For the 3 main defensin families, Insect, Plant, Vertebrate, all of the Sn values are more than 90.00% with the very high Sp (98.13%, 98.60%, and 97.08%, respectively). It indicates that our proposed iDEF-PseRAAC predictor shows more confidence for family classification. The worst predictions were made in Unclassified class. After checking the defensin sequence of Unclassified, we find 1 of the reasons is mainly concentrated on the low conservative domain for unclassified S4 subsets. And the unclassified family annotation of Defensins knowledgebase is another reason for this prediction results. For example, there are 4 Unclassified defensin proteins that should be classified into vertebrate family according to our prediction (Table 3).
The comparison between our model with previous methods.

Abbreviations: Sn, sensitivity; Sp, specificity; MCC, Matthews correlation coefficient; OA, overall accuracy.
The prediction accuracy matrix [Mi, j] of 4 defensin families based on dipeptide composition of type 5, cluster 19 (T = 5, C = 19).

As the F-score method can find the most appropriate degree among the 2 mutual restraint indicators of “recall rate” and “accuracy rate,” we can choose the most suitable dimension from the obtained features dimensions. Specifically, after the F-score method improved, our method can make the defensin family reach 92.38% more accurately, about 7 percentage points higher than the former method. All in all, although the success rate of using our method for forecasting is not super good, it is significantly better than using other methods before. This result proves that our method is feasible and effective.
Implementation of network server
To visualize our method and conveniently provide it to some basic research, a user-friendly server iDEF-PseRAAC was established to identify the defensin. Here, we provide a guide on how to use the web server, the guide content is as follows:
Step 1. Users could access this web server via http://bioinfor.imu.edu.cn/idpf
Step 2. After entering the page shown in Figure 5, you can copy and paste the defense sequence to be used for classification and search into the FASTA format input box, or you can upload the sequence file in FASTA format to the web server by clicking the upload button. As for the FASTA format, it starts with the greater-than symbol (“>”) in the first column, followed by the format of the sequence data. And if another line starting with a “>” appears, it means the end of the sequence. A specific example of the FASTA format can be viewed in the input box and the Example module.
Step 3. Click the submit button. Wait a few minutes to enter the analysis interface. The analysis report displayed by the new interface will display the analysis and prediction results of each sequence and their specific data values.

The homepage of the iDEF-PseRAAC web server is shown by a screenshot.
Through the above operations, users can easily get the results they want without going through the complicated formulas involved with the computer operation. According to the test, we can classify and predict all the defensins in addition to the relevant defensin-like peptides that have no antibacterial effect (e.g. scorpion toxins or plant S-locus proteins). 44 In addition, page termed as “Contact” offers our contact information.
Conclusions
The defensins play an important role in the innate immunity of animals and plants by combating pathogenic invading micro-organisms.45,46 So far, a very limited study has been done in this area. After the sequence reduction preprocessing and feature selecting described above, a promising defensing family prediction engine was constructed which was developed to improve prediction performance for defensin proteins in this work. To discriminate defensing families with higher precision, we have developed SVM models based on features like dipeptide composition along with reduced amino acid book. High prediction results can be obtained by the combination of the optimal reduction method and the F-score method. These results also indicate that the use of reduced amino acid books may have broad application in protein and DNA identification. However, the sequence-based protein classification is 1 of the limitations of our method. In fact, the active domain of defensin is highly conservative and the sequence motif models may be an important addition feature for prediction improvement. 47 In the future, for better understanding the analysis frame, the flowchart of this study will be displayed. 48
Footnotes
Acknowledgements
We extend our sincere gratitude to the 3 reviewers for their constructive suggestions of this article.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Nature Scientific Foundation of China (No: 61561036, 61702290), Program for Young Talents of Science and Technology in Universities of Inner Mongolia Autonomous Region (NJYT-18-B01), the Fund for Excellent Young Scholars of Inner Mongolia (2017JQ04), and Student’s Platform for Innovation and Entrepreneurship Training Program of Inner Mongolia University (201814295). The funders did not participate in research design, data collection and analysis, decision to publish, or preparation of the manuscript.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author contributions
YCZ and GFC conceived the study; YCZ and SHH performed the experiments; LY and YC analyzed the data; LZ contributed analysis tools; and YCZ, YL, and YC contributed to the writing of the manuscript. All authors read and approved the manuscript.