
Abstract
Pseudouridine (Ψ) is the first discovered and the most prevalent posttranscriptional modification, which has been widely studied during the past decades. Pseudouridine was observed in almost all kinds of RNAs and shown to have important biological functions. Currently, the time-consuming and high-cost procedures of experimental approaches limit its uses in real-life Ψ site detection. Alternatively, by taking advantage of the explosive growth of Ψ sequencing data, the computational methods may provide a more cost-effective avenue. To date, the existing mouse Ψ site predictors were all developed based on sequence-derived features, and their performance can be further improved by adding the domain knowledge derived feature. Therefore, it is highly desirable to propose a genomic feature-based computational method to increase the accuracy and efficiency of the identification of Ψ RNA modification in the mouse transcriptome. In our study, a predictive framework PSI-MOUSE was built. Besides the conventional sequence-based features, PSI-MOUSE first introduced 38 additional genomic features derived from the mouse genome, which achieved a satisfactory improvement in the prediction performance, compared with other existing models. Moreover, PSI-MOUSE also features in automatically annotating the putative Ψ sites with diverse types of posttranscriptional regulations (RNA-binding protein [RBP]-binding regions, miRNA-RNA interactions, and splicing sites), which can serve as a useful research tool for the study of Ψ RNA modification in the mouse genome. Finally, 3282 experimentally validated mouse Ψ sites were also collected in a database with customized query functions. For the convenience of academic users, a website was built to provide a user-friendly interface for the query and analysis on the database. The website is freely accessible at www.xjtlu.edu.cn/biologicalsciences/psimouse and http://psimouse.rnamd.com. We introduced the genome-derived features to mouse for the first time, and we achieved a good performance in mouse Ψ site prediction. Compared with the existing state-of-art methods, our newly developed approach PSI-MOUSE obtained a substantial improvement in prediction accuracy, marking the reliable contributions of genomic features for the prediction of RNA modifications in a species other than human.
Introduction
Pseudouridine (Ψ) is the first discovered 1 and the most abundant RNA modification 2 that has been widely studied in the past decades. To date, 13 pseudouridine synthases (PUSs) have been identified, which are responsible for the catalytic reactions of Ψ sites in various RNA types,3-7 as well as its differential functions in nuclear miRNA processing. 8 It has been shown that pseudouridylation in the rRNA and tRNA can maintain the ribosome entry sites 9 and stabilize its structure. 10 Pseudouridylation of tRNA-derived fragments were also found to impact stem cell commitment during early embryogenesis through translation control. 11 In mRNA, Ψ is contributed to the regulation of transcript stability, 12 translation efficiency, and RNA immune response. 13 The mRNA structure was proved to be recognized as the target of PUS 1, which implicates that modulation of RNA structure may be a possible mechanism to regulate mRNA pseudouridylation. 14 Pseudouridine was also found to be responded to environmental changes like serum starvation and been regulated by those signals accordingly. 15 In the mouse model, the associations between diseases and abnormal pseudouridylation have been reported.16-18 For example, the sequence-specific pseudouridylation of ribosomal RNAs is linked to the growth and metastasis of xenograft tumors. 16 The mutation of DKC1, a Ψ synthase (PUS) that affects the accumulation of telomerase RNA, 17 can lead to dyskeratosis congenita (DC) and cancer. 18
Currently, the transcriptome-wide distribution of Ψ RNA modification has been profiled by several types of high-throughput sequencing techniques, which contains Pseudo-seq, 15 Ψ-seq, 12 PSI-seq, 19 CeU-seq, 20 and ribosomal binding site (RBS)-seq. 21 The first 4 approaches were developed with similar principles, in which RNA is treated with the N-cyclohexyl-N′-(2-morpholinoethyl)-carbodiimide-metho-p-toluenesulfonate (CMC), and the reverse transcription is stopped on Ψ position by the presence of a bulky group. As a recently developed novel Ψ site-detection approach, the RBS-seq 21 is realized based on a modified version of the RNA bisulfite sequencing, which could result in better sensitivity. Although thousands of experimentally validated Ψ sites were revealed by the 5 sequencing techniques mentioned above, only 2 mouse Ψ data sets are publicly available, while both of them are identified by CeU-seq technique. Several computational models have been developed22-27 to support the prediction of mouse Ψ sites in a given RNA transcript. However, all those methods are based on sequence-derived features. The published prediction models were reported to have reasonable performances, but their accuracy still has room for improvement. Moreover, it is worth noting that, in a more recent study, Dou et al applied a feature extraction approach bi-profile Bayes (BPB) 28 to obtain sequence information from both positive and negative RNA sequences. 29 Interestingly, their results concluded that the overall performances for Ψ identification using BPB features as well as the combined features (a combination of sequence and BPB features) were not obviously enhanced, with the general accuracies ranging from 60% to 70%. Therefore, it is highly desirable to develop a high-accuracy approach for the identification of Ψ modification in mouse transcriptome, not only for taking the best use of experimentally detected data but also to further improve the real-life biological usages of Ψ predictors.
In this study, we first introduced genome-derived information to the predictive framework of Ψ RNA modification in the mouse transcriptome. The genomic feature was first introduced and implemented in research for human m6A site prediction, 30 which achieved a marked improvement in performance compared with previously sequence-based only predictors. In a more recent study, the combination of genomic and sequence features was applied to site prediction work of human Ψ RNA modification, 31 which validated the consistent performance of genome-derived information in a different type of human RNA modification. However, both these 2 studies focused on human transcriptome only, no evidence was found if genome-derived features work equally effective in a species other than human. In our study PSI-MOUSE, we extracted 38 genomic features from mouse transcripts. Combining the hand-crafted features with conventional sequence-based information, a high-accuracy mouse Ψ site predictor was established. We further trained and validated our predictive framework in the yeast genome and compared the performance with other competing methods. PSI-MOUSE also systematically checks whether the putative Ψ sites localize in regions with various post-transcriptional regulation factors, that is, RNA-binding protein (RBP)-binding regions, miRNA targets, and splicing sites, which can serve as a useful computational approach to facilitate the study of mouse Ψ RNA modification. The web-based server of PSI-MOUSE is freely accessed at www.xjtlu.edu.cn/biologicalsciences/psimouse and http://psimouse.rnamd.com.
Materials and Methods
Training and testing data
The known base-resolution mouse Ψ sites used for training and benchmarking were obtained from CeU-seq (Table 1) downloaded from Gene Expression Omnibus (GEO), we then filtered the experimentally validated Ψ sites to remain only those located on known transcripts for the extraction of genomic features. For the extraction of negative data, the unmodified U sites located on the same transcripts of positive Ψ sites were randomly selected. To fully utilize the underrepresented positive data, we match 10 negative sites for each of the positive site, from which 10 separate predictors were established with 1:1 positive-to-negative ratio, and their prediction performances were averaged during the evaluation. The known mouse Ψ sites from CeU-seq were detected under 2 independent conditions (brain and liver, see Table 1). In order to perform data set-level leave-one-out cross-validation, data set M1 was used for training first, while its performance was validated by data set M2 generated under another condition. Subsequently, data set M2 was used for training, with data set M1 for independent testing.
Base-resolution data set used for Ψ site prediction.
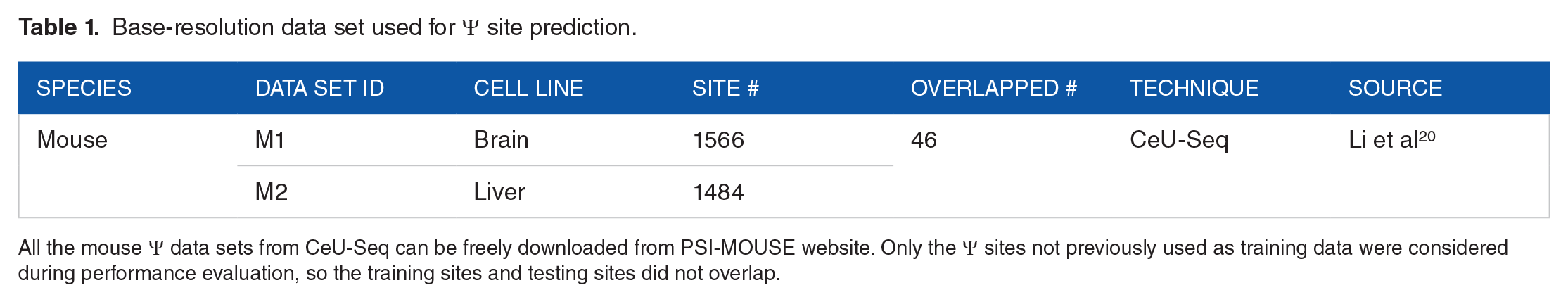
All the mouse Ψ data sets from CeU-Seq can be freely downloaded from PSI-MOUSE website. Only the Ψ sites not previously used as training data were considered during performance evaluation, so the training sites and testing sites did not overlap.
In order to faithfully compare the performance of our approach with competing methods, we further used the mouse and yeast Ψ data sets collected from Chen et al’s 23 study (Supplementary Table S1), which were also applied as benchmark data sets in many previous published studies.25-27
Feature extraction
Genome-derived features
The previous Ψ predictors were all developed by extracting sequence-based information from RNA segments. Inspired by the successful application of genomic features used in human m6A prediction, a total of 38 mouse and 22 yeast genome-derived features were generated to capture the attributes of the transcriptomic topologies for Ψ sites. The Genomic Features R/Bioconductor package with transcript annotations mm10 TxDb package and sacCer3 TxDb package 32 were used to generate Genomic Features 1-16, which are dummy variables indicating the overlapping state with the corresponding transcriptomic regions. We checked whether the uridine sites localize in a variety of types of topological features, that is, 5′UTR, 3′UTR, and intron. In order to overcome the isoform ambiguity from the annotation, only the transcript sub-regions on the primary (longest) transcripts of each gene were extracted. The second category of features (number 17-20) calculates the relative position of uridine sites in the transcript regions. The features are real numbered values between 0 and 1, where the value close to 0 indicates the relative approximation with the 5′ start of the region. The values are set to 0 if the sites do not belong to this region. Genomic features 21 to 25 calculate the length of the corresponding transcript regions where uridine sites localize. The values are also set to 0 for the sites not overlapping with the transcript region. The distances between uridine to the splicing junctions and the nearest Ψ site are captured in features 26 to 29. The clustering status of uridine sites was presented in features 30 and 31, which were defined by the number of neighboring uridine sites within the 100 bp and 1000 bp flanking regions of the target uridine sites. Besides, the RNA secondary structures around the uridine site are predicted using RNAfold with the Vienna RNA package 33 and presented in features 32 and 33. Finally, the various functional properties of the transcripts containing the Ψ RNA modification were represented by features 34 to 38, such as the number of isoforms and the transcript types of the coding. The detailed information of all genomic features we used in the Ψ site prediction was listed in Table 2.
Genome-derived features used for mouse and yeast Ψ site prediction.

Genomic features generated from both mouse and yeast genome are in bold.
Sequence-derived feature
To try to achieve the best performance in accuracy, 2 kinds of sequence-based features were also extracted and combined with genomic features mentioned above. The chemical properties of nucleotides and nucleotide density were first created and used for splice site prediction
34
and then being widely used in the predictive framework of RNA modification.35-37 For chemical properties, the 4 types of nucleotides were classified into 3 categories. First, the adenosine and guanosine have 2 rings in their structure, while the cytidine and uridine only have one ring. Second, the guanosine and cytidine have stronger hydrogen bonding than that of adenosine and uridine. And finally, adenosine and cytidine contain the amino group, while guanosine and uridine contain the keto group. Based on these 3 principles, the ith nucleotide from sequence S may be encoded by a vector

Thus, the A, C, G, U can be encoded as a vector (1,1,1), (0,1,0), (1,0,0), and (0,0,1), respectively. For the nucleotide density, the cumulative nucleotide frequency of nucleotide in ith position is calculated. The density of nucleotide in ith position is defined as the occurrences of the nucleotide
Machine-learning approach used for Ψ site prediction
Support vector machine (SVM) has been widely applied for the prediction of genomic and proteomic data in computational biology, such as the microRNA target prediction, 38 and protein phosphorylation prediction. 39 In terms of RNA modification, the SVM was also used in previous m6A, 40 m5C, 41 and m1A 42 prediction and was shown to be a robust and effective machine-learning algorithm. In this project, the R interface of LIBSVM was used to build the predictor using the kernel of radial basis function. 43 In the data set-level leave-one-out experiment, only the Ψ sites not previously used as training data were considered in independent testing, which avoided the leakage between the training and testing data. Consequently, the performance evaluated on the testing data can justly reflect the ability of the purposed predictor to identify unknown Ψ sites.
Model performance evaluation
To evaluate the performance, we calculated the receiver operating characteristic (ROC) curve (sensitivity against 1-specificity) and the area under ROC curve (AUROC) as the main performance evaluation metric. The sensitivity (Sn), specificity (Sp), Matthews correlation coefficient (MCC), and overall accuracy (ACC) were calculated as other indicators to evaluate the reliability of the model. All the prediction processing is based on R language. To compare the performance of the newly proposed model with the existing Ψ site predictors, we reproduced the machine learning scaffold of PPUS, iRNA-PseU, and PseUI by realizing each of their sequence-based encoding methods, and the predictors are learned with the training data used in our study


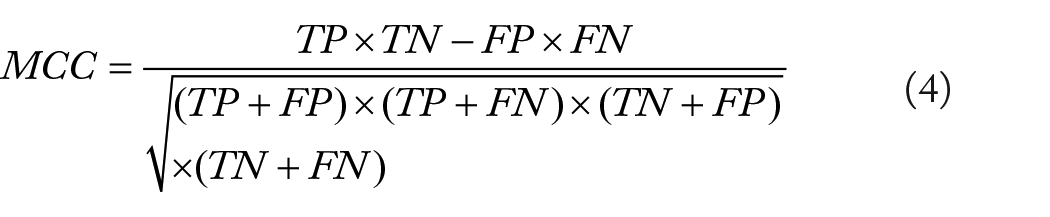

where TP represents the true positive, while TN represents the true negative; FP is the number of false positives, and FN the number of false negatives.
Estimate the probability of mouse Ψ RNA modification
In our study, the likelihood ratio (LR) is used to indicate how likely the computationally predicted result to be a true Ψ RNA modification. A LR of “100” suggests that it is an experimentally validated ψ site. The calculation of LR is showed as follows

The statistical significance of LR is assessed by an upper bound of the p value. It indicates how extreme the observed LR is among all the mouse transcriptome U sites. It is calculated from the relative ranking of the putative Ψ sites, that is, if only 0.1% of U sites have an LR score larger than a specific U site, then the upper bound of the p value of this site is .001. A transcriptome-wide prediction of mouse Ψ site was applied using PSI-MOUSE, a putative Ψ site is considered as high confidence if its LR within top 0.5% of LR (corresponding to p value < .005) of all the transcriptome U sites, followed by medium confidence (.005 < p value < .05) and low confidence (p value > .05).
Gene annotation and posttranscriptional regulation association analysis
To help academic users gain a more comprehensive understanding of the predicted mouse Ψ sites, various types of functional genomic information were automatically annotated in PSI-MOUSE for each putative Ψ site using ANNOVAR package, 44 that is, gene symbol, Ensembl gene ID, gene region, and gene type. PSI-MOUSE also checks whether the putative Ψ sites localized in regions with RBPs, miRNA-RNA targets, and splice sites, which help users to explore the potential effect of Ψ RNA modification on posttranscriptional regulation. The RBPs regions were obtained from POSTAR2, 45 the miRNA-RNA targets were downloaded from miRanda 46 and starBase2, 47 and the Canonical splice sites (GT-AG) from UCSC 32 annotations. For splicing sites analysis, specifically, we expanded 100 bp upstream region from 5′ splicing sites and 100 bp downstream region from 3′ splicing sites, the putative Ψ sites were then mapped with these regions. The Supplementary Table S2 shows detailed information related to posttranscriptional regulation analysis.
Results
As the process of RNA library preparation for existing data sets was based on polyA selection, to avoid the under-representation of the intronic Ψ sites in the data, we designed 2 modes for mouse Ψ site prediction. For the mature mRNA mode, the positive and negative Ψ sites mapped to intron regions were both filtered to only remain the sites localized on the mature mRNA transcripts. While for the full transcript mode, all Ψ sites were considered in the prediction. Consequently, the performance of the predictor was evaluated under these 2 modes, respectively. In addition, we applied feature selection to identify the most significant subset of features, which was implemented using Perturb method. 48 The functional insights of genomic features in our tool can be reflected by the feature importance plot. The feature importance scores can indicate the scientific significance of genomic markers in terms of its predictability for Ψ. According to the rank of the importance, the N most significant features were reserved in the prediction and evaluated with 5-fold cross-validation. In the process of feature selection, the Ψ sites from mouse brain data set were used as training data, while the mouse liver Ψ sites were used for testing purposes. Finally, the top 26 and 28 genomic features contribute to the best prediction performance under mouse full transcript and mature mRNA model, respectively (see Figure 1).

Feature selection of the genome-derived features for mouse Ψ site prediction. Top 26 and 28 genomic features were used in further prediction under mouse full transcript model (A) and mouse mature mRNA model (B), respectively. mRNA indicates messenger RNA.
We observed that our approach PSI-MOUSE achieved remarkable improvement in prediction accuracy compared with existing encoding methods for Ψ prediction when tested on 5-fold cross-validation and independent data sets (see Table 3). Besides, the SVM algorithm applied in our PSI-MOUSE model, we further re-built predictors using 4 other machine-learning approaches, which are Random Forest, Naïve Bayes, decision tree, and generalized linear model. When tested on independent data sets, our model, PSI-MOUSE, achieved a better performance than the competitive predictors in all conditions (Table 4). This result suggested that the features used by the prediction model contribute to most of the predictive power, and the impact of machine-learning algorithm is limited. The detailed evaluation by the sensitivity, specificity, ACC, and MCC were summarized in Supplementary Table S3 and S4 for the full-transcript model and mRNA model, respectively.
Performance evaluation of mouse Ψ site predictors (AUROC).

Abbreviation: AUROC, area under ROC curve.
Using the sequence-based encoding methods described in each existing predictor, we faithfully reproduced the iRNA-PseU, PPUS, and PseUI using the same training data of PSI-MOUSE. The 5-folds cross-validation and independent data set testing were performed to evaluate prediction accuracy under full-transcript and mature mRNA modes, respectively. When the independent data set testing was performed, the observations used for testing were excluded from the training data.
Performance on an independent data set using different machine-learning algorithms (AUROC).

Abbreviations: AUROC, area under ROC curve; DT: decision tree; GLM: generalized linear model; NB: Naïve Bayes; RF: random forest; SVM: support vector machine.
The iRNA-PseU, PPUS, and PseUI were rebuilt using the same training data of PSI-MOUSE with their own sequence-based encoding methods.
Besides, the study of iPseU-CNN, 27 iPseU-NCP, 25 and XG-PseU 26 all used data sets from Chen et al’s study to develop and validate their predictive pipelines. To faithfully compare the performance of our approach with these competing methods, we further trained and tested our predictive framework using data sets from Chen et al’s study. For mouse Ψ site prediction, data set M_944 was used for training purpose and tested on 5-fold cross-validation. Consistent with previous results, PSI-MOUSE achieved a major improvement in all conditions compared with other state-of-the-art predictors (Table 5), suggesting the reliability of the approach. For Ψ site prediction in yeast genome, data set S_628 was used to train the predictor, while its performance was tested on independent testing data set S_200. When tested on 5-fold cross-validation and independent data set, we observed that our approach, which integrated additional genomic features besides the conventional sequence features, achieved reasonable improvement in prediction performance compared with other competing predictors, respectively (Supplementary Table S5). To sum up, for the first time, we generated genome-derived features from mouse and yeast transcriptome, respectively. The predictive framework achieved satisfactory improvement compared with previous published predictors, marking the reliable contributions of genomic features for the prediction of RNA modifications in species other than human.
Performance evaluation of mouse Ψ predictors using data set M_944.
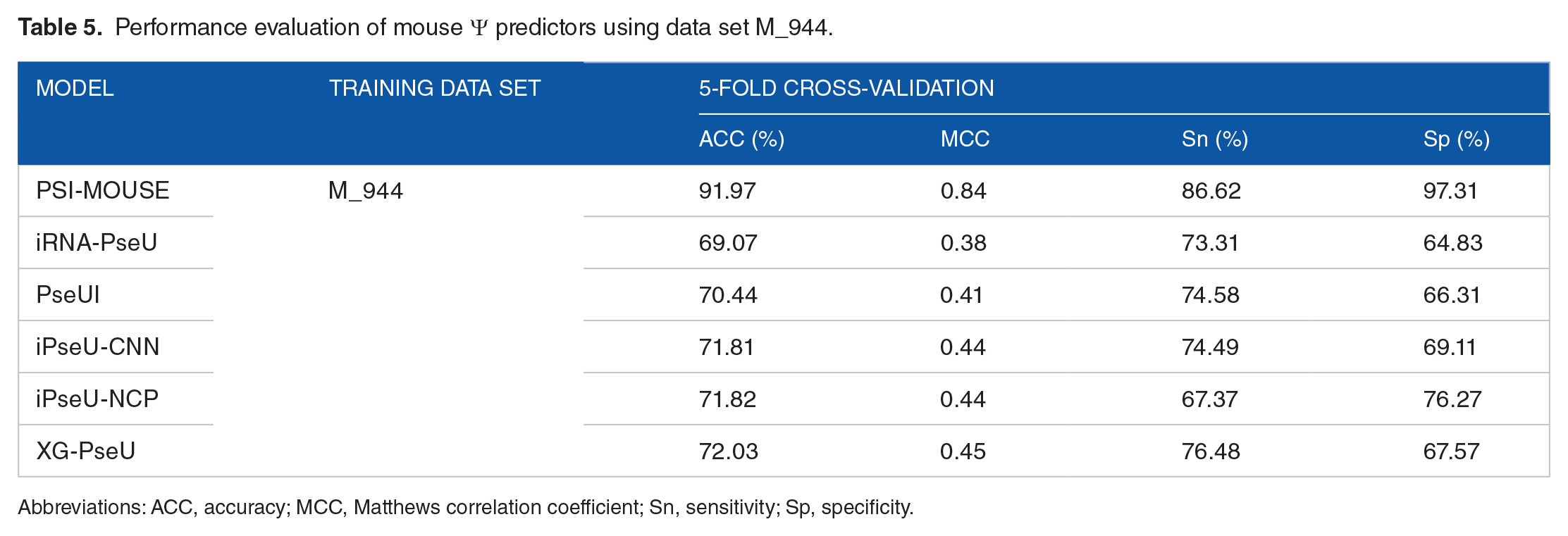
Abbreviations: ACC, accuracy; MCC, Matthews correlation coefficient; Sn, sensitivity; Sp, specificity.
Web server interface
A web server for our newly purposed model was built using Hyper Text Markup Language (HTML), cascading style sheets (CSSs), and hypertext preprocessor (PHP). All the components of PSI-MOUSE can be easily accessed through the homepage of PSI-MOUSE (see Figure 2). The webserver of PSI-MOUSE provides different prediction modes, which allows users to customize their prediction under different pipelines, that is, 2 feature sets, various combinations of annotation methods. Users can upload either genome coordinate in txt format, or the FASTA file containing RNA sequences as the input of PSI-MOUSE. The database of PSI-MOUSE collected 3282 experimentally validated mouse Ψ sites, users can also query the database by Gene and Chromosome region. All the materials presented in the database and web server can be freely downloaded. PSI-MOUSE is freely accessed at www.xjtlu.edu.cn/biologicalsciences/psimouse and http://psimouse.rnamd.com.

Screenshot of the homepage of PSI-MOUSE. The web server and database of PSI-MOUSE can be easily accessed from the homepage. The webserver of PSI-MOUSE currently does not allow us to submit prediction jobs using Safari iOS system.
Discussion
With the massive amount of data generated from various types of high-throughput sequencing techniques, many computational methods have been developed to facilitate the research of RNA modification, such as site prediction and data collection works,49-53 RNA modification-associated genetic variants analysis tools,54,55 as well as functional annotation tools.56-59 In this study, we innovatively represented 38 RNA topological features on the mouse genome, and a prediction framework PSI-MOUSE was built upon them. Compared with existing works that based on sequence-derived features only, PSI-MOUSE achieved a significant improvement in performance accuracy, indicating the successful application of genome-derived features in the mouse transcriptome. In addition, 3282 experimentally validated mouse Ψ sites with functional annotations were also collected in our work.
However, the mouse experimentally validated Ψ sites are only available in 2 conditions detected from the same sequencing technique, and thus, the prediction performance may be over-estimated. Besides, we further validated our approach using mouse and yeast Ψ data sets collected from Chen et al’s study, which were also applied in many state-of-the-art Ψ site prediction studies. We observed a remarkable improvement of prediction accuracy in the mouse genome, as well as a relatively minor bonus in the yeast genome. The genomic features derived from annotation file of yeast transcript were obviously less than that of from mouse transcript; therefore, the application of genomic features in yeast can be further studied, and the performance of yeast Ψ site predictor can still be improved by deriving more informative genomic patterns from yeast genome. In future research, PSI-MOUSE will be further updated with more experimental identified Ψ sites detected under multiple technical contexts. In addition, the PSI-MOUSE scheme can be further expanded to the prediction of other RNA modification such as m1A 60 and m7G. 61
Supplemental Material
Supplement_v0_xyz359528f8f7314 – Supplemental material for PSI-MOUSE: Predicting Mouse Pseudouridine Sites From Sequence and Genome-Derived Features
Supplemental material, Supplement_v0_xyz359528f8f7314 for PSI-MOUSE: Predicting Mouse Pseudouridine Sites From Sequence and Genome-Derived Features by Bowen Song, Kunqi Chen, Yujiao Tang, Jialin Ma, Jia Meng and Zhen Wei in Evolutionary Bioinformatics
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work has been supported by the National Natural Science Foundation of China (grant no. 31671373); XJTLU Key Program Special Fund (grant no. KSF-T-01).
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
ZW initialized the project. ZW and BS designed the research plan. ZW and KC constructed the genomic features considered in mouse pseudouridine site prediction. BS performed the development of the pseudouridine site web server. YT and BS built the website. BS and ZW drafted the manuscript. All authors read, critically revised, and approved the final manuscript.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.