
Abstract
Chinese indigenous pigs in the Taihu Lake region are well known for their high fecundity and other excellent characteristics. To better understand the characteristics of these breeds in this area as well as to provide the government and breeders the molecular basis for formulating a reasonable conservation policy, we explored the structure of haplotype blocks and genetic diversity of the 7 populations which is relevant for the management and conservation of these important genetic resources using next-generation sequencing data. In this study, a total of 131 300 single-nucleotide polymorphisms with minor allele frequencies ⩾0.05 were obtained for further analysis. In general, there are similar within-breed genetic diversities (He, Ho, Pn, Ar) among these 7 pig populations in the Taihu Lake region. Average values for the inbreeding coefficients estimates in the 7 populations are 0.110 (F1), 0.056 (F2), and 0.078 (F3). All the breeds have seen a continuous decline in Ne estimates over time with FJ and SW populations having a very similar curve. Moreover, the Ne of SMS pig breeds were smaller than other Chinese pig breeds, indicating that SMS pig breeds underwent stronger selection pressure than other Chinese pig breeds. The average genetic distances among the 7 populations in the Taihu Lake region were 0.235 (MMS), 0.240 (SMS), 0.269 (EH), 0.248 (MI), 0.221 (FJ), 0.254 (JX), and 0.212 (SW). A summary of the number of haplotype blocks and haplotype diversity was also presented. This study provide a deep understanding of the current situation of conservation in this region, thereby uncovering the pertinent insight to better formulate more reasonable preservation policies for the government departments and breeding planners to follow-up.
Introduction
The developing countries which are characterized with production environments that are low to medium input and high stress harbors most of the world’s breeds and each of these breeds are expected to have adapted to their specific environment. This expectation is strongly supported by empirical evidence implying that the genetic basis of population differentiation will be nonadditive for fitness traits and each breed will have different adaptive gene complexes. The Chinese indigenous pigs in the Taihu Lake region are well known for their high fecundity and enjoy the reputation of “national treasure.” These domestic pigs around the Taihu Lake also have many other excellent features such as disease resistance, resistant to rough feeding, and excellent meat quality. The first survey of pig resources in China regarded these animals as a single breed termed Taihu pigs. 1 However, since 1974, Taihu pigs have been divided into 7 breeds including Erhualian (EH), Meishan (MS), Fengjing (FJ), Jiaxing Black (JX), Mi (MI), Shawutou (SW), and Hengjing pig breed which is now extinct.According to the second survey of China’s swine breeds in 2011, these pigs were divided to 6 breeds (MS, FJ, JX, MI, SW and EH) with the Meishan breed further subdivided into 2 subpopulations which are called the Middle Meishan (MMS) and the Small Meishan (SMS).2,3
From a global perspective, conservation is not only about endangered breeds but also about those that are not being used efficiently. A small proportion of breeds (mainly in the developed world) are involved in planned genetic improvement programs. For other breeds, and particularly in the developing world, there is an urgent need to develop breeding programs to improve their production and productivity. In recent years, the state and government have attached great importance to the protection of local genetic resources. The main objective is to conserve the indigenous breeds with the aim of minimizing the loss of among breed diversity which includes breeding programs that ensure efficient utilization and conserving those at risk. Therefore, the program urges farms to establish an efficient strategy to maintain genetic diversity and avoid inbreeding within these local breeds, which requires an in-depth investigation of population structure and phylogenetic relationships of these breeds. Genetic diversity contributes to the prioritization process as a tool (revealing migration routes, refuges, and historical responses) and as an object of conservation concern. 4
We can consider the implications of the relationship between genetic diversity and conservation at many levels: genes, individuals, populations, varieties, subspecies, species, genera, and so on. Genetic diversity provides a retrospective view of evolutionary lineages of taxa, a snapshot of the current genetic structure within and among populations, and a glimpse ahead to the future evolutionary potential of populations and species. Genetic diversity is also one of 3 forms of biodiversity recognized by the International Union for Conservation of Nature (IUCN) as deserving conservation, along with species and ecosystem diversity. Genetic diversity has been defined as the variety of alleles and genotypes present in a population and this is reflected in morphological, physiological, and behavioral differences between individuals and populations. 5 From a functional point of view, genetic diversity can be classified as neutral, deleterious, or adaptive. 6 Since the beginning of the 1990s, the development of appropriate tools has resulted in a leading role for molecular markers in the characterization of genetic diversity. At this level, genetic diversity is usually measured by the frequencies of genotypes and alleles, the proportion of polymorphic loci, the observed and expected heterozygosity, or the allelic diversity. 7
Recent studies have extensively evaluated genetic variation and population structure of Chinese pig breeds using not only high-density single-nucleotide polymorphism (SNP) markers8,9 but also whole-genome sequencing data. 10 These investigations focused primarily on genetic diversity and detected the signatures of positive selection between Chinese and Western pig breeds. Wang et al 11 studied genetic diversity and population structure of Chinese indigenous pig breeds in the Taihu region to provide basis for the division of breeds. Chen et al 12 reported on genetic diversity and population structure in Chinese indigenous pig breeds in Zhejiang Province. Also, many studies have extensively analyzed linkage disequilibrium (LD) features and haplotype blocks in livestock species, especially in pigs. 13 The genome structure especially LD and haplotype blocks can provide fundamental information on the genome organization of these pig breeds and gives us a reference for formulating a new conservation strategy. However, to our knowledge, research on the genetic properties and relationships, especially for the structure of the haplotype block of the 7 indigenous pig populations in the Taihu Lake region from the perspective of conservation, is lacking. Therefore, in this study, we investigated the genetic diversity and haplotype blocks of the 7 Chinese indigenous populations in the Taihu Lake region to reveal current status of conservation and relationships of these pig populations. By studying the current indigenous pig breeds in the Taihu Lake region, we can provide a theoretical basis for our next step to better protect their genetic diversity and formulate conservation policies.
Materials and Methods
Ethics statement
All experimental procedures were approved by the Institutional Animal Care and Use Committee of Shanghai Jiao Tong University, and all methods involved pigs were in accordance with the agreement of Institutional Animal Care and Use Committee of Shanghai Jiao Tong University (contract no. 2011-0033).
Population and sequencing data
A total of 445 pigs (75 Small Meishan, 97 Medium Meishan, 36 Mizhu, 42 Erhualian, 91 Jiaxing Black, 72 Shawutou, and 32 Fengjing) from the 6 Chinese indigenous breeds in the Taihu Lake region were selected, including 252 pigs (69 Small Meishan, 50 Medium Meishan, 36 Mizhu, 31 Erhualian, 29 Jiaxing Black, 21 Shawutou, and 16 Fengjing) from Wang et al. 11 All DNA samples were genotyped according to the GGRS protocol 14 (http://klab.sjtu.edu.cn/GGRS/). Briefly, high-molecular-weight genomic DNA samples were extracted from ear tissue, digested with AvaII and then ligated with a unique adapter barcode. Next, the samples were pooled and enriched to construct a sequencing library. Finally, the sequence libraries (fragments ranging from 300 to 400 bp [base pairs], including the adapter barcode sequence) were sequenced on an Illumina HiSeq2000 (the sequencing process is given in detail by the manufacturer, Illumina) instrument with a paired-end (2 × 100 bp) pattern. The SNPs were identified and genotyped using SAMtools, 15 and these variants were retained for further analysis according to the following criteria: (1) SNP test scores are greater than or equal to 20 (ie, the accuracy of more than 99%), (2) the calling rates of SNP are greater than or equal to 90%, (3) the minor allele frequency (MAF) was greater than or equal to 5%, and (4) the detected SNP is the only one that appears on a fixed chromosome. Before these genotyped SNPs are phased by FASTPHASE 16 for further analysis, the missing genotypes were imputed using iBLUP 17 with the command line “perl iBLUP.pl genotype.vcf 445 89 0.1,” in which 445 is the total number of samples, 89 is the minimum detected number of samples, and 0.1 is the LD threshold. iBLUP is a genotype imputation method that imputes missing genotypes using identity-by-descent and LD information. 17 A total of 131 300 SNPs with MAFs ⩾0.05 were obtained in our study.
Genetic diversity within populations
The allelic richness (Ar), proportion of polymorphic markers (Pn), expected heterozygosity (He) and observed heterozygosity (Ho) were used to investigate the genome-wide genetic variability within these 7 populations. Ar was calculated using ADZE v1.0. 18 Pn, He, and Ho were calculated using PLINK v1.07. 19
Marker-based inbreeding coefficients were estimated using the GCTA software. 20 Three different metrics were obtained using the -ibc option of the program: based on (1) the variance of the additive genotype (F1), (2) the excess of homozygosity (F2), and (3) the correlation between uniting gametes (F3). 20
The historical effective population size (Ne) was estimated by the software of SNeP v1.1, 21 which can estimate Ne trends across generations using multilocus SNP data. This approach estimates historical effective population size based on the relationship between LD, Ne, and recombination rate, as well as corrects for sample size simutaneously:

where
Genetic relationship among populations
Genetic distance
To estimate the genetic distances among populations, all 131 300 SNPs were used to calculate the average proportion of alleles shared,

where IBS1 and IBS2 are the numbers of loci that share 1 or 2 alleles at 1 locus. The genetic distance (D) between all pairwise combinations of individuals was calculated as follows:
Haplotype construction and haplotype diversity
A haplotype is a contraction of the phrase haploid genotype and is a stretch of DNA that is inherited as a unit. In diploid genomes, haplotypes are a set of closely linked nucleotides present on a chromosome that are inherited together. Thus, haplotypes are stretches of DNA in LD that are not broken up by recombination.
Haplotype blocks are estimated following the default procedure in HAPLOVIEW (v4.1). HAPLOVIEW (v4.1) was also used to define the haplotype blocks present in the genome. The method followed for block definition was previously described by Gabriel et al.
24
Haplotype diversity is defined as
Data availability
All the SNP data we used were uploaded to our Web site (https://jbox.sjtu.edu.cn/l/XH2s6V). Supplemental File S1 contains all the figures of the NJ tree within the 7 pig populations, respectively. Supplemental File S2 contains the results of the haplotype blocks of the other 6 populations (MMS, MI, EH, FJ, SW, and JX).The authors affirm that all data necessary for confirming the conclusions of the article are present within the article, figures, and tables.
Results
Genetic diversity within populations
He Ho Fis Pn Ar
The within-breed genetic diversity of the 7 pig populations is presented in Table 1. Among the 7 populations, FJ had the largest He and Ho, whereas the value for Ho in the EH population was the lowest. Overall, the values of Ho were always greater than the values of He among the 7 populations and the Fis values were all negative which indicates excess of heterozygosity. The Pn values were all the same (0.999) in these 7 indigenous pig populations, which means that these populations have the similar proportions of polymorphic markers and also suggests that these populations were regarded as a single breed termed Taihu pigs. As for genetic diversity measured by allelic richness, the 7 populations had similar values of 1.99, almost closer to 2 indicating that the 7 populations have higher allelic richness.
Sample sizes and genetic diversities of the 7 pig populations in the Taihu Lake region.
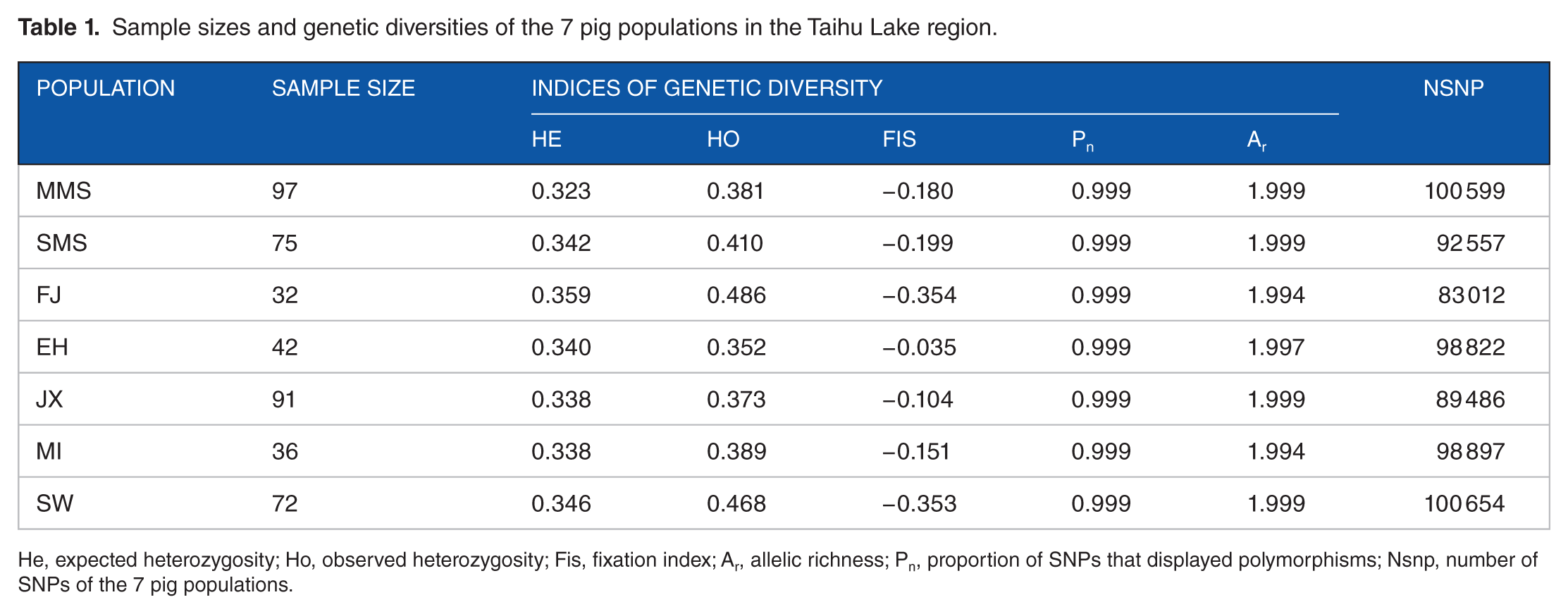
He, expected heterozygosity; Ho, observed heterozygosity; Fis, fixation index; Ar, allelic richness; Pn, proportion of SNPs that displayed polymorphisms; Nsnp, number of SNPs of the 7 pig populations.
Inbreeding coefficient
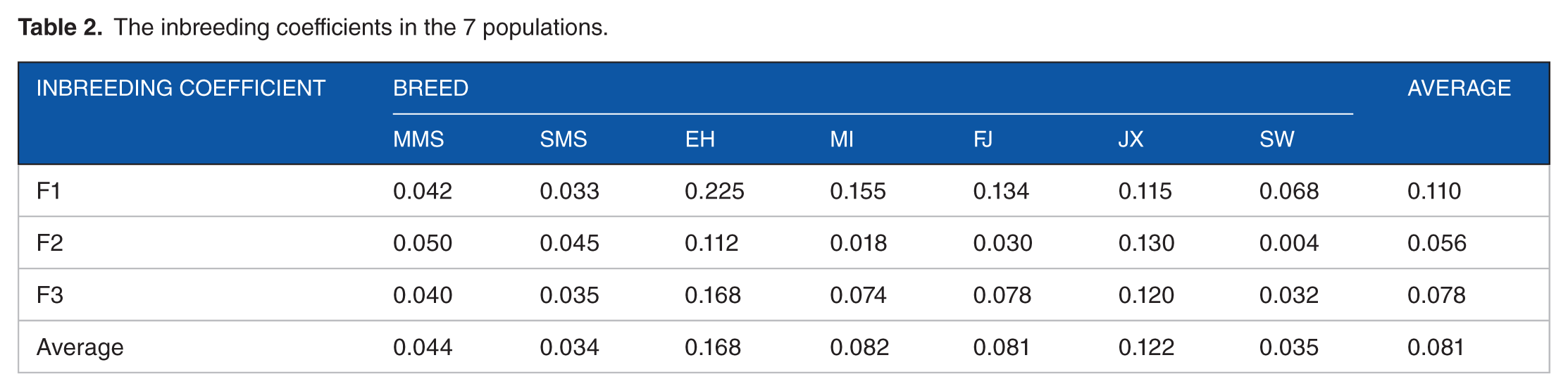
Average values for the positive estimates in the 7 populations were 0.110 (F1), 0.056 (F2), and 0.078 (F3) (Table 2). Although the estimates from various approaches were different, however, they all showed similar results. SMS, SW, and MMS had the lowest inbreeding degree compared with MI, FJ, and EH. Overall, the average inbreeding coefficient for all populations in the Taihu Lake region was 0.081, which implies that there is an acceptable conservation effect for the Chinese indigenous pigs in the Taihu Lake region. The correlations of the inbreeding coefficients estimated by the 3 methods were all higher with values of 0.75, 0.95, and 0.88, respectively (Figure 1).
The inbreeding coefficients in the 7 populations.
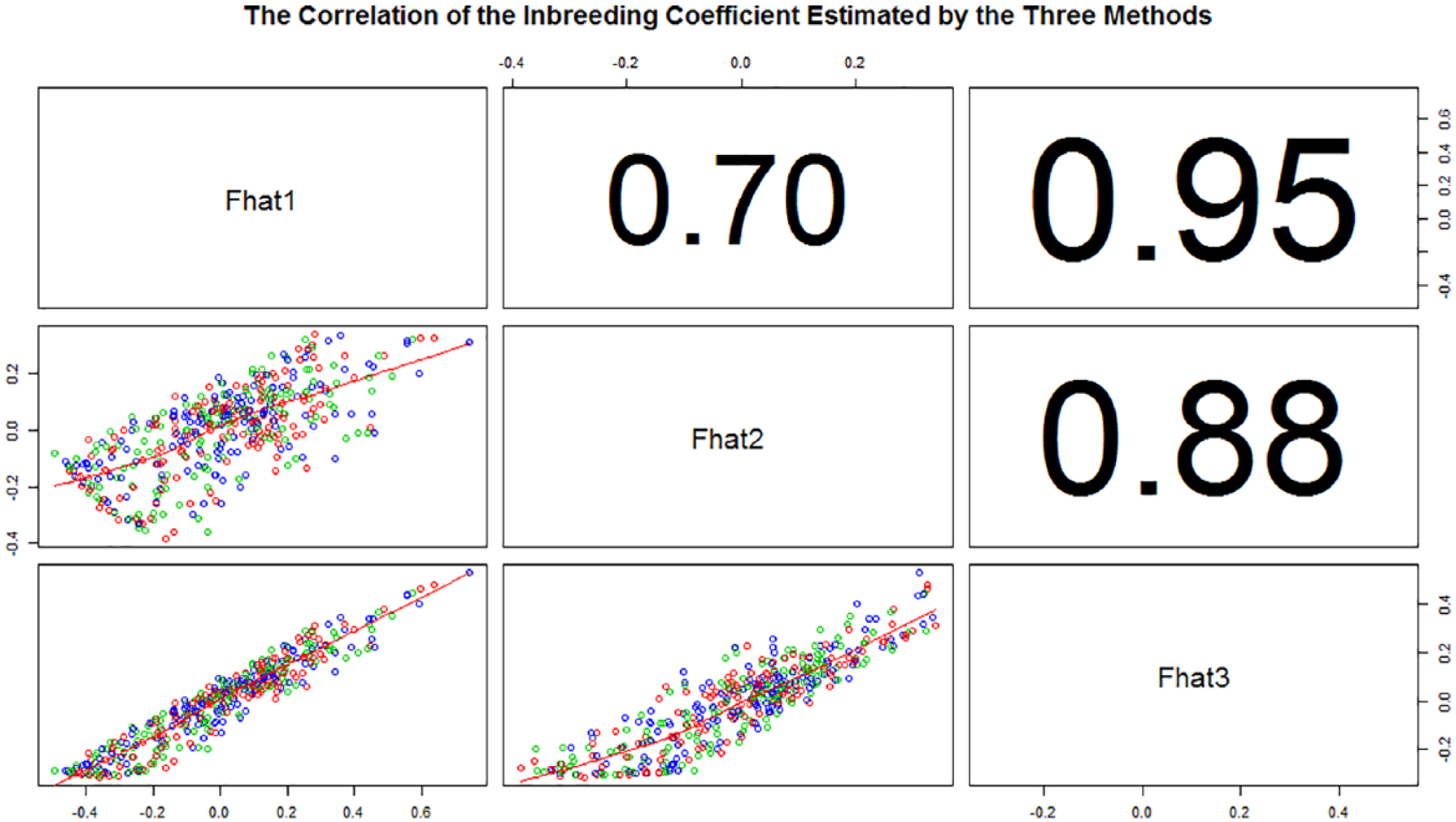
The correlation of the inbreeding coefficient estimated by the 3 methods.
Effective population size
The tendency of effective population size (Ne) of each pig breed along the generations is shown in Figure 2. The past Ne was reflected by LD over shorter recombinational distances and the longer distances provided recent ancestry. 25 In general, all the breeds have seen a continuous decline on Ne estimates over time and FJ and SW populations have very similar curves and results. Also, the Ne of SMS pig breeds are smaller than other Chinese pig breeds, indicating that SMS pig breeds might undergo stronger selection pressure than other Chinese pig breeds.
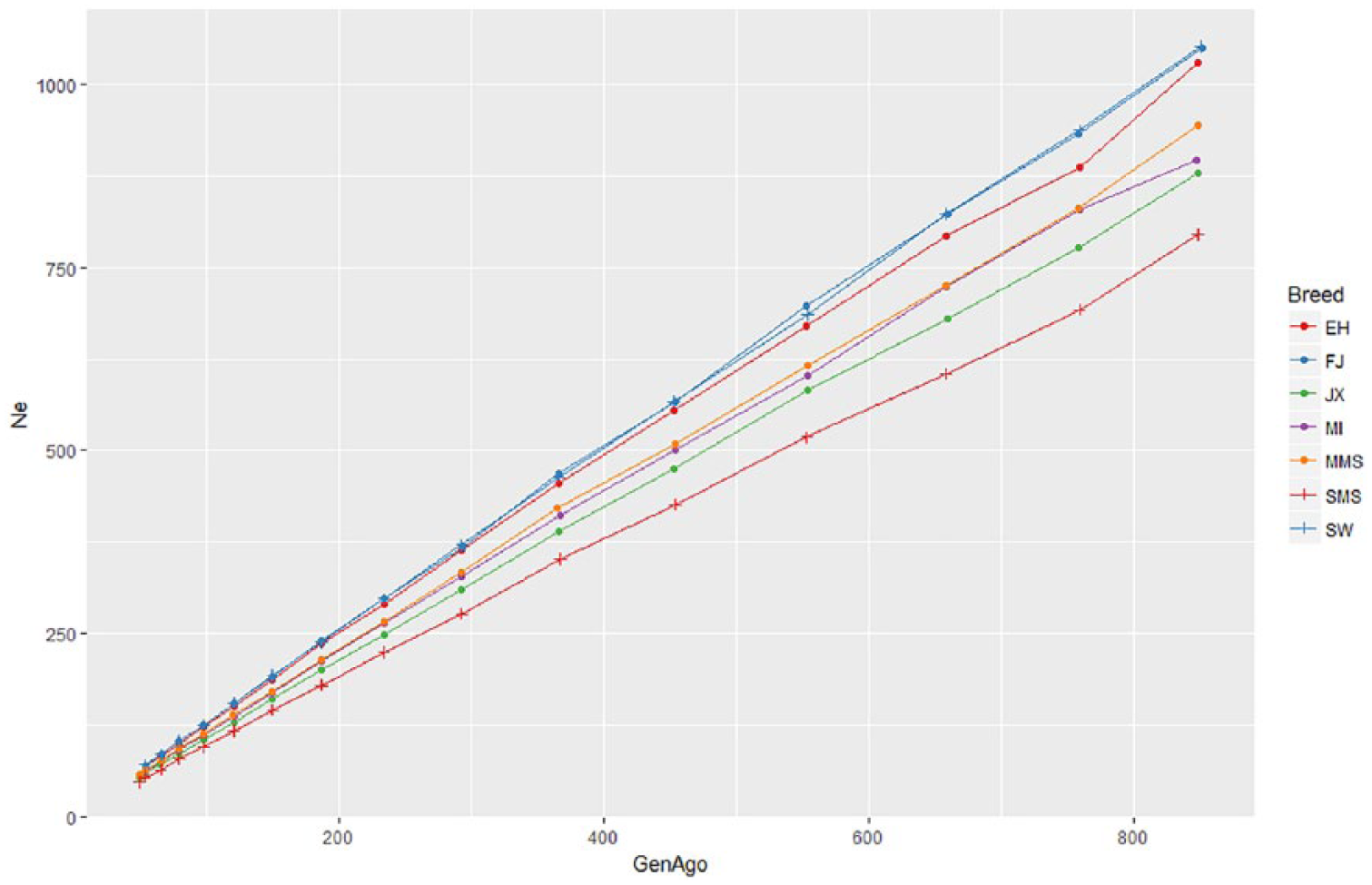
The tendency of Ne of the 7 indigenous pig populations.
Genetic relationship among populations
Genetic distance
The average genetic distances among the 7 populations in the Taihu Lake region were 0.235 (MMS), 0.240 (SMS), 0.269 (EH), 0.248 (MI), 0.221 (FJ), 0.254 (JX), and 0.212 (SW). All these 7 populations had similar genetic distance within their own groups and it is obvious that SW has the nearest genetic distance of the 7 populations, which is consistent with our previous results in the inbreeding coefficient.
The NJ trees were also constructed using genome-wide genotypes for more intuitive expression of genetic distances within the 7 pig populations (Supplemental Files S1 and S2).
The analysis of the haplotype blocks and haplotype diversity
A summary of the number of haplotype and haplotype diversity is presented in Table 3. We also give a summary of the distribution, size, number, and SNPs involved in the haplotype blocks per chromosome of SMS population in Table 4 and the statistics of the haplotype blocks of the other 6 populations are presented in Supplemental File S3. From Table 3, it can be seen that there are more haplotypes in MS (MMS, SMS) population compared with the other populations. Also, there are no significant differences among the haplotype diversities and the average of haplotype diversity was about 0.418, which means that there is a similar conservation status among these 7 populations in the Taihu Lake region.
The number of haplotype and haplotype diversity among the 7 populations.

The block structure per chromosome in MMS.

In the MMS population, a total of 14 960 haplotype blocks spanning 7851 kb of the genome were detected (Table 4). The average block size was 0.52 kb, ranging from 0.02 to 194.80 kb (chr13, 166282257-166477058, 4 SNPs). In total, 35 491 SNPs (35.28% of all SNPs used in MMS) formed blocks with a range of 2 to 9 SNPs per block. The autosomes showing the longest and shortest haplotypic structures in the genome were chr1 with 1342 blocks spanning 929.27 kb and chr18 with 446 blocks covering 409.14 kb. One of the important reasons for this is that it can be related to the length of chromosomes in pigs. Also, chromosome 1 had the highest density (8.94%) of SNP in haplotype blocks, whereas the lowest density (2.91%) was observed in chromosome X.
We also made a statistics of common haplotypes across populations. As shown in Table 5, most common haplotypes (283) were found to occur between MMS and SMS and the least common haplotypes (63) were between FJ and EH. The numbers of common haplotypes had no significant differences for every 2 populations. However, the population-specific haplotypes can better represent unique characters in one population and thus indicates that we are supposed to pay more attention to the population-specific haplotypes in future conservation programs.
The statistics of common haplotypes across populations.

Discussion
There are many ways to measure the genetic variation and the loss of its diversity. With the application of molecular marker technology to the study of livestock and poultry diversity, it is very important for a specific conservation population and population genetic diversity indicators to measure the genetic variation and conservation of genetic diversity. The sensitivity of different genetic diversity indicators is different. Hence, the actual situation is crucial to be used as selection criteria for genetic diversity indicators.
Heterozygosity is one of the major genetic variations in natural populations. It is often one of the first “parameters” that one presents in a data set. It can tell us a great deal about the structure and even history of a population. High heterozygosity means lots of genetic variability, and low heterozygosity means little genetic variability. Often, we will compare the observed level of heterozygosity with what we expect under Hardy-Weinberg equilibrium. If the observed heterozygosity is lower than expected, we tend to attribute the discrepancy to forces such as inbreeding. Allelic richness is the number of alleles per locus rarefied to match the number of observations in the population with the lowest sample size. 26 This measure obviously depends on sample size, and to compare samples of different sizes, the number of alleles per locus is often replaced by allelic richness. Allelic richness provides complementary information to gene diversity (expected heterozygosity). Situations can be given of populations with the same heterozygosity but different allelic richness and vice versa. However, the consequences of these different population compositions can be different in terms of potentiality of the population for adaptation and evolution. Allelic richness and gene diversity can also behave differently in terms of genetic differentiation between subpopulations in the context of a subdivided population. El Mousadik and Petit 26 proposed a coefficient of allelic richness differentiation (ρST) and found that this parameter gives higher values than gene diversity differentiation in an analysis of allozymes in argan trees. Also, a locus could be defined as monomorphic if the most common allele frequency is 100%, 99%, or 95% of all sampled alleles. As loss of rare alleles is expected to be one of the most immediate results of reduced population size, either the 100% or 99% criterion may be better estimates in endangered species. This may appear a straightforward measure but different studies vary in what criteria are used for scoring a locus as polymorphic.
Another aspect of interest while studying a population under selection pressure is to study the level of inbreeding. Traditional estimation of the inbreeding coefficient based on pedigree data 27 is dependent on the completeness and accuracy of the available pedigree records. Currently, using the information provided by molecular markers (genome-wide SNP chip panels), we can estimate this coefficient with or without pedigree information. Several methods have been described for this purpose.20,28,29 Individuals with the same inbreeding coefficient could be classified as inbred when they were sampled from a population with few inbred individuals and as outbred when they were sampled from a population where inbreeding was more frequent. Effective population size (Ne) is another important population genetic parameter which can describe the amount of genetic drift in populations. It has been subject to much research to estimate Ne over the past 80 years. The methods to estimate Ne from LD were developed about 40 years ago. However, only the most recent advances in DNA technology have made the calculation of Ne depending on large amounts of genetic marker data available. 21
Genetic distance is a measure of the genetic divergence between species or between populations within a species, whether the distance measures time from common ancestor or degree of differentiation. 30 Populations with many similar alleles have small genetic distances. This indicates that they are closely related and have a recent common ancestor. Genetic distance is useful for reconstructing the history of populations. Genetic distance is also used for understanding the origin of biodiversity. For example, the genetic distances between different breeds of domesticated animals are often investigated to determine which breeds should be protected to maintain genetic diversity. 31
Substantial evidence has already accumulated that the genome can be parsed into haplotype blocks of variable length. Haplotype blocks, together with the corresponding tag SNPs and common haplotypes determined by haplotype block–partitioning algorithms, can be used in genome-wide association studies, as well as in the fine-scale mapping of complex disease genes. Understanding the patterns of haplotype blocks is useful to develop appropriate management and conservation programs, to maintain overall genetic diversity and avoid inbreeding. Allelic diversity is an alternative criterion to measure genetic diversity, and some authors32,33 consider that this parameter is the most relevant in conservation programs, as high number of alleles imply a source of single-locus variation for important traits such as the major histocompatibility complex, which is responsible for the recognition of pathogens. It is also important from a long-term perspective because the limit of selection response is determined by the initial number of alleles 34 and it is more sensitive to bottlenecks than expected heterozygosity.
The protection and preservation of the current breeds or populations depend on the phenotype, origin, and distribution and lack of molecular genetic basis. In this study, we can essentially understand their genetic differences using genome-wide high-density markers to investigate haplotype structures and genetic diversity in domestic animal populations. Through the calculation of Ar and Pn, we confirmed that the genetic diversity of the indigenous pig breeds in China is at a relatively high level. This may be due to the lower selective pressure and the higher genetic diversity of their ancestors. The haplotype structure and genetic diversity of the indigenous pig breeds in the Taihu Lake region were evaluated using 131 300 SNPs that were relatively evenly distributed in the genome. This will further deepen our understanding of the characteristics of Chinese indigenous pig germplasm resources and provide a molecular basis for the subsequent development of conservation and policy formulation.
As a result of environmental conditions and breed selection procedures used across decades, a number of indigenous pig breeds have been developed over time in the Taihu Lake region. Our results depict the haplotype blocks and genetic diversity of the 7 populations pig populations in the Taihu Lake region, which is relevant for the management and conservation of these important genetic resources in indigenous pig breeds.
Conclusions
In this study, we analyzed the haplotype structure and genetic diversity of the 7 local pig populations in the Taihu Lake region to better achieve the utilization and protection of their genetic resources. It is proved that the genetic diversity of the 7 Chinese indigenous pig populations in the Taihu Lake region is at a high level as a whole, but it is still necessary to further improve the conservation effect. Furthermore, we provided some derivations and perspectives on conservation strategies in a subdivided meta-population and discussed how to contribute to the sustainable livestock systems in highly variable and challenging environments. In brief, we conducted a comprehensive survey of the nucleotide variability of the 7 Chinese indigenous pig populations in the Taihu Lake region on a genome-wide scale and believe that the findings presented will lay a good foundation for the development of a national plan for the conservation and utilization of these pig populations.
Supplemental Material
Supplemental_Material – Supplemental material for Exploring the Structure of Haplotype Blocks and Genetic Diversity in Chinese Indigenous Pig Populations for Conservation Purpose
Supplemental material, Supplemental_Material for Exploring the Structure of Haplotype Blocks and Genetic Diversity in Chinese Indigenous Pig Populations for Conservation Purpose by Qing-bo Zhao, Hao Sun, Zhe Zhang, Zhong Xu, Babatunde Shittu Olasege, Pei-pei Ma, Xiang-zhe Zhang, Qi-shan Wang and Yu-chun Pan in Evolutionary Bioinformatics
Footnotes
Acknowledgements
The authors also greatly appreciate the 2 anonymous reviewers for their diligent work and associate editor, whose comments and suggestions gave a great contribution to the improvement of the manuscript.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the National Natural Science Foundation of China (grant nos 31772552, U1402266, 31672386, 31472069).
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
YP and QW designed and supervised the study, whereas QZ analyzed the data and wrote the manuscript. HS and ZX collected the samples and all authors read and edited the manuscript.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.