
Abstract
Genetic variations in the human genome represent the differences in DNA sequence within individuals. This highlights the important role of whole human genome sequencing which has become the keystone for precision medicine and disease prediction. Morocco is an important hub for studying human population migration and mixing history. This study presents the analysis of 3 Moroccan genomes; the variant analysis revealed 6 379 606 single nucleotide variants (SNVs) and 1 050 577 small InDels. Of those identified SNVs, 219 152 were novel, with 1233 occurring in coding regions, and 5580 non-synonymous single nucleotide variants (nsSNP) variants were predicted to affect protein functions. The InDels produced 1055 coding variants and 454 non-3n length variants, and their size ranged from -49 and 49 bp. We further analysed the gene pathways of 8 novel coding variants found in the 3 genomes and revealed 5 genes involved in various diseases and biological pathways. We found that the Moroccan genomes share 92.78% of African ancestry, and 92.86% of Non-Finnish European ancestry, according to the gnomAD database. Then, population structure inference, by admixture analysis and network-based approach, revealed that the studied genomes form a mixed population structure, highlighting the increased genetic diversity in Morocco.
Keywords
Introduction
Individual genome analysis is now a reality, owing to technological advances that have made it more accessible and inexpensive. The outcome of whole genome sequencing is crucial since it provides a reasonably accurate picture of genetic history and its impact on health. This technology has been used on projects allowing the analysis and discovery of human genome variation, such as the International HapMap Project, 1 the 1000 Genomes Project, and the International Genome Sample Resource. 2
Notwithstanding the constant updating of the reference genome and the abundance of human genome projects, some populations are still underrepresented; the latest version of the Genome Aggregation Database (gnomAD) contains 76 156 whole genomes with 27% belonging to the African/African American Population. 3 Thus, efforts still need to be increased to the characterisation of African populations especially the Middle East and North Africa regions which are underrepresented in the public databases. 4
Morocco has always been a crossroad of various cultures due to its geographical location in the northwest corner of Africa, it dominates the Atlantic and Mediterranean oceans and has land borders with Algeria, the Spanish enclaves of Ceuta and Melilla, and Mauritania. Due to these various regional influences, the genetic makeup of the Moroccan population is a complex mixture of ancestral Maghrebi lineages, along with northeast and West African, European, and West Asians to different degrees.5,6 However, to date, there is a lack of publications describing the genetic variability of Moroccan individuals. The main objectives of the present study are the analysis of 3 Moroccan genomes, the identification of novel variants, the allocation of mitochondrial DNA haplogroups to each genome, and the structure analysis of the studied genomes.
Materials and Methods
Data source
For this study, we considered the raw sequencing results of 3 Moroccan genomes previously sequenced. 7 Those samples were randomly chosen from healthy volunteers from different Moroccan regions. Data is available through the link http://www.ncbi.nlm.nih.gov/bioproject/660888.
Genetic variant discovery and annotation
Paired-end reads of each sample were mapped to the GRCh38 reference genome using the Burrows-Wheeler Aligner. 8 The mapping result, BAM files, were sorted by chromosomal coordinates and duplicate reads were marked using Samtools v1.10 9 and Picard MarkDuplicates 10 respectively, to deliver the final BAM files. Single nucleotide variants (SNVs) and insertions/deletions (InDels) were jointly called across the 3 samples using the Genome Analysis Toolkit 11 via the HaplotypeCaller. Variant call accuracy was estimated using the Variant Quality Score Recalibration approach. All steps and parameters follow the protocol recommended by GATK. The multi-allelic sites were split into bi-allelic sites and each variant was then annotated with the Variant Effect Predictor. 12 We added population frequencies using gnomAD 13 (v 3.1.2). ClinVar was used for the interpretations of the clinical significance of these variants to disease (v 1.7). 14 We also used SIFT 15 and Polyphen 16 to predict the possible impact of the amino acid substitution. A variant is damaging if the SIFT score is less than 0.05 and the PolyPhen score is greater than 0.908. For the pathway analysis of novel variants, we used the SNPnexus web server. 17
Population structure analysis and mitochondrial DNA haplogroup identification
We used ADMIXTURE 18 software to get an overview of the variation of Moroccan genomes across several populations. For this purpose, we downloaded a public Affymetrix Human Origins dataset described in an analysis of the ancestral population of ancient human genomes. 19 This dataset contains the genotype and single nucleotide polymorphism information of 1963 people from different regions of Africa, Europe, America, Oceania, Asia, and Eurasia. We merged these data with Moroccan samples using plink, 20 then filtered out SNPs that have a high level of Linkage Disequilibrium, by using an r2 threshold of 0.1. For the choice of the k value, which is the number of ancestral populations, we used ADMIXTURE’s cross-validation procedure for values of k from 4 to 17 and chose k = 14 for the final interpretation. We used pong 21 for admixture graphs visualisation. We performed mean pairwise Fst analysis by the R package ADMIXTOOLS. 22 We analysed population structure using NetSruct_Hierarchy, 23 a programme based on a network approach to construct population structure trees. Regarding the identification of mitochondrial DNA (mtDNA) haplogroups for the 3 Moroccan genomes, we used the haplogrep2 24 software.
Results
Genetic variation in Moroccans
Compared to the human reference genome (GRCh38.p14), variant calling results revealed, in the 3 Moroccan genomes, 7 430 183 variants in the autosomal chromosomes, and 6 379 606 SNV (Table 1). The transition-to-transversion Ti:Tv ratio was significantly higher at 2.18 for SNVs in the exome than for all the SNVs (1.98). When evaluated against the dbSNP database, 3.4% of the SNVs were absent, from which 1225 occurred in coding regions. As shown in Table 2, most of the SNVs were found in intergenic or intronic regions by the percentages 34% and 50% respectively. The remaining 16% includes 20 564 variants at transcription factor binding sites (TFBSs), 103 049 overlapping with the UTRs, and 21 092 missense variants.
Summary of the SNV and the exonic variants.
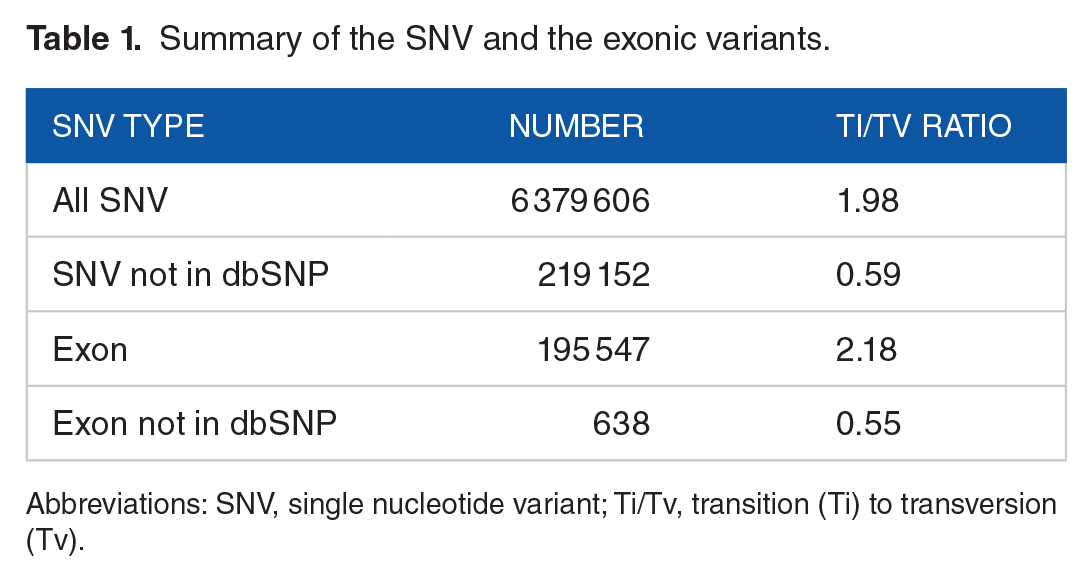
Abbreviations: SNV, single nucleotide variant; Ti/Tv, transition (Ti) to transversion (Tv).
Classification of the SNV variants.
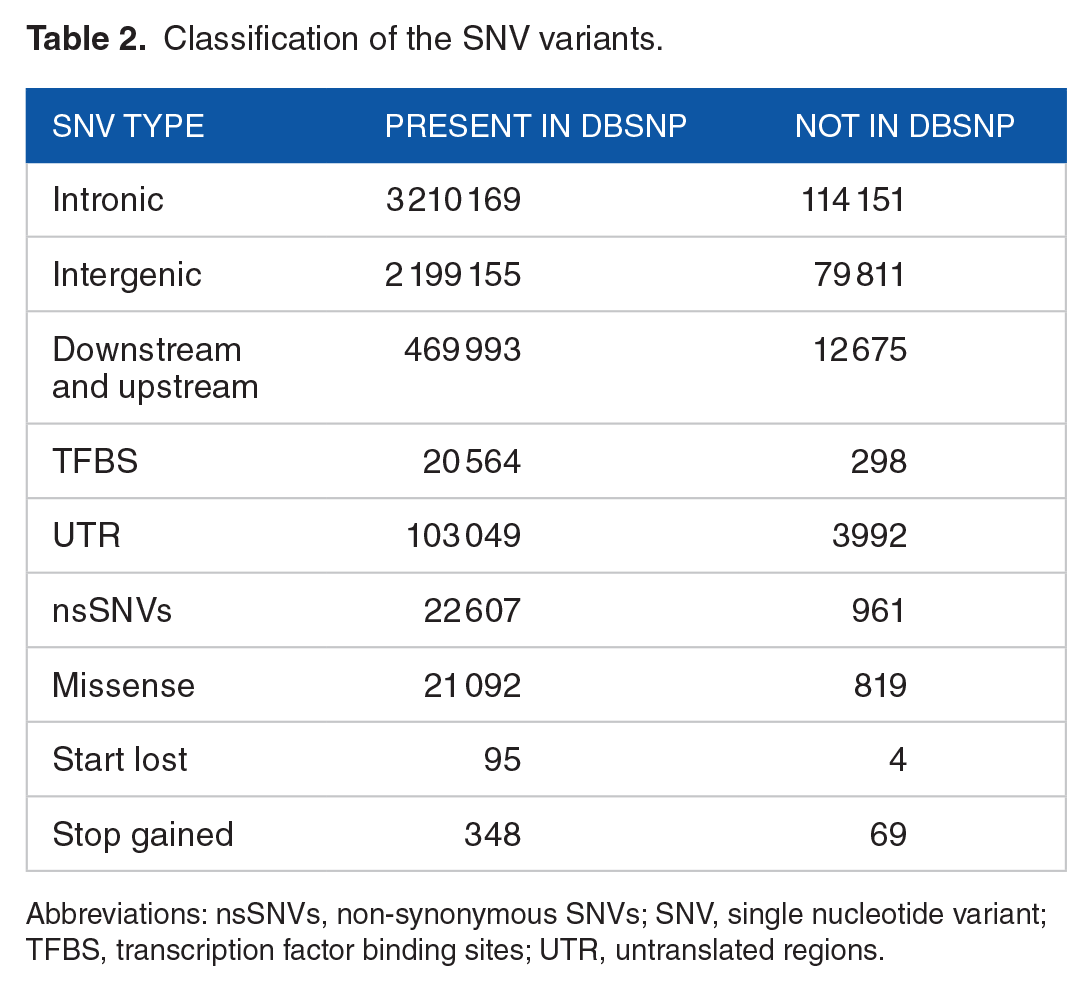
Abbreviations: nsSNVs, non-synonymous SNVs; SNV, single nucleotide variant; TFBS, transcription factor binding sites; UTR, untranslated regions.
Regarding the non-synonymous SNVs (nsSNVs), we found 5580 that are consistently predicted to be damaging by both SIFT and PolyPhen. According to ClinVar, those variants are found within 3912 genes, including 1085 harbouring at least one likely pathogenic variant. The ClinVar pathogenic variations are interpreted for Mendelian disorders and could have significant clinical impacts, especially for recessive disorders. 25
Among the 7 430 183 variants, 1 050 577 were identified as short InDels with 486 786 insertions and 563 791 deletions. Of these InDels, 6767 (0.64%) were not represented in the dbSNP. The InDels length, illustrated in Figure 1, varies between −49 and 49 bp where the average standard deviation values are 2.74 ± 3.74 bp for insertions and −3.205 ± 4.42 bp for deletions (Figure 1). The longest insertion and deletion identified are of 49 bp and found in the non-coding regions.

Size distribution of InDels.
We detected 339 610 (32%) of the InDels in the intergenic region and 428 268 (40.7%) in the intron region. Besides this, 1.76% of the InDels are in the 3′ and 5′ untranslated regions, with the majority occurring in the 3′ UTR and about 8.4% of the InDels are equally divided between upstream and downstream regions.
In the coding region, we found 481 codon insertions and 574 deletions. We also noticed a high expression of InDels with a size that is divisible by 3 bp (3n), and only 454 (43%) of the coding indels are a non-3n length causing a frameshifting (Figure S1). Indeed, purifying selection is more likely to exclude mutants with frameshifting indels from the population than those without. 26
Moroccan novel and shared variants
We used gnomAD, which gathers and coordinates exome and genome sequencing data from 76 156 whole genomes to identify unique variants in the Moroccan population. Using this, we found that 96.3% of the variants found in the 3 genomes, among 7 153 985, are shared between Morocco and the other populations, while 3.9% are unique variants (N = 276 198). Moreover, about 216 715 Moroccan variants were not present in the dbSNP with 1233 coding variants. The functional impact of those variants on the protein sequences was estimated using PolyPhen and SIFT. 47% of the nsSNVs using SIFT were predicted to affect protein function with deleterious effects, and 33% were classified as probably damaging using PolyPhen.
We examined the variants, of all types, that are shared by the 3 analysed genomes with the existing variants in the gnomAD database, to provide a general picture of the 3 Moroccan genomes’ profiles compared to different populations. We found that the variants pool of the 3 Moroccan genomes is similar to African ancestry and Non-Finnish European ancestry, by 92.78% and 92.86% respectively. However, the furthest ancestries from Morocco, in the percentage of common variants, are Amish and East Asian ancestries with 80.86% and 83.04% of common variants respectively. This seems logical considering the geographical situation of Morocco but is not sufficient to conclude a genetic mixture of these studied genomes.
Variants distribution per genome
The analysis of the 3 Moroccan genomes separately showed an average of 4 627 840 variants per genome with 3 892 653 SNVs and 735 187 InDels (Table 3). Each Moroccan genome contains approximately 1 086 782 distinct variants, not shared with the other genomes (Figure 2). From the set of SNVs and InDels annotated to be splice-site, stop-gain mutations, or to result in frameshifts, we identified, in 663 genes, 779 variants that were likely to result in loss-of-function (LoF). An average of 440 LoF variants were seen in each genome, consistent with several whole genome sequencing that reported between 200 and up to 800 LoF variants per healthy individual.27,28
Distribution of autosomal variants per genome.
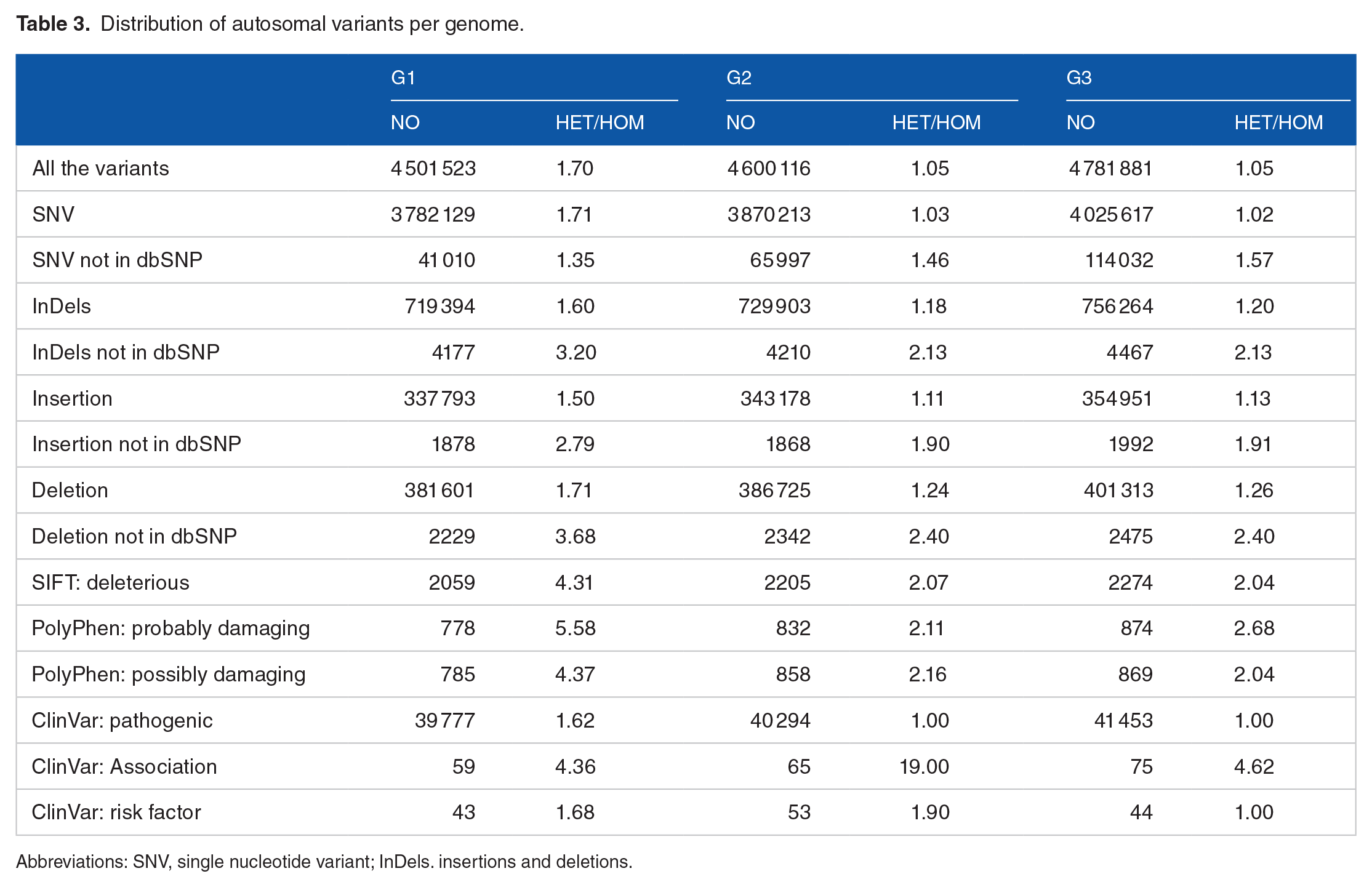
Abbreviations: SNV, single nucleotide variant; InDels. insertions and deletions.
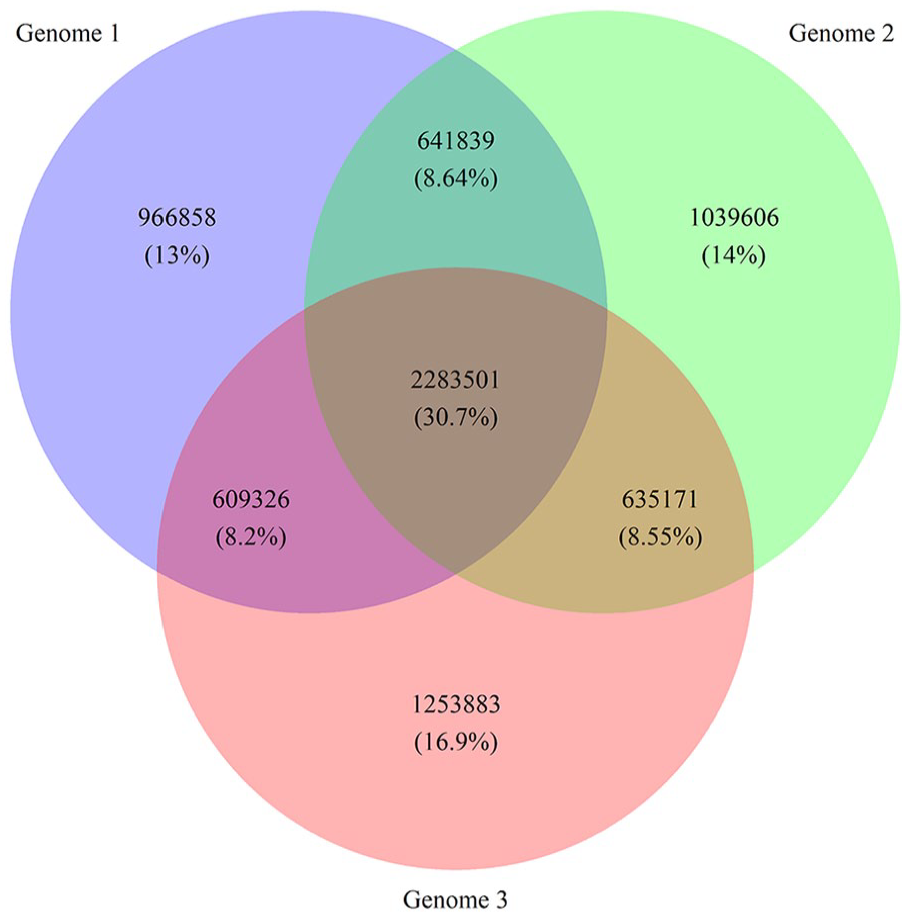
Comparison of the 3 Moroccan genomes. The Venn diagram shows the variations present and shared between the genomes.
In addition, the heterozygosity ratio, which represents the heterozygote to the non-reference homozygous ratio (Het/Hom), was higher in the 3 analysed genomes in this study, as shown in Table 3, with the values of 1.70, 1.80 and 1.97 for G1 (Genome 1), G2 (Genome 2) and G3 (Genome 3) respectively, reflecting a higher level of genetic diversity.
We also remark that the same ratio was very high for novel variants because most of the novel variants are rare and often presented as heterozygotes. The highest Het/Hom ratio was found among pathogenic or deleterious variants, consistent with negative selection against these variants to reach high frequencies.
A total of 2 283 501 variants are shared between the 3 genomes with only 0.78% (17 890) not present in gnomAD. Of those, 26 variants occurred in coding regions, with 8 not present in dbSNP. We further investigate the gene pathways for those novel coding variants using the SNPnexus web server. 17 Only the results with P-value < .05 are considered and are represented in Table S1. Two variants were involved in specific pathways and affected the DBSPP, KRTAP10-10/KRTAP10-4 genes.
Mitochondrial DNA analysis
Mutational analysis in mtDNA revealed some pathogenic mutations observed in genome 2, such as G15927A, which is claimed to be associated with asthma. 29 According to ClinVar results, the mutation A11467G, found in the same sample, genome 2, is linked with mitochondrial diseases. As for the mutations found in the other genomes, 1 and 3, they are benign, presenting no effect on health.
Regarding the identification of the mitochondrial haplogroup of each genome, we used haplogrep2 software; The 3 genomes belong to the same haplogroup, H2a2a1.
Admixture analysis, pairwise Fst estimation and network-based approach
The cross-validation error results, illustrated in Figure S2, suggest that a number of source populations higher than 11 can model the data adequately and smaller values of k lead to under-fitting. To examine the outcome’s sensitivity to k selection, we ran ADMIXTURE with different k values, from 4 to 17, and we have plotted only the results of k superior or equal to 7 (Figure 3) since they have smaller values of cross-validation errors compared to k between 4 and 7 (Figure S2). We chose k = 14 as the final interpretation since it has the lowest cross-validation error and it also demonstrates a progressive adjustment of ancestral components’ proportions as predicted by geographic locations.

Admixture plot comprising clustering solutions from k = 7 to k = 17 of the 3 Moroccan genomes combined with 1963 individuals from several populations; Africa, Europe, Eurasia, America, Oceania and different regions from Asia.
Considering k = 14, the admixture analysis result (Figure 4) revealed ancestral components slightly different for the 3 samples. We analysed the repartition of each genome by comparing it with the repartition of the other regions.

Admixture results at k = 14 zoomed in on the composition of the 3 Moroccan genomes.
The predominant inferred ancestral component of genome 1 is represented by the pink cluster, with a value of 57.1%. This cluster is occupied mainly by the Hadza population with a value of 100%; we also notice that the ancestral compositions of Somali and Masai populations are mainly composed of the pink cluster with values of 57% and 55.6%, respectively.
Regarding genome 2, it has a more heterogeneous composition; its ancestral components are distributed over 3 significant clusters; blue (26.4%), brown (21.2%), and dark green (17.1%). The blue cluster occupied 95.3% of the Chukchi population composition and 89.4% of the Eskimo population. However, dark green is the main component of several populations such as Moroccan Jewish, Adygei, Ossetian, Druze, French and Arabic regions of West Asia.
Genome 3 is assigned the majority to the beige cluster (42%), this cluster is mainly present in the composition of 4 African populations; Yoruba, Mendenka Gambian, and Bantu, with the percentages of 99.9%, 97.9%, 96.1% and 74%, respectively, and it is also the main component of an European population, Mende, with a percentage of 96.8%, and a region of East Asia; named Esan, with a value of 99.9%.
To conduct Fst pairwise analysis, we used a subset of the dataset used for admixture analysis, 19 only African and European populations, merged with the 3 Moroccan genomes, were included in the Fst calculation. The final dataset consisted of 618 individuals distributed among 50 populations. We visualised the obtained results using a heatmap (Figure 5). The genetic analysis of the 3 Moroccan genomes revealed the lowest genetic distance when compared to the Datoga population, a community located in northern Tanzania. This was indicated by a mean Fst value of 0.166. Similarly, the Masai and Somali populations also exhibited a moderate genetic variation when compared to the Moroccan genomes, with Fst values of 0.180 and 0.181, respectively.

Heatmap plot of Fst values between Moroccan analysed genomes and diverse populations from Africa and Europe. Shown values are the product of Fst estimations by 1000. Negative values indicate that more nucleotide substitutions occur within than between populations.
The same dataset used in Fst analysis, containing only African and European populations, was used to infer a population structure tree using NetStruct. The topology of the inferred PST is composed of 3 main clusters (blue, orange and green) (Figure 6). The blue cluster is mainly composed of African populations and contains 6 subbranches. Most of populations represented in the green cluster are from Europe. The 3 analysed genomes form a distinct population represented by the orange cluster.

Population structure of individuals from Africa and Europe. Each circle represents a cluster of individuals. The composition of a cluster is shown in the dashed square.
Discussion
The African population harbours the most genetic variation and diversity and thus has the highest Het/Hom ratio of SNP compared to the other populations.30 -32 In this study, the Het/Hom ratio estimated is 1.71, this value aligns with the reference ratio of 2. 33 This ratio is strongly associated with human ancestry and defines genetic variation. 34
Previous results reported on these 3 Moroccan genomes demonstrated that they share both African and European ancestries based on principal components analysis. 7 To enhance the reliability of the obtained conclusion, we unveiled detailed ancestral contributions and confirmed the connections between the 2 types of genetic ancestry. In addition, we identified a total of 1233 coding variants found to be novel compared to both dbSNP and gnomAD databases. The functional prediction of those variants showed that 278 (28%) caused damage to the protein structure and, therefore, could affect actual phenotypes. The identification of those variants could enrich the databases, such us gnomAD, with Moroccan genomes variants, and could also be useful in studies of rare variants and diseases. Furthermore, 30% of the variants are shared between the 3 genomes, with 8 novel coding variants with corresponding genes involved in various diseases. In addition, we identified a total of 779 LoF variants in which functional analysis could aid in disease-related gene prioritisation. 35
To draw any further conclusion regarding the impact or novelty of those variants, we must consider some important points; those individuals, at the time of sampling, did not have any genetic disease, meaning that most of the pathogenic variants could be benign. The second point is that to access the number of novel variants, a threshold must be applied regarding the depth of each variation to reduce false-positive ones.
The mtDNA analysis revealed the haplogroup of each genome, as well as the pathogenic mutations. Haplogroup H has been assigned to all 3 genomes; it is the most frequent clade in North Africa. 36 In a study conducted in the North of Morocco 37 about mitochondrial genetic variability, the haplogroup H was assigned to 58 Moroccans among 200. Numerous multifactorial disorders have been linked to genetic variations in mitochondrial DNA. 38 Haplogroup H was identified as a risk factor for Ischemic Cardiomyopathy 39 and was associated with keratoconus in Saudi Arabian patients. 40
Finally, we analysed the ancestry composition within the studied genomes, considering data from diverse populations. Although the profile of the 3 genomes is different, their components seem to be originated mainly from African populations, especially for genome 1, which has a similar profile to East African regions such as Hadza, Masai and Somalia, and also genome 3, which has a profile from West Africa, because it is similar to that of Yoruba, Mandenka and Gambian regions. However, few studies have analysed the genetic inference of the African population.41,42 Until today, no study has explored the admixture of the Moroccan population because of the lack of data. The results obtained in this study are a primary study of the genetic variants within and among the Moroccan population. A larger number of Moroccan genomes is needed to deduce conclusions about the population history of Morocco. Hence the whole human genome sequencing of diverse populations across Africa is needed, and it should be combined with historical DNA information; this approach allows researchers to recognise and comprehend signs of ancient admixture; For example, comparing modern genomes with historical DNA could identify the presence of ancestral components unique to a specific periods or geographic regions. 43 Additionally, persistent ancient genes could be associated with diseases, as reported by a study conducted by researchers from the National Institute of Mental Health, 44 persistent genes, inherited from ancestors, are linked to the development of a neanderthal-like brain, this connection may have implications for psychiatric health conditions, like schizophrenia.
Therefore, we envisage that increasing the number of Moroccan genome sequences and filtering candidate variants by population allele frequencies should help detect genetic diversity and discover potential disease-associated. 45
Conclusion
Africa is still neglected in genetic studies, despite its importance in human evolution, large population, and genetic diversity. We provide the analysis of the whole genomes of 3 Moroccan people. We also present admixture inference of the genomes under study when compared to various populations, indicating that their genetic history is a mix of Africa, Europe, and Arabic regions of West Asia. Future research on Moroccan genomes at a greater scale will be necessary to map their entire genetic makeup at much finer frequencies. More samples from Morocco and northwest Africa, in general, are needed to, on one hand, make the reference of the human genome more representative, and also to study these populations’ stratification, to identify the variants linked to population-specific diseases.
Supplemental Material
sj-docx-1-evb-10.1177_11769343241229278 – Supplemental material for A Comprehensive Analysis of 3 Moroccan Genomes Revealed Contributions From Both African and European Ancestries
Supplemental material, sj-docx-1-evb-10.1177_11769343241229278 for A Comprehensive Analysis of 3 Moroccan Genomes Revealed Contributions From Both African and European Ancestries by Nasma Boumajdi, Houda Bendani, Souad Kartti, Tarek Alouane, Lahcen Belyamani and Azeddine Ibrahimi in Evolutionary Bioinformatics
Footnotes
Acknowledgements
This work was carried out under National Funding from the Moroccan Ministry of Higher Education and Scientific Research to AI and scholarship of excellence from the National Center for Scientific and Technical Research in Morocco. This work was also supported, by a grant to AI from the Institute of Cancer Research of the foundation Lalla Salma, and also by a grant from Biocodex Microbiota Foundation.
Authors Contributions
NB and HB contributed to the conceptualisation, data collection and analysis, and writing the initial draft and revisions. SK and TA contributed to the conceptualisation and draft revision. LB revised and validated the last draft. AI conceived and supervised the study, contributed to writing revisions and validation. All authors read and approved the final version of the manuscript.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.