
Abstract
Our study searched all available sequences of Paphiopedilum from NCBI (National Center for Biotechnology Information) and tested for their species resolution capability in single as well as in combination forms. A total of 28 loci were applied for analyses in the study. From the nuclear genome, the highest resolution was of LFY, followed by ACO, DEF4, and RAD51. These 4 loci were found to be even better than the popular region ITS for Paphiopedilum identification. Among the chloroplast regions, the intergenic spacer atpB-rbcL gave the highest species resolution (76.7%), followed by matK, trnL, rpoC2, and ycf1. The divergence of CHS, XDH, 18S, Nad1, ccsA, rbcL, and ycf2 was very low and should not be used as identifying markers for Paphiopedilum. In addition, 2-locus combinations could improve significantly the resolving capability for the genus, in which 14/36 data sets could be resolved completely (100%) with interspecies relationships. The indel information was also effective supporting data for molecular discrimination of species.
Introduction
Paphiopedilum is one of the largest genus of the Slipper orchid group Cypripedioideae.1,2 Their flowers have the special slipper shapes and attractive colors which are the important characteristic to discriminate among individuals. The overexploitation and illegal trade, especially at the vegetative stage, make this plant come into endangered states.2,3 Therefore, a demand for exact identification of these species at any stage of their life without the necessity of the fertile part is critical for conservation.
The single region 5ʹ end of cytochrome c oxidase 1 (CO1) from the mitochondrial genome has been proved to effectively identify animal individuals.3,4 Universal sequences for identification of plants are also being searched from different genomes, mainly from nucleus and chloroplast. However, until now, the effective molecular identification of plants remains challenging because some loci have just been optimal for some particular groups of taxa. Thus, to respond to the demand of plant discrimination, separate sequences for identifying specific taxonomic groups have been done. There were a number of studies screening for potential molecular sequences to authenticate Slipper orchids. The 5 loci rpoB, rpoC1, rbcL, and matK from the chloroplast genome and ITS from the nuclear genome have been used to test for species resolution of Indian Paphiopedilum species by Parveen et al 5 in 2012. The 2 plastid loci, rpoC2 and the intergenic spacer region between atpF and atpH genes were used to generate molecular identification system for some species of Cypripedium genus. 6 The phylogenetic tree of 25 species of subfamily Cypripedioideae in the research of Guo et al 7 has shown well-resolved monophyletic branches for all studied species based on sequence analyses of 6 maternally inherited chloroplast genes (matK, rbcL, rpoC1, rpoC2, ycf1, and ycf2) and 2 low-copy nuclear genes (LFY and ACO). ITS was also used for identification and evaluation of variety relationship of 16 species and 2 varieties of genus Paphiopedilum in Vietnam. 8 Many other molecular regions have already been screened for discriminating a wide range of plants. However, the resolution results were not similar in different researches due to the differences in selected taxa, the differences in the number of studied species as well as the sequence length used, and the differences in methods.
In this study, we aim to evaluate all available submitted sequences relating to Paphiopedilum species from NCBI (National Center for Biotechnology Information) for species resolution of this genus, using a combination of measurements of tree-based method, indel fragments, variable sites, and even sequence length to search for potential loci which can be used in identification of individuals of the genus. This study may contribute to the use of proper molecular sequences as effective barcoding markers for identification of specific Paphiopedilum orchids, serving for conservation and diversity research of the plant.
Materials and Methods
Collected sequences of Paphiopedilum from GenBank
All sequences belonged to Paphiopedilum species from GenBank were selected and downloaded into separate loci. From that we obtained 2743 accession numbers of 37 loci (Additional file 1). The sequence names were then shortened with only scientific species name and their accession number for easy analyses.
The number of sequences and the number of species between loci were different from each other. Among of them, 7 loci which have contained only 1 to 2 sequences, ie, GLO, DEF, LOAE, IFR, DFR, psaB, and psbA were not used in further tests. The 2 loci, psaA-ycf3 and ndhJ, which have had descriptions with the UNVERIFIED name were also removed. The trnK sequences were determined in the same locus with other matK sequences (as matK gene locates inside the region of trnK gene) and then we integrated them into one data set of matK, with a total of 266 accessions. Due to the high variation in length of matK sequences, the matK set was divided into 2 subsets (more discussion in the section “Results and Discussion”). The 5S region showed the extreme diversity of conspecific distances after primary aligning that could lead to the paraphyletic relationships and give low species resolution, so we excluded it from our testing. As to some recommendations that the ITS2 was a good barcode in comparison with the whole-length locus ITS,9,10 in this study, we isolated the data set of ITS2 from ITS to separately examine this short region. Finally, 28 loci were applied for further study: ITS, ITS2, LFY, ACO, DEF4, RAD51, CHS, XDH, and 18S in the nuclear genome; Nad1 in the mitochondrial genome; and accD, ccsA, matK(1), matK(2), rbcL, rpoB, rpoC1, rpoC2, trnL, ycf1, ycf2, the intergenic spacers atpB-rbcL, atpF-atpH, atpH-atpF, trnH-psbA, trnL-trnF, trnQ-rps16, and trnS-trnFM in the chloroplast genome. In a total number of 107 species, the number and the composition of species were different among different loci. The species coverages of each locus are shown in Table 1. The summary of selected loci of Paphiopedilum from NCBI is shown in Additional file 1.
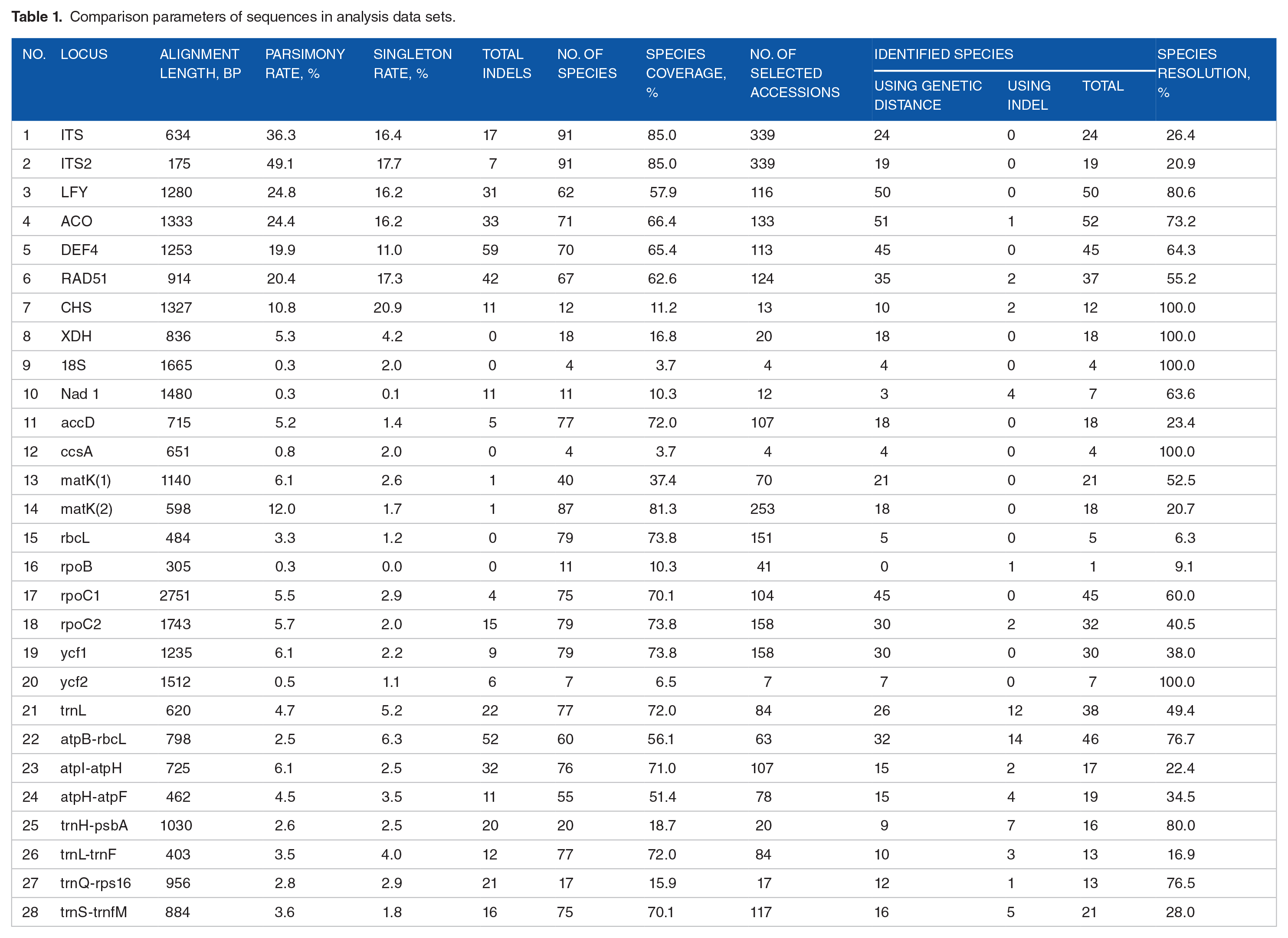
Comparison parameters of sequences in analysis data sets.
Multiple sequence alignment
Sequences at each locus were aligned using SeaView software version 4.4.0. 11 Alignments were then manually optimized, especially with noncoding regions in chloroplast genome and genes in nuclear genome which are highly divergent and contain many indel fragments. We manually noted all these information as they are also helpful in species resolution. The parsimony and singleton calculations for each data set were performed by Mega 7 program. 12 In the process of alignment, sequences that were too short and too divergent were removed from data sets. The output of alignment analyses is shown in Table 1.
Evaluation of species resolution
Phylogenetic tree for each locus were generated by Neighbor-Joining method using Mega 7 software with Kimura-2-parameter (K2P) model. 12 Evaluation of species resolution was performed using tree-based method and a combination of different measurements. First, species with just only one accession were considered to be distinguishable if the sequence was unique from others. In the phylogenetic tree, this accession would be shown as monophyletic branch. 13 Second, when multiple accessions were collected per species, these all accessions would be grouped into one monophyletic branch in the phylogenetic tree. Third, in converse, if conspecific individuals were not grouped together but separated in paraphyletic branches, then the species was considered as identification failure. A further description of insertions, deletions, and repeats should be included as if there was any difference between them that could help to authenticate them from the others.14,15 Finally, in case of undiscrimination by K2P distance, different species were grouped in the same branch in the phylogenetic tree. This means that sequences of these accessions were identical. These heterospecies sequences in the same branch would be also more observed with indel information.
In barcoding studies as well as phylogenetic researches, the identification of orthologs and paralogs give much influence on the exact species resolution. Although orthology is one of the most important criteria to evaluate the relationship between individuals, paralogous sequences which are the results of duplicate events and divergence are not correlated with speciation. The presence of paralogs in the same sample may lead to an overestimation of the number of unique species under identification. 16 In practice, this problem can be eliminated by identifying and avoiding paralog contamination before sequence analysis. First, if paralogs really exist in different size, the polymerase chain reaction products using universal primers will be expressed as ghost bands on gel in the electrophoresis step. Second, even the paralogs have the same length as the real gene, and we cannot recognize them under electrophoresis but the sequencing would be shown with ambiguities, double peaks, or noise. These are 2 simple ways to remove paralogs from our analysis. In this in silico study, all the accessions were selected from the available source of GenBank. In case of interspecies comparative, it was considered that the sequences were orthologs. In case of intraspecies relationship analysis, the accessions might be orthologs or paralogs, especially between different clones of the same samples. Instead of deleting the clone sequences, we decided to keep all of them and use as one of the factors affecting the species resolution. The locus with high variation between either intraspecies orthologs or homologous paralogs would result in low species resolution and could not be used as candidate molecular identification markers.
Results and Discussions
Multiple sequence alignment and divergence of the loci
The nuclear loci ITS, ITS2, LFY, ACO, DEF4, RAD51, CHS (except XDH and 18S regions) had the parsimony rates (10.8%-49.1%) and singleton rates (9.7%-20.9%) significantly higher than all loci in plastid genome (0.3%-12% in parsimony rate, 0%-6.5% in singleton rate; Table 1). The total indels in chloroplast protein-coding regions, in general, were very low in comparison with most other chloroplast regions and the nuclear genes (except XDH and 18S genes; Figure 1C), which explained the different alignment capabilities of these regions as mentioned below.

Species resolution of single loci of Paphiopedilum sequences and other analyzed information about parsimony rate, singleton rate, indel fragment, number of species, number of sequence, and alignment length per locus.
To have a good discrimination effect, first, the selected sequences should have a proper length that is not too long for simple amplification and not too short for containing enough divergence information. Second, these sequences should be easy to be amplified, sequenced, and aligned. Third, the divergence of the sequence should be high enough for distinguishment at the species level but not too variable at underspecies level. 3 Finally, the high species resolution is a critical criterion. In our study, as the sequences were selected from GenBank, we did not analyze the amplification and sequencing rates. However, the sequence length, the alignment capability, the divergence range, and the resolution rate were all taken into account. The rpoC2 and ycf1 regions had rather long sequences (2751 and 1743 bp [base pairs], respectively) which were not suitable to be molecular markers. Therefore, we had to cut the 2 ends of these loci to keep the most variable fragments with suitable length (1188 bp of rpoC2 and 1235 bp of ycf1) for our study.
The coding-gene regions in the chloroplast genome were easy to be aligned. The noncoding intergenic spacers in chloroplast and the nuclear loci took more time for alignment due to their great indels and polynucleotide repeats inside their sequences. However, they were all successfully aligned. Exceptionally, the LFY region contains quite long sequences (from 904 to 3404 bp) in which we could just align the half top fragment (about 1280 bp) for analysis. The other half end fragment which was too complicated to be aligned was removed.
The capability of species identification with single regions
Some of the tested regions achieved 100% species resolution, ie, CHS, XDH, 18S in the nuclear genome and ccsA, ycf2 in the chloroplast genome. However, these loci included extremely low number of species (12, 18, 4, 4, and 7, respectively; Table 1). In addition, their divergent levels were too low (Figure 1C). For these reasons, the results obtained from these loci were not confident enough to evaluate the species discrimination. The similar situations were also obtained from Nad1 region (with 11 species), trnH-psbA region (with 20 species), and trnQ-rps16 region (with 17 species; Table 1). Then, these 8 regions were removed from our further analyses.
Although the number species of CHS, XDH, 18S, Nad1, ccsA, rbcL, ycf2, trnH-psbA, and trnQ-rps16 loci were very low and could not used for analysis, we still included these regions into this step of the study because we wanted to examine their variability for future studies. The results showed that the divergence of CHS, XDH, and 18S of the nuclear genome; the Nad1 of the mitochondrial genome; and the ccsA, rbcL, and ycf2 of the chloroplast genome were very low and should not be used as identifying markers. However, trnH-psbA and trnQ-rps16 had a rather high total indel fragments and should be examined more with a larger number of studied sequences in future.
In the nuclear genome
The ITS2 locus has been highly noticed as an alternative barcode instead of the full-length ITS region due to ITS high divergence and short length for easy to amplify, to sequence, and to align.17,18 In this study, indeed ITS2 with a really short sequence (175-282 bp) could achieve just a little lower resolution than ITS (634 bp; Table 1). However, because the identification capability of ITS was still higher than ITS2, and the length of ITS (about 634 bp) was still suitable for amplification and sequencing, and moreover we did not meet any problem in alignment of this region, we still favored the whole ITS rather than the ITS2 in obtaining better resolution.
Among the analyzed regions in the nucleus, ITS and ITS2 loci gave the lowest resolution rate (26.4% and 20.9%). The highest ability was of LFY region which could resolve 50/62 species (80.6%; Figure 1D), followed by ACO, DEF4, and RAD51 (73.2%, 64.3%, and 55.2%, respectively). However, on contrast, the informative parsimony rates of both ITS and ITS2 sequences were higher than the remaining nuclear genes in the study. The reason might be that the insertions and deletions of LFY, ACO, DEF4, and RAD51 regions were all much higher than of ITS regions (Figure 1C). Besides, the number of sequences per species of ITS regions was very high (Figure 1B) which related to the variable sequences within species. The difference in species number and sequence number between the loci limited the comparison of all the loci simultaneously. For these reasons, to more exactly compare the resolution, we performed further pair tests between ITS locus with each of LFY, ACO, DEF4, and RAD51 regions in the condition that 2 tested loci contained the same species and the same sequence number (Additional file 2). The results now revealed that all 4 regions LFY, ACO, DEF4, and RAD51 still showed better resolution than ITS (Figure 2). Although ITS has been widely used in many barcoding as well as phylogenetic studies,18,19 in this research, we found that other nuclear genes were even better than ITS.

Comparison of resolution between ITS region and each of LFY, ACO, DEF4, RAD51 regions. The comparisons with the same species number showed that other nuclear genes were better than ITS.
In the chloroplast genome
To the matK region, the lengths between sequences were so different from 589 to 1275 bp. For this reason, after primary aligning, we decided to separate this data set into 2 smaller data sets: the matK(1) including 140 sequences with 1140 bp in length and the matK(2) including all selected 253 sequences from GenBank with 589 bp in length (Table 1). The species resolution of matK(1) was 52.5% which was much higher than matK(2) (20.9%; Figure 1D). The reasons might be either the difference of variation information caused by different length or the difference of species numbers between 2 subsets. Therefore, we performed a further test to compare the resolution of 2 matK sets in the condition of the same number of sequences and the same number of species as well. The results showed that the longer one which was matK(1) gave the higher success rate in species identification (data not shown). MatK sequences were easy to be aligned and the length of 1140 bp was suitable for amplification as well as for enhancing the species distinguishment. We recommend using the longer matK locus in molecular identification of Paphiopedilum species.
Among the chloroplast regions, the intergenic spacer atpB-rbcL gave the highest species resolution (76.7%), followed by matK(1), trnL, rpoC2, and ycf1 (52.5%, 49.4%, 45.9%, and 38%, respectively). The coding regions rbcL, rpoB, accD, and rpoC1 were highly conservative along their sequences and resulted in low resolution percentages (6.3%, 9.1%, 20.8%, and 23.5%, respectively). Except atpB-rbcL, other noncoding regions had low discrimination rates (atpH-atpF 36.4%, trnS-trnFM 28%, atpI-atpH 22.4%, and trnL-F 16.9%; Table 1). We made a series of further tests of the same species number between matK and other coding loci to more compare their resolutions (Additional file 2). To keep the most number of the same species for testing, matK was analyzed with the length 716 bp. MatK has been popularly used in identifying studies of Orchidaceae,5,20,21 in our tests, although the length of matK was just 716 bp, this region was really a potential chloroplast marker which had the better resolution than both trnL and ycf1 regions, and just a little lower than rpoC2 due to the decrease in ITS length (Figure 3). Ycf1 has been recently considered as “more variable than matK” 22 and highly evaluated as the most variable and parsimony-informative among the 5 chloroplast genes (matK, rbcL, rpoC1, rpoC2, and ycf2) on plants.7,23 However, in our study on Paphiopedilum, species distinguishment of this region was lower than matK and rpoC2. Besides matK, the locus rpoC2 could be a potential marker for identification of Paphiopedilum.

Comparison of resolution with the same species between pairs of regions matK, rpoC2, trnL, and ycf1.
In the study of Parveen et al, 5 matK received 100% and ITS got 50% species resolution among the 3 other loci rpoB, rpoC1, and rbcL. However, because the study just resolved only 8 species of Paphiopedilum, the result seemed to need more analysis. When Guo et al 18 focused on 9 regions including 8 from the chloroplast and one is the ITS, in this study, we looked inside a larger range of molecular markers of about 28 loci for identification capability Paphiopedilum species. The results supported potential region matK and developed some new potential identification markers as LFY, ACO which were just used for the aim of phylogenetic relationship analysis in Guo et al.7,24
The use of indel information in species identification
In the chloroplast genome, the noncoding regions seemed to have more complexity than the coding regions based on high number of insertions and deletions. These characteristics contributed to the more difficulty in sequence alignment. However, because we could pass all the alignments in previous step, indel fragments on contrast became useful information for discrimination at species level. Specifically, in our study, atpB-rbcL locus could identify a total of 46 distinguished species, in which 13 over 46 species have been obviously separated using indel information (Table 1). Besides, trnH-psbA marker could separate 16/20 species and one-third of these 16 species were discriminated using insertions and deletions. In addition, trnL locus could identify 12 out of a total of 26 distinguished species using indel fragments. Moreover, insertions and deletions were also used in such other molecular regions (ACO, RAD51, CHS, Nad1, rpoB, atpI-atpH, atpH-atpF, trnH-psbA, trnL-F, trnQ-rps16, trnS-trnFM) of our study (Table 1). Although the indel information was not used by bioinformatics tools and by some researchers, it has been proved as effective supporting data for species authentication in a few previous studies.6,7,14 We totally agree with the use of indel information as the fifth character state in molecular identification of species, besides the 4 traditional characters—A, T, G, and C.
Capability of species identification with multiple region combinations
As no single locus could discriminate all species of Paphiopedilum in the study, the 9 recommended potential loci derived from single-locus tests, ie, LFY, ACO, DEF4, RAD51 from the nuclear genome and matK, rpoC2, trnL, ycf1, and atpB-rbcL from the chloroplast genome were chosen for further testing with the combination of 2 loci as a unique sequence for identification (Table 2). For species with more than one congeneric sequence, we chose only one representative sequence to calculate the interdistance between species.
Comparison of species resolution of single-locus sequences and 2-locus combinations.
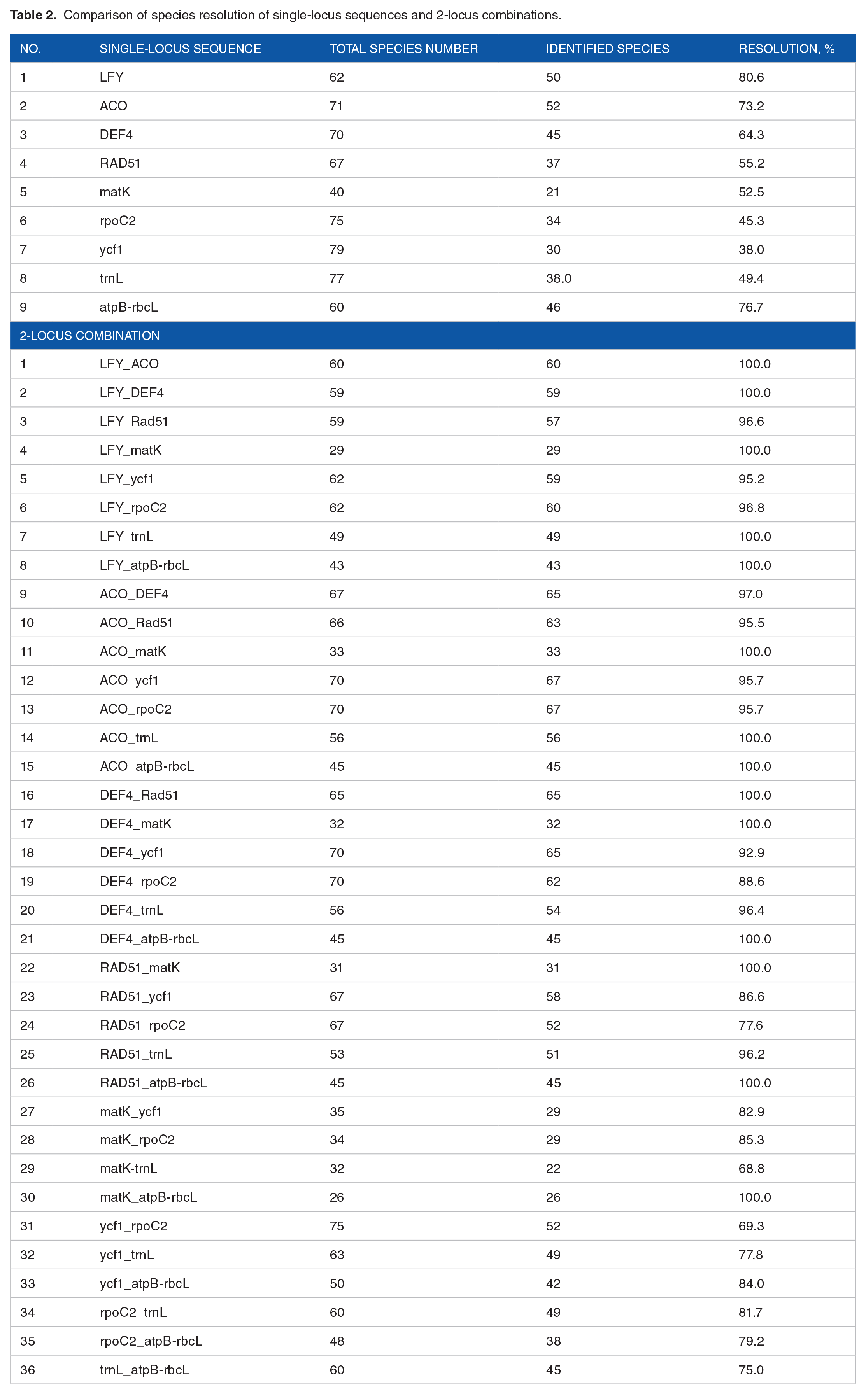
The resolutions of combination sequences were all higher than that of the single loci. In 2-locus combination, 14/36 data sets could be resolved completely (100%) with interspecies relationships (Figure 4). LFY, matK, and atpB-rbcL gave the best results in which 5/8 combinations obtained 100% resolution, followed by ACO, DEF4, RAD51, and trnL (4/8, 4/8, 3/8, and 2/8, respectively). The results revealed high potential for discrimination ability of these combination barcodes.

Species resolution of single loci and 2-locus combinations. Percent species resolutions showed more potential in a combination of sequences for identification study.
Conclusions
Our study revealed that the 4 loci, LFY, ACO, DEF4, and RAD51, in the nuclear genome were better than ITS in identification of Paphiopedilum species, and the intergenic spacer atpB-rbcL had the highest resolution from the chloroplast genome. Along with matK, atpB-rbcL, and rpoC2 in the chloroplast genome had high variability and should be considered as high-potential loci of species resolution for Paphiopedilum genus. In addition, the combination of potential loci could improve significantly the resolving capability. Besides, the indel information could be used as effective supporting data for molecular discrimination of species.
Because of the limitation of available sequence information for Paphiopedilum and the differences of sequence number among different loci, our evaluation still limited when comparing directly among loci. Therefore, we suggest a further study on both sequencing and in silico analysis for more potential loci from other locations of this plant genome. Furthermore, continuous efforts based on these potential loci should be done on a higher number of samples to enhance identification ability for many useful applications in conservation and biodiversity foundation for this valuable orchid Paphiopedilum.
Supplementary Material
Supplementary Material, Supplementary_Material – In Silico Study on Molecular Sequences for Identification of Paphiopedilum Species
Supplementary Material, Supplementary_Material for In Silico Study on Molecular Sequences for Identification of Paphiopedilum Species by Huyen-Trang Vu, Phuong Huynh, Hoang-Dung Tran and Ly Le in Evolutionary Bioinformatics
Footnotes
Acknowledgements
The computer resources were provided by Computational Biology Center of International University, Vietnam National University.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project was funded by the Asian Office of Aerospace Research and Development (AOARD) under grant FA2386-17-1-4032.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
H-TV analyzed and interpreted the results and was a major contributor in writing the manuscript. PH performed the bioinformatics calculation. H-DT gave advice on the manuscript. LL gave advice and gave final approval of the version to be published. All authors read and approved the final manuscript.
Availability of Data and Materials
All the sequences used in the study were downloaded from GenBank. Details of accession numbers are given in Additional file 3.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.