
Abstract
Confidence in pairwise alignments of biological sequences, obtained by various methods such as Blast or Smith-Waterman, is critical for automatic analyses of genomic data. In the asymptotic limit of long sequences, the Karlin-Altschul model computes a P-value assuming that the number of high scoring matching regions above a threshold is Poisson distributed. Using a simple approach combined with recent results in reliability theory, we demonstrate here that the Karlin-Altshul model can be derived with no reference to the extreme events theory.
Sequences were considered as systems in which components are amino acids and having a high redundancy of Information reflected by their alignment scores. Evolution of the information shared between aligned components determined the Shared Amount of Information (SA.I.) between sequences, i.e. the score. The Gumbel distribution parameters of aligned sequences scores find here some theoretical rationale. The first is the Hazard Rate of the distribution of scores between residues and the second is the probability that two aligned residues do not lose bits of information (i.e. conserve an initial pairing score) when a mutation occurs.
Introduction
Almost all sequence alignments methods compute a score s(a, b) between two compared sequences a and b. This score is a measure of similarity between the two sequences and help to distinguish biologically significant relationship from chance similarities (Smith and Waterman, 1981; Altschul et al. 1990; Waterman, 1995). These methods assign scores to insertions, deletions and replacements, and compute an alignment of two sequences that corresponds to the least costly set of such mutations. Assignment of a similarity measure begins with a matrix of similarity scores for all possible pairs of residues. Identities and conservative substitution have positive scores, while unlikely replacements have negative scores (Dayhoff et al. 1978; Henikoff and Henikoff, 1992; Bastien et al. 2005a). The score of the computed alignment is the sum of the elementary scores for each pair of aligned residues. All these methods allow the introduction of gaps in the alignment to maximize the final score and to taking account of deletion events in DNA (Waterman, 1995).
Because of the exponential increase of the number of sequence in each database and the large number of sequenced genomes, confidence in alignment score probabilities is critical to perform a rapid and accurate discrimination between alignments. The two main probability models compare the score s(a, b) with a score computed using random sequence A and B.
The first method proposed by Karlin and Altschul (1990) is an estimate of the probability of an observed local ungapped alignment score according to an Extreme Value Distribution, (or EVD; for review: Coles, 2001) in the asymptotic limit of long sequences. The Karlin-Altschul formula is the consequence of interpreting the number of highest scoring matching regions above a threshold by a Poisson distribution (Karlin and Altschul, 1990). As a consequence, if s is the score obtained after aligning two real sequences a and b (with m and n their respective lengths), the probability of finding an ungapped segment pair with a score lower than or equal to s, follows a particular Gumbel distribution (named EVD type I):
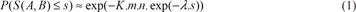

The second method uses Monte Carlo simulations to investigate the significance of a score s calculated from the alignment of two real sequences a and b (Lipman and Pearson, 1985; Pearson, 1998; Comet et al. 1999; Bacro and Comet, 2001; Bastien et al. 2004). This is done by shuffling b and compute the Z-Value Z(a, b*) = (s −
From a theoretical point of view, the only thing that can be measured between two biological sequences primary structures is the Shared Amount of Information (SAI) between them. It is well-known that the SAI decreases in average with time because sequences accumulate mutations during this period. The SAI between two sequences can be defined by two ways. The first is to define it by a mathematical distance between the two sequences. Using the Information Theory, it has recently been demonstrate that the Mutual Information I(a; b) between the two sequences a and b is a better measure of the SAI than all mathematical distances one can compute between a and b (Bastien et al. 2005b). Indeed, Mutual Information between events i and j is measured as s has a natural quantum. This follows from the fact that evolution of s is due to mutation from one amino acid to another. This last phenomenon is a discrete one. A natural way to model this phenomenon is to use a Poisson distribution for the number of Mutual Information one residue pairs can lost when a mutation of one of the two residues occurs. For one given residue pairs, we estimate the CF. at the point s = 0. This is done by using the probability that no change in s = 0 occurs during some un-specified evolutionary process, given that on average a decrease of η occurs. Expansion of η in terms of s then gives the C.F. in terms of s, and hence the cumulative distribution P(s).
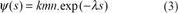
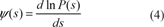

Derivation of the Conservation Function for Sequence Alignments
Derivation of the C.F. for residues
First, let us consider two sequences a and b of length m and n and having a Mutual Information equal to zero, that is to say s(a, b) = 0. So, each aligned pair of residue have an elementary score set up to zero. If one considers that no positive change is possible, then the probability of an N bit-decrease of the MI between the two residues is given by a Poisson distribution,
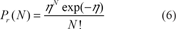
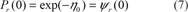

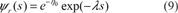
Derivation of the C.F. for sequences
For two sequences a and b of length m and n, we have mn possible pairs of residues, which can lead to negative substitution and so P(N) = Pr(N)
mn
. Using equation (5) and (9), we can state the following formula for the C.F. for two sequences:

This formula is identical to the C.F. of the Karlin-Altshul formula, where the parameter k is identified to exp(–η0), the probability for a residue to have no Negative Substitution, knowing the fact that there is no Positive Substitution.
Discussion
This interpretation of the parameter k can be checked quantitatively by comparing the magnitude order of k in sequence comparison simulation and the value of exp(–η0) obtained from BLOSUM62 (Henikoff and Henikoff, 1992), the most popular substitution matrix. The reason for comparing magnitude order instead of more accurate values comes from the fact that values obtained from BLOSUM62 are rounded to the integer (Henikoff and Henikoff, 1992) and that the subsequent error made on exp(–η0) is quite large.
Estimation of the C.F. at one point is made using its definition, that is to say


Future development of this method includes the exact computation of each residue C.F. and of gap penalty so as to obtain a rapid calculation of the alignment score statistics without pre-computed parameters Altschul et al. (2001) and without expensive simulations (Aude and Louis, 2002; Bastien et al. 2007). Whereas Karlin and Altschul (1990) derived their formula by considering maximum of sums of random elementary scores and obtained an extreme value distribution, we demonstrate here that this formula can arise from a different way considering the elementary SAI shared by residues. The elementary Information losses lead to a global loss of Mutual Information at the sequence level and are characterized by the Gumbel formula (1).
Footnotes
Acknowledgements
The author wishes to thank E. Maréchal for fruitful discussions and useful comments on a draft version of this paper. OB was supported by the Fondation Recherche Médicale and the Agence Nationale de la Recherche, as part of the PlasmoExplore project. Author would like to thank anonymous reviewers for their valuable comments.