
Abstract
Low molecular weight metabolites are important plant hormones and signaling molecules, and play an important part among the processes of plant development. Their activities may also be affected by the chemical modifications of methylation performed by SABATH. In this study, a total of 24 and 21 SABATH genes in Arabidopsis and rice, respectively, were identified and taken a comprehensive study. Phylogenetic analysis showed that AtSABATH and OsSABATH genes could be classified into 4 major groups and 6 subgroups. Gene expansion analysis showed that the main expansion mechanism of SABATH gene family in Arabidopsis and rice was tandem duplication and segmental duplication. The ratios of nonsynonymous (Ka) and synonymous (Ks) substitution rates of 12 pairs paralogous of AtSABATH and OsSABATH genes indicated that the SABATH gene family in Arabidopsis and rice had gone through purifying selection. Positive selection analysis with site models and branch-site models revealed that AtSABATH and OsSABATH genes had undergone selective pressure for adaptive evolution. Motif analysis showed that certain motifs only existed in specific subgroups or species, which indicated that the SABATH proteins of Arabidopsis and rice appear divergence in different species and subgroups. Functional divergence analysis also suggested that the AtSABATH and OsSABATH subgroup genes had functional differences, and the positive selection sites which contributed to functional divergence among subgroups were detected. These results provide insights into functional conservation and diversification of SABATH gene family, and are useful information for further elucidating SABATH gene family functions.
Keywords
Introduction
A large number of low molecular weight metabolites are ubiquitously produced by plants. The metabolites, such as salicylic acid (SA), jasmonic acid (JA), gibberellic acid (GA), and indole-3-acetic acid (IAA), are important plant hormones and signaling molecules, and these not only play an important part in diverse biological processes of plant growth and development but also have a critical role in plant interactions with the environment. Recently, studies showed that their activities may also be affected by the chemical modifications of methylation, whereas the SA, JA, GA, and IAA could undergo the same type of novel modification: methylation of their free carboxyl group, 1 and the methylate are methyl esters such as methyl jasmonate, methyl salicylate, and methyl benzoate. In addition, those products often contribute to the characteristic scents or flavors of plants, which render them appealing to humans or animals, and also involve in the regulation of plants’ diverse developmental processes, such as seed germination, root growth, leaf abscission, and flower or fruit development.2,3
Depending on the methyl group provided by S-adenosyl-
The SABATH methyltransferases, mostly the carboxyl methyltransferases, 9 and many characterized SABATH methyltransferases also play an important part in plant growth and development. Whereas, lots of SABATH methyltransferases were first identified in Arabidopsis and rice. For instance, the jasmonic acid carboxyl methyltransferase (JMT), a critical enzyme for jasmonate-regulated plant responses, could provide good defense against fungi. 10 Gibberellic acid carboxyl methyltransferase (GAMT) has a function in regulating seed germination. 11 Farnesoic acid carboxyl methyltransferase (FAMT) could provide good defense against insect herbivores. 12 Indole-3-acetic acid carboxyl methyltransferase (IAMT) plays an important role in regulating plant development and auxin homeostasis. 13 Then, in other species, many SABATH methyltransferases have been successively discovered. Cinnamate/p-coumarate carboxyl methyltransferases (CCMT), 14 loganic acid methyltransferases (LAMT), 15 and anthranilic acid methyltransferase (AAMT) 16 were found in sweet basil (Ocimum basilicum), Catharanthus roseus and maize (Zea mays), respectively, and all of them are related to plant resistance. 5 PgIAMT1, which was discovered in white spruce (Picea glauca), may take effect in embryogenesis via regulating the homeostasis of IAA. 1 PpSABATH1, identified from the moss (Physcomitrella patens), which could catalyze S-methylation of thiols, has a role in tolerance to toxic thiols through detoxification. 9
Moreover, the SABATH methyltransferases also contain nitrogen methyltransferases such as 7-methylxanthine methyltransferase (MXMT), 3,7-methylxanthine methyltransferase (DXMT), and xanthosine methyltransferase (XMT), 1 which involved in caffeine biosynthesis isolated from tea or coffee plants. 17 Besides, due to the resulting reaction, products of such SABATHs are nitrogen-containing compounds that are toxic to herbivorous insects, and many nitrogen methyltransferases may also have roles in plant defense. 18
The SABATH gene family was discovered in Arabidopsis first 8 and then was identified in rice. 19 Although the evolutionary relationship of SABATHs has been preliminarily analyzed, the factors that influence evolution hadn’t been studied in-depth. By virtue of the fast biochemical assay system for the initial screening of compounds for individual SABATH proteins, 59 potential substrates were found to be existing in plants, 12 whereas most members of SABATH proteins do not catalyze a single substrate. 20 Thus, functions of the SABATH maybe diverse, whereas the reason of functional divergence hasn’t been detected. It is well known that the Arabidopsis thaliana as dicotyledon is the first established model plant worldwide. Rice as one of the most important food crops is considered as the model organism of monocotyledon for genetic and molecular studies. 21 With the development of high-throughput technologies, more and more complete plant genome and complete chloroplast genome have been reported.22,23 The Arabidopsis Information Resource (TAIR) and International Rice Genome Sequencing Project (IRGSP) 24 or the Rice Annotation Project (RAP) database provide a abundant research platform for searching unknown genes and gene families, exploring their functions, and studying their molecular phylogenetic evaluation. Therefore, under genome-wide comprehensive analysis, we could try to detect more bioinformatics on SABATH gene family clearly through this key enzyme in dicotyledon and monocotyledon model species of Arabidopsis and rice, respectively, including the elementary biological information, the phylogenetic relationship, the functional divergence, and so on.
Here, using bioinformatics resources and tools, we comprehensively analyzed SABATH gene family in Arabidopsis and rice after the members of this gene family were redefined. We constructed a phylogenetic tree to evaluate the evolutionary relationships of SABATH genes in Arabidopsis and rice. We also analyzed the gene expansion mechanism using Plant Genome Duplication Database (PGDD). Then, we calculated the ratio of nonsynonymous (Ka) and synonymous (Ks) substitution for the paralogs to test the driving force of duplicated genes. With site models and branch-site models, the positive selection of SABATH genes in Arabidopsis and rice was test under Phylogenetic Analysis by Maximum Likelihood (PAML) program. Finally, the functional divergence of SABATH genes in Arabidopsis and rice was analyzed with the DIVERGE program.
Methods
Identification of the members of SABATH family in Arabidopsis and Rice
With TAIR database (http://www.arabidopsis.org/), we obtained the nucleotide and amino acids sequences of 24 AtSABATH genes, which were found by John C D’Auria. 8 The SABATH amino acid sequences from other species (Table S1) were downloaded from the National Center for Biotechnology Information (NCBI) database. Then, those sequences and 24 AtSABATH amino acid sequences were set as queries to search the RAP database (http://rapdb.dna.affrc.go.jp/) with the BLASTP program. The e-value was set 10−10 as cutoff to the homologue recognition, and if the sequence satisfied e ⩽ 10−10, it was selected as a candidate protein. And then, we used the tool of Pfam 25 to detect the SABATH domains of all these candidate proteins to further identify all of the predicted SABATH members in rice. If there was SABATH domain in the candidate proteins, it belonged to the SABATH gene family. Finally, the basic information such as amino acids sequences, molecular weight (Mw), and theoretical isoelectric point (pI) of AtSABATH and OsSABATH were obtained from TAIR and RAP databases.
Multiple sequence alignment and phylogenetic analysis
Multiple sequence alignment of the SABATH amino acid sequences from AtSABATH and OsSABATH is based on the method described previously, 5 with the DNAMAN program (Lynnon Corporation, San Ramon, CA, USA) and online program of Gblock (http://www.phylogeny.fr/one_task.cgi?task_type=gblocks). The phylogenetic tree was constructed using Bayesian inference implemented in MrBayes26,27 with the amino acid sequences of the AtSABATH and OsSABATH. The program of ProtTest was used to choose the best model of JTT + I + G + F for the phylogenetic tree construction. 28 The phylogenetic tree was represented with the help of the Treeview1.61 software. 29
Gene genomic distribution and segmental duplication analysis
Genomic distribution of SABATH genes in Arabidopsis and rice on chromosomes was performed using Chromosome Map Tool, according to the annotation information in TAIR and RAP databases. The segmental duplication of SABATH genes in Arabidopsis and rice was analyzed based on the PGDD (http://chibba.pgml.uga.edu/duplication/). 30
Gene structure analysis and motif detection
The gene structure was investigated with the online Gene Structure Display Server (http://gsds1.cbi.pku.edu.cn/) based on each coding sequence (CDS) and corresponding genomic sequence. Conserved motifs in AtSABATH and OsSABATH proteins were performed with the method described previously, 5 using program MEME with the following criteria: any number of repetitions of a motif and expected e-values less than 2 × 10−30.31,32
Ka and Ks calculation
The paralogs for SABATH genes in Arabidopsis and rice were inferred from the phylogenetic tree. The program of PAL2NAL 33 was used to estimate nonsynonymous (Ka) and synonymous (Ks) substitution rates, and the ratio of nonsynonymous and synonymous (Ka/Ks) substitution rates of each paralogous gene pair. Meanwhile, the Ka/Ks ratios for the paralogous genes can also be calculated with a sliding window of 20 aa.5,34
Detection of positive selection
The approach of Yang and coworkers35,36 was applied to test the positive selection of the SABATH genes in Arabidopsis and rice with the codeml program of PAML v4.9a 37 under the site model and branch-site model.
In site models analysis, we used the M0, M1a, M2a, M3, M7, and M8 models to identify codons that were influenced by positive selection and also to detect positively selected sites (Supplementary command 1). M0 (one ratio) hypothesizes the different sites which have the same evolution rate, whereas the M3 (discrete) hypothesizes a discrete distribution with 3 ratios of purifying selection, neutral evolution, and positive selection (p0, p1, and p2). 38 The M2a is a positive selection model, whereas M1a is a neutral selection model; M8 hypothesizes a beta and ω distribution model, whereas M7 is a beta distribution model. We employed the program codeml to calculate the dN/dS (nonsynonymous/synonymous) ratio and to detect the variation in the ω parameter among sites by comparing the likelihood ratio test (LRT) between the site models: M0 (one ratio) vs M3 (discrete), M1a (neutral) vs M2a (selection), and M7 (beta) vs M8 (beta + ω).
Branch-site model hypothesizes the different evolutionary rates to vary among different sites and branches simultaneously. 37 The improved branch-site model was used to compare the ratio of nonsynonymous and synonymous substitution rates between branches, and to test the positive selection amino acid sites of AtSABATH and OsSABATH. 39 All the branches were divided into foreground and background, and the branches on the foreground were tested for positive selection; the other branches on the tree were used as the background. For each branch, the ratio of nonsynonymous and synonymous substitution rates was calculated with the Null Model (ω = 1) (Supplementary command 2) and Alternative Model (ω > 1) 5 (Supplementary command 3). Then, we identified the positive selection sits by comparing the LRT between Null Model and Alternative Model. If LRT suggested the presence of codons under positive selection on the foreground branch, the codon was probably from the site class of positive selection.5,40 Bayes Empirical Bayes (BEB) method was used to estimate the Posterior probabilities (Qks). 37
Estimation of functional divergence
The functional divergence analysis between SABATH subgroups genes in Arabidopsis and rice was performed with DIVERGE version 3.0 software. 41 The significant changes in the site-specific shift was estimated based on the maximum likelihood procedures, 38 and the neighbor-joining tree is reconstructed with AtSABATH and OsSABATH amino acid sequences under MEGA 6.0. 42 Then, the coefficients of Type-I and Type-II functional divergences (θI and θII) between 2 clusters were calculated. The Type-I (θI) and Type-II functional divergences (θII) were based on evolutionary rate 43 and differences in biochemical properties of amino acids, respectively. 44 The coefficients of Type-I functional divergence (θI) greater than 0 indicates site-specific-altered selective constraints, and the coefficients of Type-II functional divergence (θII) greater than 0 demonstrates a radical shift of amino acid physiochemical property occurred after gene duplication or speciation. 43
The amino acid sites related to functional divergence could also be detected by the posterior probabilities (Qk). A high possibility that the evolutionary rate or the radical change in the amino acid property of a site was different between 2 clusters will be with a large posterior probability (Qk). 43 In addition, we empirically used Qk > 0.8 and 1.0 as cutoff in the identification of the Type-I and Type-II functional divergence-related residues between gene groups, respectively. 38
Results
Sequence feature of SABATH genes in Arabidopsis and Rice
According to the approach of identification of the members of SABATH family in Arabidopsis and rice, we blasted the rice database with the aa sequences of 24 Arabidopsis and 15 other species SABATH genes. The results showed that there were 27 SABATH in rice (Table S2). However, 5 members didn’t have the motifs that must be necessary for the SABATH (Figure S1) and 1 member was a pseudogene. 19 Thus, a total of 24 and 21 SABATH members of SABATH gene family in Arabidopsis and rice were identified. The gene length of AtSABATH and OsSABATH were found to vary from 1261 bp (At3g44840) and 1474 bp (Os01g0700300) to 3472 bp (At5g55250) and 8025 bp (Os06g0313440) (Tables S3 and S4). The length of AtSABATH and OsSABATH proteins varied from 348 aa (At3g44840 and At3g44860) and 286 aa (Os06g0243300) to 415 aa (At5g04370) and 419 aa (Os06g0242900) (Tables S3 and S4), respectively. The molecular weights of the AtSABATH and OsSABATH proteins range from 38.51 kDa (At3g44860) and 32.40 kDa (Os11g0257700) to 47.26 kDa (At5g04370) and 47.53 kDa (Os06g0242900) (Tables S3 and S4), respectively, and the theoretical isoelectric points were predicted to range from 4.7698 (At1g15125) and 4.9176 (Os11g0256600) to 8.9032 (At5g04370) and 9.4957 (Os06g0243300) (Tables S3 and S4), respectively.
Phylogenetic analysis of SABATH gene family in Arabidopsis and Rice
To detect the evolutionary relationship among SABATH gene family in Arabidopsis and rice, we constructed an unrooted phylogenetic tree using the SABATH amino acid sequences of Arabidopsis and rice (Figure 1). Base on the phylogenetic tree, and the SABATH gene family of Arabidopsis and rice were divided into 4 major groups, where Groups A and B were also divided into 2 subgroups (Figure 1). The bootstrap values for all the subgroups were high, which indicates that the genes in each subgroup might share a similar origin (Figure 1). In addition, 6 pairs of paralogous genes were identified from SABATH gene family in Arabidopsis such as At5g38020 and At3g21950 in subgroup B1; At5g56300 and At4g26420 in subgroup D; At5g38100 and At5g38780, At5g37970 and At5g37990, At3g44870 and At3g44860, At1g66700 and At1g66690 in subgroup C (Figure 1). Six pairs of paralogous genes of SABATH gene family in rice were identified, which were Os06g0315000 and Os06g0315300 in subgroup A1; Os06g0311800 and Os06g0313320, Os11g0256600 and Os11g0257700, Os06g0242900 and Os06g0243300, Os06g0241400 and Os06g0242000 in subgroup A2; and Os01g0700300 and Os01g0701700 in subgroup B2 (Figure 1).

The phylogenetic tree for the SABATH gene family in Arabidopsis and rice. The tree was constructed using Bayesian inference implemented in MrBayes based on the 45 amino acid sequences of the AtSABATH and OsSABATH under the model of JTT + I + G + F.
Gene genomic distribution and expansion
To get insight into the expansion mechanism of SABATH gene family in Arabidopsis and rice, we detected their genomic distribution and segmental duplication. According to the annotation information, the AtSABATH genes were distributed on all the 5 Arabidopsis chromosomes (Figure 2). Six and ten AtSABATH genes were present on chromosomes 1 and 5, respectively. Whereas, 5 genes were on chromosome 3, 2 AtSABATH genes were on chromosome 4, and only 1 gene was on chromosome 2 (Figure 2). Meanwhile, OsSABATH genes were dispersed on chromosomes 1, 2, 4, 5, 6, and 11 (Figure 3). There were 13 OsSABATH genes on chromosome 6. Two and three OsSABATH genes were present on chromosomes 1 and 11, respectively. Only 1 gene was present on the chromosomes 2, 4, and 5 (Figure 3). Furthermore, At1g66700/At1g66690/At1g66720, At3g44870/At3g44860, At5g37970/At5g37990, At5g04380/At5g04370, and At5g38100/At5g38780 in Arabidopsis and the genes of Os06g0315000/Os06g0315300, Os06g0311800/Os06g0313320, Os11g0256600/Os11g0257700, Os06g0242900/Os06g0243300, Os06g0241400/Os06g0242000, and Os01g0700300/Os01g0701700 in rice were located in the same intergenic region, respectively. According to the phylogenetic tree, these genes showed a close evolutionary relationship, suggesting that they arose through a tandem duplication event. 45 In addition, based on the PGDD and phylogenetic tree, the pair of At5g38020 and At3g21950 in Arabidopsis and the pair of Os02g0719600 and Os06g0311800 in rice are highly conserved, indicating that these 2 genes were formed through segmental duplication. 45

Genomic distribution of Arabidopsis SABATH genes on chromosomes.

Genomic distribution of rice SABATH genes on chromosomes.
Gene structure analysis
Genes’ structural features of SABATH gene family in Arabidopsis and rice are listed in Figure 4. Structure analyses revealed that the number of exons of all the SABATH genes in Arabidopsis and rice varies from 1 to 9. Only 1 gene was intronless (Os05g0102000). The average exon number in the groups was 3 or 4. In addition, the exons which have the same splicing phase at both ends are called symmetric exons, whereas the excess of symmetric exons and phase 0 introns is likely to recombination fusion, protein domain exchange, and exon shuffling.46,47 Through analyzing the 167 exons, there were 63 exons that were symmetric with phase 0 introns, only 1 exon was symmetric with phase 1 introns, and no exon was symmetric with phase 2 introns. Although, for the 122 introns, the number of phase 0 was 111, phase 1 was 7 and phase 2 was 4. Thus, our analyses of the gene structures indicated diversity among the SABATH genes in Arabidopsis and rice.

The structural features of each SABATH gene in Arabidopsis and rice.
Conserved domains and motif analysis
Using Pfam program, we found that all the SABATH proteins identified in Arabidopsis and rice included the SABATH conserved domain sequences. Then, the domain sequences were aligned using the DNAMAN program. The results revealed that most of the members of SABATH family in Arabidopsis and rice, including a functional domain that was conserved among O-MTs,48,49 contain the binding sites (the motifs I and III) of SAM (Figure 5), a well-known methyl donor in plant cells. 10 With the MEME program, 16 conserved motifs in AtSABATH and OsSABATH amino acid sequences were identified (Table S5), and the frequency of all those conserved motifs varied from 6 to 45 (Table S5). The number of amino acids in each SABATH motifs ranged from 11 to 35, and the number of motifs in each SABATH proteins varied from 8 to 13. Among all the motifs, motifs 1, 2, 3, 4, and 7 were widespread in AtSABATH and OsSABATH proteins (Table S6). Besides, the conserved motif 3 matched the functional domain motif I, whereas the conserved motif 2 matched the functional domain motif III (Figure 5). The normal expression sequences of 16 motifs are listed in Table S5. Although many motifs were shared by Arabidopsis and rice, there were still species-specific motifs (motifs 8, 12, and 16 in Arabidopsis and motif 13 in rice) (Table S6).

(A) The multiple sequence alignments of the Arabidopsis and rice SABATH proteins’ conserved domain of O-methyltransferases including the binding sites (the functional domain motifs I and III, which were defined via protein X-ray crystallography were indicated) of SAM (S-adenosyl-
Driving forces for genetic divergence
Gene duplication is one of important incident for gene family expansion and plays a major role in functional diversity during evolution. 50 Thus, to detect whether Darwinian positive selection participated in promoting gene divergence after duplication, the ratio of nonsynonymous (Ka) and synonymous (Ks) substitution rates (Ka/Ks) was calculated with the CDS of paralogous AtSABATH and OsSABATH. Generally, Ka/Ks ratio <1, =1, and >1 indicate negative or purifying selection, neutral evolution, and positive selection, respectively. 51 The ratios of Ka/Ks for all the 12 SABATH paralogous pairs in Arabidopsis and rice were <1 (Table S7), suggesting that the SABATH genes have undergone purifying selection pressure in Arabidopsis and rice. In the meantime, the Ka/Ks ratios for all the paralogous genes were also calculated with sliding window of 20 aa. If the regions of all the paralogous genes had gone through positive selections, the ratio of Ka/Ks was >1 in those regions, whereas the proportion of such regions genes was few (Figures 6 and 7). However, the regions with value >1 were in the majority only in the pair of At5g56300 and At4g26420, as a whole of Ka/Ks was still <1 (Figure 6). Most regions appearing with the ratios of Ka/Ks <1 in paralogous genes also suggested that the AtSABATH and OsSABATH genes in had gone through purifying selection (Figures 6 and 7).

The Ka/Ks ratios for AtSABATH 6 paralogous pairs proteins with a sliding window of 20 amino acids.

The Ka/Ks ratios for OsSABATH 6 paralogous pairs proteins with a sliding window of 20 amino acids.
Positive selection on AtSABATH and OsSABATH genes
To preliminarily test the evolutionary mechanism of SABATH gene family in Arabidopsis and rice, we examined the hypothesis of positive selection of AtSABATH and OsSABATH genes under PAML program36,37 with site models and branch-site models according to the phylogenetic tree (Figure 1).
The parameter estimates, log likelihood, and the LRT tests of site models are shown in Table 1. M0 and M3 were first compared to examine how dN/dS ratios differed among codon positions. Under model M0, the value of log likelihood was ι = –42921.859158, with an estimate of ω = 0.25066. Meanwhile, under model M3, the value of log likelihood was ι = –41868.048450, with 3 values of ω (ω0 = 0.03053, ω1 = 0.18380, and ω2 = 0.18380) (Table 1), suggesting that the predominant force acting on the evolution of the SABATH gene family in Arabidopsis and rice was relaxed purifying selection. Moreover, the value of twice the log likelihood differences between model M3 and model M0 was statistically for all codons tested, revealing that the overall selective constraint levels fluctuated across the SABATH gene family group lineages in Arabidopsis and rice. Then, we compared the M2a vs M1a and M8 vs M7 to test whether positive selection promoted divergence between genes. 38 Both of the values of log likelihood were ι = –42463.032156 under the M1a and M2a for AtSABATH and OsSABATH genes. The values of log likelihood under M7 and M8 for AtSABATH and OsSABATH genes were ι = –41836.150234 and ι = –41836.150365, respectively (Table 1). In both cases, the value of 2ΔlnL was close to 0; there was no statistical significance, and no site was tested under positive selection at the level of 95% (Table 1).
Tests for positive selection among codons of AtSABATH and OsSABATH genes using site models.
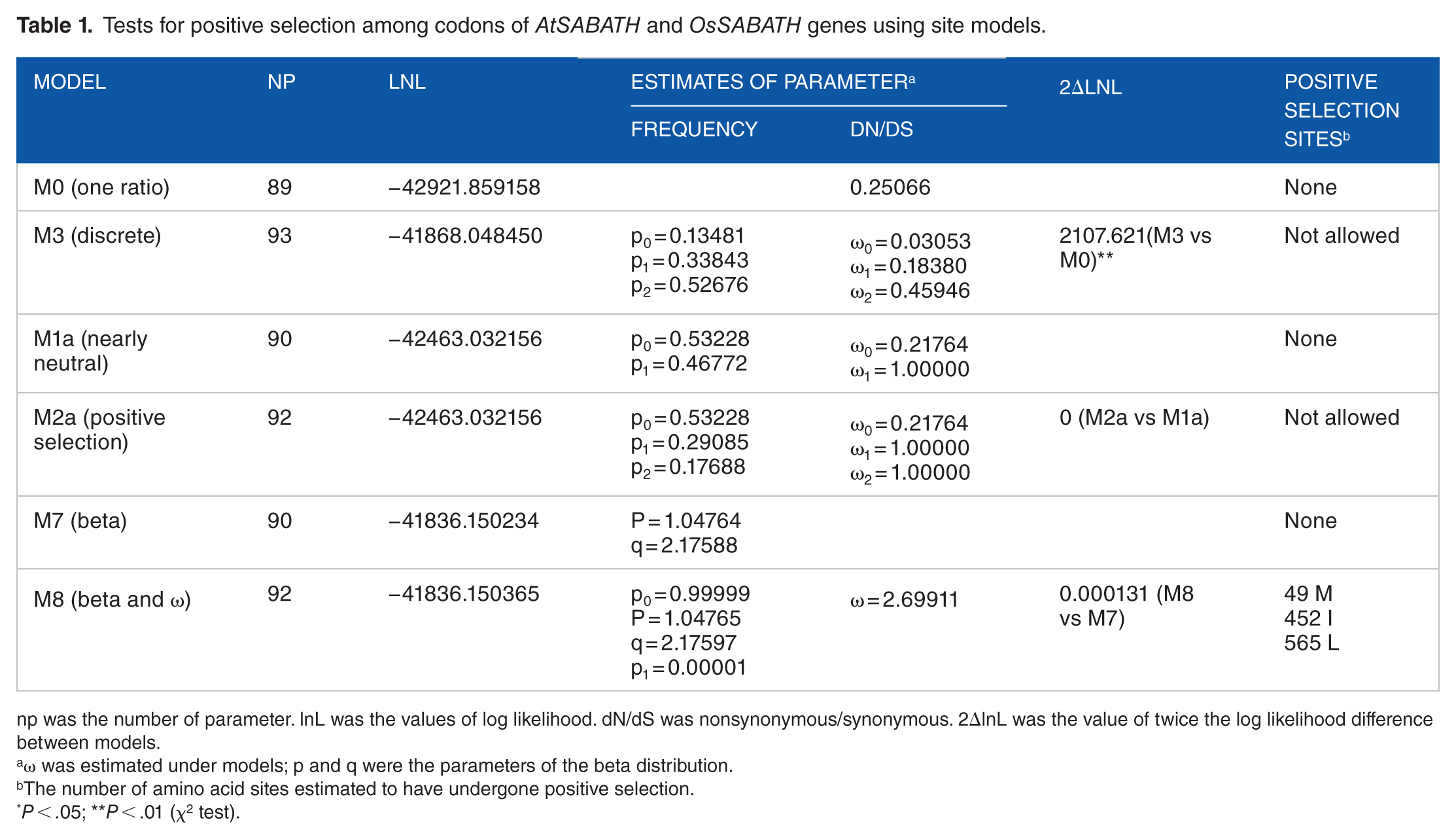
np was the number of parameter. lnL was the values of log likelihood. dN/dS was nonsynonymous/synonymous. 2ΔlnL was the value of twice the log likelihood difference between models.
ω was estimated under models; p and q were the parameters of the beta distribution.
The number of amino acid sites estimated to have undergone positive selection.
P < .05; **P < .01 (χ2 test).
In branch-site models analysis, Null and Alternative models were compared to detect the affecting sites under positive selection in particular lineages. The parameter estimates, log likelihood, and the LRT tests of those models of those 2 models are listed in Table 2. When each subgroup was set as foreground branch, the difference between Null and Alternative models was strongly statistically significant (P < .01) (Table 2). It indicated that different SABATH lineages in Arabidopsis and rice may have different evolutionary rates. In addition, when the group A1 was set as foreground branch, 1 site was examined under positive selection at a level of 99%. Meanwhile, 2 sites were detected under positive selection at a level of 95%, when the group B2 was set as foreground branch. The results suggested that groups A1 and B2 could be confronted with strong positive Darwinian selection, because significant positive sites were detected at .01 and .05 significance levels (Table 2).
Selective pressure analyses of SABATH genes in Arabidopsis and rice by branch-site model.
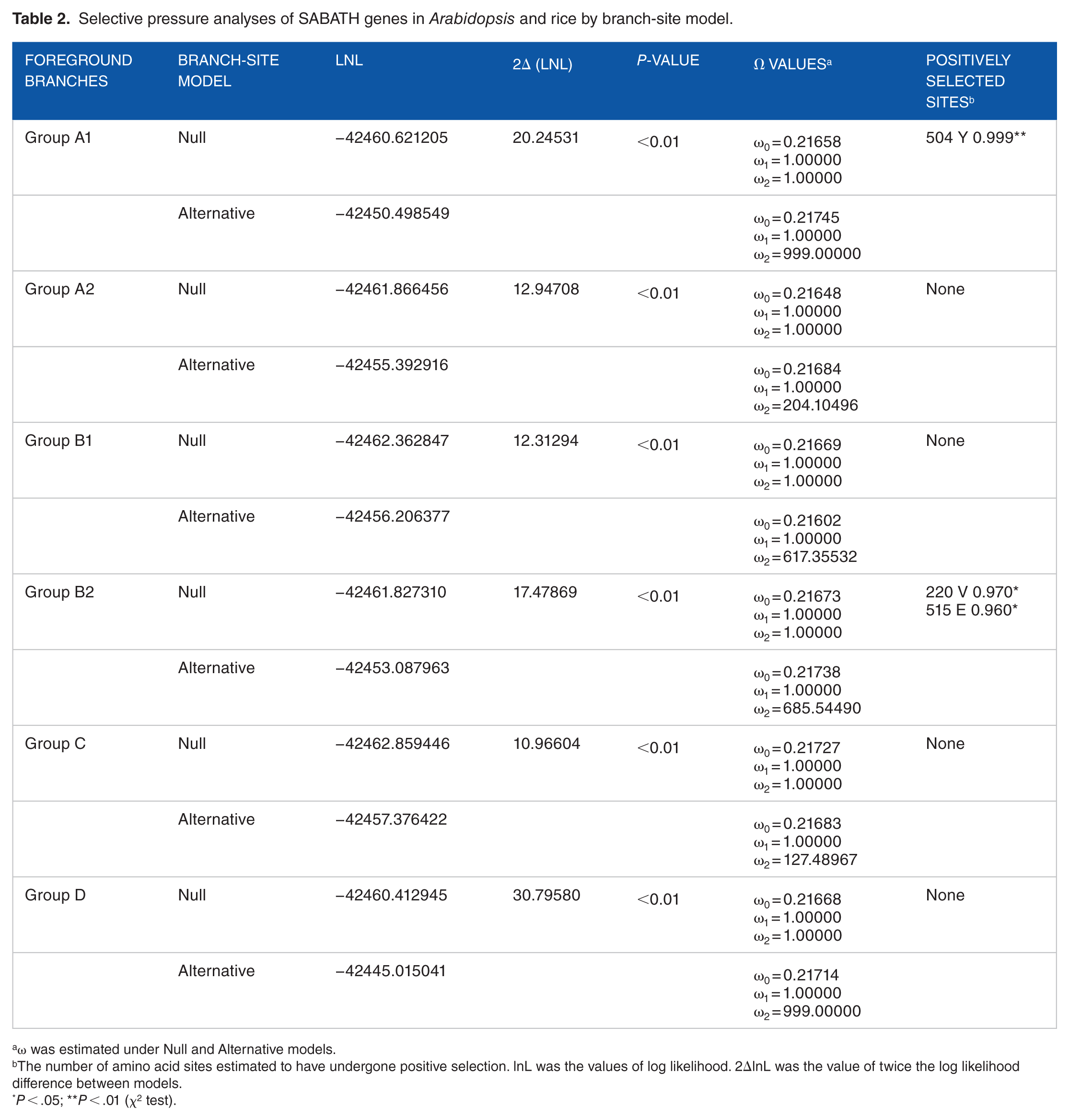
ω was estimated under Null and Alternative models.
The number of amino acid sites estimated to have undergone positive selection. lnL was the values of log likelihood. 2ΔlnL was the value of twice the log likelihood difference between models.
P < .05; **P < .01 (χ2 test).
Functional divergence analysis of AtSABATH and OsSABATH proteins
With the software of DIVERGE, we could evaluate the shifted evolutionary rate and altered amino acid property, which lead to the functional divergence after gene duplication.43,44 The AtSABATH and OsSABATH amino acid sequences were also divided into 6 major clusters in accordance with the neighbor-joining tree (Figure S2). We also carried out Posterior probability (Qk) to test the amino acid sites affecting functional divergence between the AtSABATH and OsSABATH clusters.
Using the DIVERGE program, we found that all the coefficients for the Type-I functional divergence was greater than 0 through comparing among AtSABATH and OsSABATH subgroups (Table S8). The coefficients for the Type-I functional divergence of 15 group pairs, including Group A1 vs Group A2, Group A1 vs Group B1, Group A1 vs Group B2, Group A1 vs Group C, Group A1 vs Group D, Group A2 vs Group B1, Group A2 vs Group B2, Group A2 vs Group C, Group A2 vs Group D, Group B1 vs Group B2, Group B1 vs Group C, Group B1 vs Group D, Group B2 vs Group C, Group B2 vs Group D, and Group C vs Group D, ranged from 0.048777 to 0.597337. In addition, the Type-I functional divergence (θI) of Group A1 vs Group A2, Group A1 vs Group B2, Group A2 vs Group B2, Group A2 vs Group C, Group A2 vs Group D, Group B1 vs Group B2, Group B1 vs Group C, and Group B2 vs Group C were statistically significant (Table S8). It revealed that some amino acid sites may have occurred significant site-specific changes between these group pairs, which bring about a subgroup-specific functional divergence during their evolution. However, for the values of Type-II functional divergence (θII), there were no pairs greater than 0 (Table S9), which advised that most residues in the AtSABATH and OsSABATH gene family hadn’t happened obviously as physical and chemical properties change.
Based on past experience, 5 Qk > 0.8 and 1.0 were set as threshold to estimate the Type-I and Type-II functional divergence-related positive selection sites between gene groups, respectively. According to the elaborate result of posterior probabilities analysis, the positive selection sites that were related to functional divergence in group pairs were different in the distribution and the number. For the Type-I functional divergence, when Qk > 0.8, all of the group pairs contained positive selection sites except Group A 1 vs Group B1, Group A1 vs Group D, and Group A2 vs Group B1 (Table S8). Besides, for the Type-II functional divergence, when Qk > 1.0, only 3 group pairs (Group A1 vs Group A2, Group A2 vs Group B2, and Group B1 vs Group B2) contained positive selection sites (Table S9). The result illustrated that these positive selection sites may play an important role in functional divergence of AtSABATH and OsSABATH during the evolutionary process. The positive selection sites affecting the Type-I and Type-II functional divergence between groups detailed distribution are demonstrated in Figures 8 and 9.

Site-specific prediction for Type-I functional divergence between groups of AtSABATH and OsSABATH.

Site-specific profile for predicting critical amino acid residues responsible for the Type-II functional divergence between groups of AtSABATH and OsSABATH.
Discussion
The plant SABATH gene family is a group of key enzyme for regulating the plant growth. Meanwhile, the substrates and products of SABATH methyltransferases play an important role in developmental processes of higher plants. Further studies on this family can help not only illustrating the SABATH genes’ vital function in developmental processes of high plants but also elucidating the evolutionary relationships between different species. It has been shown that 24 AtSABATH genes and 41 OsSABATH genes exist in the Arabidopsis and rice genome, respectively.8,19 However, due to some of the shorter proteins may be from inaccurate annotation in rice genome, 52 the members of SABATH gene family in rice weren’t accurate. 19 Thus, it’s necessary to redefine the rice SABATH gene family. With new method, after BLAST analysis, and correcting with the tool of Pfam, and also according to the length of SABATH amino acid, 8 a total of 21 OsSABATH genes were detected, finally.
Generally, gene duplication events were important for gene family evolution and occurred via 3 major mechanisms: segmental duplication, tandem duplication, and transposition events.45,53 In addition, one of the main functions of phylogenetic study was to identify putative paralogs that normally display different functions and orthologs that usually retain the same function. 21 Based on the phylogenetic tree, 6 pairs of paralogs genes in AtSABATH and also 6 pairs of paralogs genes in OsSABATH were identified (Figure 1). More than half of SABATH genes in Arabidopsis and rice were contained in paralogous pairs (50% for Arabidopsis and 58% for rice). That was to say, more than half of SABATH genes in Arabidopsis and rice had undergone duplication. It indicated that lots of SABATH genes in Arabidopsis and rice have undergone gene family expansion and functional diversity during evolution. Furthermore, through genomic distribution and segmental duplication analysis, it also suggested that some SABATH genes in Arabidopsis and rice had arose through a tandem duplication event or segmental duplication.
Besides, according to the ratio of Ka and Ks of paralogous AtSABATH and OsSABATH genes, there was no ratio >1, and the result of sliding window showing that the ratio of Ka and Ks was >1 in regions of all the paralogous genes was less, which indicated that the AtSABATH and OsSABATH genes had gone through purifying selection. Meanwhile, codeml program in PAML was applied to test the hypothesis of positive selection. In sites model, through the comparison of models M0 and M3, models M2a and M1a, and models M8 and M7, we found that M3 better than M0, M2a not better than M1a, and M8 not better than M7, which indicated that different sites bare different selection pressures, and SABATH gene family in Arabidopsis and rice also undergo purifying selection. In branch-site models, the subgroups were divided into foreground groups and background groups to detect positive selection among sites and branches. The results showed that only 3 positive sites and 2 lineage groups were found to be under positive selection. It also suggested that AtSABATH and OsSABATH genes mainly undergo neutral evolution and purifying selection.
Functional divergence analysis indicated that different subgroup showed functional divergence during their evolution. Using program MEME, we found that some motifs only existed in specific subgroup or species. It also revealed that the SABATH proteins may demonstrate functional differences in different species and subgroups. Whereas, according to the groups indicated from the phylogenetic tree (Figure 1) and the conserved motifs, most SABATH proteins in the subgroup had similar motif orders and compositions (Table S6), suggesting that the proteins in uniform subgroup may possess similar feature during plant evolution. In addition, we also found that gene structure in the same subgroup was similar to each other, which indicated that the genes in same subgroup might possess similar functions (Figure 4).
Conclusions
In this study, we redefined 21 members of SABATH genes in rice according to the members of SABATH gene family in Arabidopsis and the rice genome database. Then, the AtSABATH and OsSABATH genes were analyzed with comprehensive methods, containing SABATH function domain and conserved motif characterization, gene structure and expansion mechanism explanation, phylogenetic, positive selection and functional divergence analysis.
We showed that 45 AtSABATH and OsSABATH genes could be divided into 6 subgroups and 12 pairs of paralogous genes, which were detected with the phylogenetic tree. The SABATH genes in Arabidopsis and rice have confronted purifying selection pressure because the ratios of Ka/Ks for the 12 paralogous were <1. Chromosome locations and PGDD analysis showed that the main expansion mechanism of SABATH genes in Arabidopsis and rice was tandem duplication and segmental duplication. Conserved motif analysis revealed that some group-specific motifs maybe attribute to functional divergence of AtSABATH and OsSABATH genes. Functional divergence analysis also manifested that the AtSABATH and OsSABATH genes have experienced functional divergence during evolution. Positive selection analysis with site models and branch-site models suggested that SABATH genes in Arabidopsis and rice have undergone positive selection. These research results offered extensive information about AtSABATH and OsSABATH, and are valuable for in-depth study of the SABATH gene family functions in plant.
Supplemental Material
Fig_S1_xyz2077906db024b – Supplemental material for Genome-Wide Comprehensive Analysis of the SABATH Gene Family in Arabidopsis and Rice
Supplemental material, Fig_S1_xyz2077906db024b for Genome-Wide Comprehensive Analysis of the SABATH Gene Family in Arabidopsis and Rice by Bin Wang, Min Li, Yijun Yuan and Shaofang Liu in Evolutionary Bioinformatics
Supplemental Material
Fig_S2_xyz20779fc99717f – Supplemental material for Genome-Wide Comprehensive Analysis of the SABATH Gene Family in Arabidopsis and Rice
Supplemental material, Fig_S2_xyz20779fc99717f for Genome-Wide Comprehensive Analysis of the SABATH Gene Family in Arabidopsis and Rice by Bin Wang, Min Li, Yijun Yuan and Shaofang Liu in Evolutionary Bioinformatics
Supplemental Material
Supplementary_commands – Supplemental material for Genome-Wide Comprehensive Analysis of the SABATH Gene Family in Arabidopsis and Rice
Supplemental material, Supplementary_commands for Genome-Wide Comprehensive Analysis of the SABATH Gene Family in Arabidopsis and Rice by Bin Wang, Min Li, Yijun Yuan and Shaofang Liu in Evolutionary Bioinformatics
Supplemental Material
Supporting_tables – Supplemental material for Genome-Wide Comprehensive Analysis of the SABATH Gene Family in Arabidopsis and Rice
Supplemental material, Supporting_tables for Genome-Wide Comprehensive Analysis of the SABATH Gene Family in Arabidopsis and Rice by Bin Wang, Min Li, Yijun Yuan and Shaofang Liu in Evolutionary Bioinformatics
Footnotes
Acknowledgements
The authors are grateful to the National Engineering Laboratory for Resource Development of Endangered Crude Drugs in Northwest China. The authors also acknowledge the support from the Jiangxi Provincial Education Department, the Major Project of Shaanxi Province, China. This study was carried out with the support of the Jiangxi Province Project Education Fund (Project No: GJJ170434) and State Key Laboratory Breeding Base of Nuclear Resources and Environment Foundation (Grant No: RGET1503).
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the Jiangxi province project Education Fund (Project No: GJJ170434) and State Key Laboratory Breeding Base of Nuclear Resources and Environment Foundation (Grant No: RGET1503).
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
BW wrote the first draft, with ML, SL, and YY providing analysis data on SABATH gene family. All authors contributed to and critically revised the manuscript.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.