
Abstract
Objectives
Feline infectious peritonitis (FIP), caused by genetic mutants of feline enteric coronavirus known as FIPV, is a highly fatal disease of cats with no currently available vaccine or US Food and Drug Administration-approved cure. Dissemination of FIPV in affected cats results in a range of clinical signs, including cavitary effusions, anorexia, fever and lesions of pyogranulomatous vasculitis and perivasculitis, with or without central nervous system or ocular involvement. The objectives of this study were to screen an array of antiviral compounds for anti-FIPV (serotype II) activity, determine cytotoxicity safety profiles of identified compounds with anti-FIPV activity and strategically combine identified monotherapies to assess compound synergy against FIPV in vitro. Based upon clinically successful combination treatment strategies for human patients with HIV and hepatitis C virus infections, we hypothesized that a combined anticoronaviral therapy approach featuring concurrent multiple mechanisms of drug action would result in an additive or synergistic antiviral effect.
Methods
This study screened 90 putative antiviral compounds for efficacy and cytotoxicity using a multimodal in vitro strategy, including plaque bioassays, real-time RT-PCR viral inhibition and cytotoxicity assays.
Results
Through this process, we identified 26 compounds with effective antiviral activity against FIPV, representing a variety of drug classes and mechanisms of antiviral action. The most effective compounds include GC376, GS-441524, EIDD2801 and EIDD1931. We documented antiviral efficacy for combinations of antiviral agents, with a few examined drug combinations demonstrating evidence of limited synergistic antiviral activity.
Conclusions and relevance
Although evidence of compound synergy was identified for several combinations of antiviral agents, monotherapies were ultimately determined to be the most effective in the inhibition of viral transcription.
Introduction
Feline infectious peritonitis (FIP) is a highly fatal disease of cats with no effective vaccine or US Food and Drug Administration-approved treatment. Although the pathogenesis has not been fully elucidated, FIP is generally understood to develop as a result of specific mutations in the viral genome of the minimally pathogenic and ubiquitous feline enteric coronavirus (FECV), creating the virulent FIP virus (FIPV).1,2 These FECV mutations result in a virus–host cell tropism switch from intestinal enterocytes to monocytes/macrophages. These two biotypes (FECV and FIPV) are generally considered to exist as two serotypes (I and II); however, this convention has recently been questioned, and suggestions have been made that the ‘serotypes’ be considered as distinct viruses based on their spike protein differences and expected biologic and clinical outcomes. 3 Regardless of whether these two serotypes represent distinct viruses, both are capable of causing FIP, with serotype I being more prevalent in nature. 4 Although serotype I is more prevalent, it is less well studied than serotype II owing to challenges in propagating this virus in vitro.
Productive monocyte/macrophage infection by FIPV, variably widespread anatomic dissemination and immune-mediated perivasculitis results in the highly fatal systemic inflammatory disease, FIP. 5 As a result of viral dissemination, FIP may present with clinical signs reflecting inflammation in a variety of anatomic sites potentially including the abdominal cavity and associated viscera, thoracic cavity, central nervous system and/or eye.6–9 FIP remains a devastating viral disease of cats owing to its high mortality rate, challenges in establishing a precise etiologic diagnosis, and the current lack of available and effective treatment options.8,10 The development of an effective vaccine for FIP has been complicated by the role of antibody-dependent enhancement in FIP disease pathogenesis, where the presence of non-neutralizing anticoronaviral antibodies have been shown to exacerbate FIP disease.11–13
Recent antiviral clinical trials in both experimentally and naturally FIPV-infected cats have shown promise in treating and curing FIP through the use of GS-441524, a nucleoside analog and metabolite of remdesivir (Veklury; Gilead Sciences), or GC376, a 3C-like protease inhibitor of FIPV (Anavive Lifesciences).14–16 There have been particular treatment challenges for cats with more complicated and multisystemic forms of FIP, including those with neurologic or ocular involvement. A recent clinical trial of GS-441524 at higher dosages for the treatment of neurologic FIP established the possibility of long-term resolution of disease for some of these more complicated forms of FIP. 17 Despite the recent clinical successes, these antiviral compounds are currently unavailable for legal clinical veterinary use in cats with FIP.
Using a collection of compounds selected based on their proven efficacy in interfering with the replication of other RNA viruses, we identified a subset of compounds with variable anti-FIPV activity against serotype II and characterized their safety and efficacy profiles in vitro. Of these compounds, several have been used as treatments for retroviral, hepatitis C or other coronaviral infections, including current investigations as therapies for COVID-19.18–21 Given the success of combined anti-retroviral therapy (cART) against HIV-1 and combination therapies against hepatitis C virus, 22 it would seem feasible that concurrently targeting FIPV at different steps of the virus lifecycle with a combined anticoronaviral therapy (cACT) may offer a greater level of sustained and more complete success than has been achieved with monotherapies alone. The inclusion of an antiviral agent in cACT capable of penetrating the blood–brain barrier and/or blood–ocular barrier and achieving pharmacologically relevant tissue concentrations may facilitate system-wide eradication of FIPV.
Here, we describe a set of in vitro assays facilitating the identification of effective and safe anticoronaviral compounds. We hypothesized that the combinatorial use of two or more effective antiviral monotherapies with differing mechanisms of action would facilitate the identification of additive or synergistic combinations providing superior anticoronaviral efficacy vs their use as sole agents (monotherapy).
Materials and methods
FIPV inoculum for in vitro experiments
Crandell-Rees feline kidney cells (CRFK; ATCC) were cultured in T150 flasks (Corning), inoculated with serotype II FIPV (WSU-79-1146; GenBank DQ010921) and propagated in 50 ml Dulbecco’s Modified Eagle’s Medium with 4.5 g/l glucose (Corning) and 10% fetal bovine serum (Gemini Bio). After 72 h of incubation at 37°C, extensive cytopathic effect (CPE) and large areas of cell clearing/detachment were noted. Flasks were then flash frozen at −70°C for 8 mins, thawed briefly at room temperature, and the cells and supernatant were then centrifuged at 1500 g for 5 mins, followed by a second centrifugation step at 4000 g for 5 mins, in order to isolate cell-free viral stocks. Supernatant containing the viral stock was divided into 0.5 and 1.0 ml aliquots in 1.5 ml cryotubes (Nalgene) and stored at −70°C. After freezing, a single tube was thawed, and the viral titer established using both bioassay (tissue culture infectious dose-50 [TCID50]) and real-time quantitative RT-PCR (RT-qPCR) methods (below).
The TCID50 was determined using a viral plaquing assay. CRFK cells were grown in a 96-well tissue culture plate (Genesee Scientific) until the CRFK cells achieved approximately 75–85% confluency. Serial 10-fold dilutions were made of FIPV stock and 200 µl samples of each dilution were added to 10-well replicates. At 72 h post-infection, the cells were fixed with methanol and stained with crystal violet (Sigma-Aldrich). Individual wells were evaluated visually for virus-induced CPE, scored as CPE positive or negative, and the TCID50 was determined based upon the equation log10TCID50 = [total number of wells CPE positive/number of replicates] + 0.5 to reflect infectious virions per milliliter of supernatant. 23
Quantification of FIPV by RT-qPCR
Cell-free viral RNA was isolated from the viral stock using the QIAamp Viral RNA Mini Kit (Qiagen), following the manufacturer’s instructions. The isolated RNA was treated with DNase (TURBO DNase; Invitrogen) and subsequently reverse transcribed using the High-Capacity RNA-to-cDNA Kit (Applied Biosystems) following the manufacturers’ protocols. The copy number of FIPV and feline GAPDH cDNA were determined using Applied Biosystem’s QuantStudio 3 Real-Time PCR System and PowerUp SYBR Green Master Mix, following the manufacturer’s protocol for a 10 µl reaction. Each PCR reaction was performed in triplicate with water template as a negative control and plasmid DNA as a positive control. A control reaction excluding reverse transcriptase was included in each real-time PCR assay set. cDNA templates were amplified using the FIPV forward primer, 5′-GGAAGTTTAGATTTGATTTGGCAATGCTAG, and the FIP reverse primer, 5′-AACAATCACTAGATCCAGACGTTAGCT (terminal portion of the FIPV 7b gene). 15 Real-time PCR for the feline GAPDH housekeeping gene was performed concurrently using the primers, 5 GAPDH, 5′-AAATTCCACGGCACAGTCAAG, and 3 GAPDH, 5′-TGATGGGCTTTCCATTGATGA. Cycling conditions for both FIPV and GAPDH amplicons were as follows: 50°C for 2 mins and 95°C for 2 mins, followed by 40 cycles of 95°C for 15 s, 58°C for 30 s and 72°C for 1 min. The final step included a dissociation curve to evaluate specificity of primer binding. FIPV and GAPDH copy numbers were calculated based on standard curves generated in our laboratory. Copies of FIPV cDNA determined via RT-qPCR were normalized per 106 copies of feline GAPDH cDNA.
Development of anti-helicase chemical fragments
In general, the drugs examined and described in this study were pre-existing antiviral agents. In contrast, the helicase enzyme of FIPV was cloned, expressed and used as a target for coronavirus and enzyme-specific viral discovery. The target DNA sequence of AviTag-FIP Helicase-HisTag was optimized and synthesized. The synthesized sequence was cloned (Dr Adeyemi Adedeji) into vector pET30a with Avi-His tag for protein expression in Escherichia coli. E coli strain BL21(DE3) was transformed with recombinant plasmid. A single colony was inoculated into 1 l of auto-induced medium containing antibiotic, and the culture was incubated at 37°C at 200 rpm. When the OD600 reached about 3, the temperature of the cell culture was changed to 15°C for 16 h. Cells were harvested by centrifugation. Cell pellets were resuspended with lysis buffer, followed by sonication. The precipitate after centrifugation was dissolved using denaturing agent. Target protein was obtained by one-step purification using a nickel (Ni) column. Target protein was sterilized by a 0.22 µm filter. Yield was 7.2 mg at 0.90 mg/ml, and was stored in phosphate buffered saline, 10% glycerol and 0.5 mM l-arginine at pH 7.4. The concentration was determined by Bradford protein assay, with bovine serum albumin as the standard. The protein purity and molecular weight were determined by sodium dodecyl sulfate polyacrylamide gel electrophoresis with Western blot confirmation.
Surface plasmon resonance (SPR) fragment screening was performed on a ForteBio Pioneer FE SPR platform. A HisCap sensor chip, which contains a nitrilotriacetic acid surface matrix, was used. Channels 1 and 3 were charged with 100 µM NiCl2, followed by injection of 50 µg/ml of FIP protein. Channel 2 was left free of protein, as well as NiCl2, as a reference. Channel 1 was immobilized to a density of ~8000 response units (RU), while channel 3 contained about 12,000 RU. Channel 1 was used. The buffer used for immobilization was 10 mM HEPES (pH 7.4), 150 mM NaCl and 0.1% Tween-20. For the assay, dimethyl sulfoxide (DMSO) was added to a final concentration of 4%. The proprietary compound library was diluted into the same buffer without DMSO, to a final DMSO concentration of 4% DMSO. Library compounds were screened at a concentration of 100 µM using the OneStep gradient injection method. Hits were selected based upon RU and kinetics and utilized for cell-based screening.
Viral plaquing assay
To screen compounds for antiviral activity, infected CRFK cells were treated with compounds in six-well replicates and compared with positive control wells (infected cells), negative controls (uninfected cells) and treatment controls (infected cells treated with a known effective antiviral compound) run concurrently on each tissue culture plate. CRFK cells were grown in 96-well tissue culture plates (Genesee Scientific) containing 200 µl of culture media. At ~75–85% cell confluency, the media in the uninfected control wells was aspirated and replaced with 200 µl of fresh media. The media in the infected wells was aspirated and replaced with media inoculated with FIPV at a multiplicity of infection (MOI) of 0.004 infectious virions per cell. At 1 h post-infection, each putative antiviral compound was added to six FIPV-infected wells and six uninfected control wells (to screen for compound cytotoxicity). All compounds were initially screened at 10 µM, except for the ‘chemical fragment’ compounds supplied by M Olsen (Midwestern University), which were assessed at 50 µM. The tissue culture plates were incubated for 72 h at 37°C and subsequently fixed with methanol and stained with crystal violet. Plates were scanned for absorbance at 620 nm using an ELISA plate reader (FilterMax F3 [Molecular Devices] and Softmax Pro [Molecular Devices]). The individual well absorbance values along with the average absorbance value and SEM for the six-well experimental replicates were recorded for each treatment condition.
For agents that demonstrated antiviral efficacy in the initial screening at 10 or 50 µM (protected from virus-associated CPE), the half-maximal effective concentration (EC50) was determined by performing a progressive two-fold compound dilution series in the viral plaquing assay. For EC50 determination, CRFK cells were grown in 96-well tissue culture plates similarly to that performed for the antiviral screening assay. Aside from the uninfected control wells, all remaining wells were infected with the FIPV as described above in two-fold dilution series ranging from 20 µM to 0 µM in six-well replicates. The number of dilution steps ranged from six to 14 and was compound dependent. Six-well replicates of uninfected CRFK cells served as a control for normal CRFK cells; six-well replicates of CRFK cells infected with FIPV served as untreated, FIPV-infected control wells; and six-well replicates of FIPV-infected CRFK cells treated with GS-441524 served as control wells for protection against virus-induced cell death based on published data regarding the efficacy of GS-441524 use in vitro in CRFK cells. 16
Tissue culture plates were incubated for 72 h and subsequently fixed with methanol, stained with crystal violet and scanned for absorbance at 620 nm using an ELISA plate reader. The EC50 was calculated by plotting a non-linear regression equation (dose–response curve) using Prism 8 (GraphPad).
Viral RNA knock-down assay
RT-qPCR assays were used to quantify compound inhibition of viral RNA production. CRFK cells were cultured in a six-well tissue culture plate (Genesee Scientific). At approximately 75–85% cellular confluency, the culture media was replenished with fresh media and the cells were infected with FIPV serotype II at a MOI of 0.2. One hour post-infection, FIPV-infected wells were treated with one (monotherapy), two or three (cACT) antiviral compounds performed in triplicate. Compound dosage was based upon the compounds’ EC50 and ranged from 0.001 to 20 µM. Triplicate wells of FIPV-infected and untreated CRFK cells acted as virus-infected controls. All cell culture wells were subsequently incubated for 24 h, and cell-associated total RNA was isolated using the PureLink RNA mini kit (Invitrogen). The RNA was treated with DNAse (TURBO DNase; Ambion), reverse transcribed to cDNA using a High-Capacity RNA-to-cDNA Kit (Applied Biosystems) and FIPV cDNA and feline GAPDH cDNA were measured using RT-qPCR, as described above. Fold reduction in viral titer was determined by dividing the normalized average FIPV RNA copy number for untreated, FIPV-infected CRFK cells into the normalized average FIPV RNA copy number for treated CRFK cells with the compound(s) of interest. The expected additive effect was determined by adding the fold reduction for each monotherapy treatment used in combination. Foldover additive effect was determined by dividing the predicted additive effect into the combined fold reduction value for the particular combined therapy of interest.
Determination of cytotoxicity safety profiles
Compound cytotoxicity in feline cells was assessed using the commercially available kit (CellTox Green Cytotoxicity Assay; Promega) according to the manufacturer’s instructions. Untreated CRFK cells were used as negative controls and cells treated with a cytotoxic solution provided by the manufacturer was used as the positive toxicity control. Briefly, in addition to the control wells, CRFK cells were plated in 96-well tissue culture plates (Genesee Scientific) in four-well replicates with 5, 10, 25, 50 or 100 µM concentrations of the compound of interest and were incubated for 72 h. After 72 h, the kit DNA binding dye was applied to all wells, incubated at 37°C, shielded from light for 15 mins and the fluorescence intensity determined at 485–500 nmEx/520–530 nmEm using a plate reader (FilterMax F3 [Molecular Devices] and Softmax Pro [Molecular Devices]). Compound cytotoxicity at a particular concentration was assumed to be proportional to the intensity of fluorescence based on the selective penetration and binding of the dye to the DNA of degenerate, apoptotic or necrotic cells. The cytotoxicity range was determined by setting the fluorescence value for cells treated with the positive control reagent as 100% and the untreated feline cells as 0% cytotoxicity. The mean fluorescence value for the four wells containing each compound concentration were then interpolated as a percentage (percent cytotoxicity) ranging from 0% to 100%. Clofazimine cytotoxicity was determined by dosing monolayers of CRFK cells in a 96-well plate with serial dilutions of the compound starting at a maximum dose of 100 µM, incubated for 72 h and then stained with crystal violet and scanned for absorbance at 620 nm using an ELISA plate reader. The 50% cytotoxic concentration (CC50) was calculated by plotting a non-linear regression equation (dose–response curve) using GraphPad Prism version 8.
Results
Compound screening
In order to identify compounds with anti-FIPV activity, a compilation of 90 compounds (Figure S1 in the supplementary material) from differing drug classes and with a variety of reputed mechanisms of antiviral action were screened for anticoronaviral activity in in vitro assays. Compounds screened included nucleoside polymerase inhibitors (NPIs), non-NPIs, protease inhibitors (PIs), NS5A inhibitors, a set of anti-helicase chemical ‘fragments’ 24 and a set of compounds with undetermined or multiple antiviral mechanisms of action. From this group of 90 compounds, 26 different compounds were determined to possess antiviral activity against FIPV, including NPIs, PIs, NS5A inhibitors and three compounds with undetermined mechanisms of action (termed ‘other’; Figure 1). The antiviral compounds that demonstrated efficacy against FIPV included toremifene citrate, daclatasvir, elbasvir, lopinavir, ritonavir, nelfinavir mesylate, K777/K11777, grazoprevir, amodiaquine, EIDD1931, EIDD2801, clofazimine and GS-441524 sourced from three different China-based manufacturers (Figure 2). We tested several nucleoside analog compounds provided by Gilead Sciences structurally related to the nucleoside analogs GS-441524 and remdesivir for their antiviral properties and found several with potential (included in the above-reported 26 identified compounds), but we did not pursue these agents further. As a result, the total number of antiviral agents carried forward for further analyses was 14. This total includes the previously identified 3-C protease inhibitor, GC376 (Anavive Lifesciences).

Compounds screened by mechanism of action. (a) Pie graph depiction of all compounds screened. (b) Compounds identified during screening to possess anti-feline infectious peritonitis virus (FIPV) activity in vitro

Example screening plaque assay using crystal violet staining to identify anti-feline infectious peritonitis virus (FIPV) activity at 10 µM. The top left row shows control wells with Crandell-Rees feline kidney (CRFK) cells only and no drug or FIPV. The top right row shows a positive control using GS-441524 with known complete protection of CRFK cells against FIPV-induced cell death. The entire bottom row of wells represents CRFK cells infected with FIPV and no drug treatment. The remaining rows are screening wells with the left half assessing for cytotoxicity at 10 µM (no FIPV infection) and the right half assessing for anti-FIPV activity at 10 µM for any given compound. Loss of staining indicates cell loss. Daclatasvir and velpatasvir demonstrated anti-FIPV activity evidenced by increased crystal violet staining (relatively intact cell monolayers) relative to FIPV-only control wells (bottom row of plate). Ombitasvir, pibrentasvir and ravidasvir demonstrated an absent-to-minimal antiviral effect, with ravidasvir also demonstrating cytotoxicity at 10 µM based on the dramatic well clearing seen on the left half of the plate without FIPV
Determining antiviral efficacy
The EC50 (antiviral efficacy) was determined for 10 antiviral compounds. For these compounds, the EC50 ranged from 0.04 µM to 13.47 µM (Figure 3). One of the antiviral agents – daclatasvir – demonstrated unacceptable cytotoxicity at 20 µM and was removed from further testing. GS-441524 sourced from China (HY-103586; MedChemExpress) was shown to have a comparable EC50 relative to previously published values for GS-441524 sourced from Gilead Sciences, 15 and the EC50 for GC376, previously reported as 0.04 µM, 25 was determined in our laboratory to be 0.19 µM.

(a) Half-maximal effective concentration (EC50) values and (b) representative non-linear regression analyses for two of the compounds with anti-feline infectious peritonitis virus (FIPV) activity. Serial dilutions of each compound with anti-FIPV activity were performed to identify the EC50. GS-441524 results shown here represent the compound sourced from MedChemExpress (MCE)
Cytotoxicity safety profiles
Cytotoxicity safety profiles (CSPs) were determined by using the Promega CellTox Green Cytotoxicity Assay for 11 different antiviral compounds in CRFK cells. At 5 µM, seven of the tested compounds demonstrated essentially no cytotoxicity, while two of the antivirals – amodiaquine and toremifene – had 11% and 12% cytotoxicity, respectively (Figure 4). The CC50 for GC376 has been reported previously as >150 µM. 25 The CC50 for clofazimine was determined to be 8.3 µM. Interestingly, based upon the Promega CellTox Green Cytotoxicity Assay, the cytotoxicity of both EIDD compounds was essentially undetectable up to 100 µM. However, visual inspection of the EIDD-treated tissue culture wells just prior to fluorescent dye application revealed evidence of agent-associated CPE. The untreated CRFK cells featured an adherent spindloid morphology in a closely packed monolayer, while the EIDD-treated wells demonstrated an overt decrease in cellular confluency with variable cell morphology, including rounding and detachment of cells (CPE). The discordance between the subjective visual assessment of EIDD-treated wells and the results of the fluorescence assay (CellTox Green) remains enigmatic. It is possible that the agent-associated reduction in cell number in EIDD-treated wells resulted in loss and degradation of nucleic acid necessary for fluorescence binding and detection in the CellTox Green assay. These results suggest that the EIDD compounds may be associated with a greater degree of agent-associated cytotoxicity than the CellTox Green assay results indicate.

Cytotoxicity safety profiles and representative graphs. (a) Compounds with demonstrated anti-feline infection peritonitis virus (FIPV) activity and the percent cytotoxicity at 5, 10, 25, 50 and 100 µM. (b) Representative percent cytotoxicity bar graphs ± SD for two of the compounds with anti-FIPV activity. Percent cytotoxicity values were determined by normalizing cytotoxicity to the positive toxicity control wells (100% cytotoxicity) and untreated Crandell-Rees feline kidney control cells (0% cytotoxicity)
Quantification of compound inhibition of viral RNA production with monotherapy
An RT-qPCR assay was used to measure each antiviral compound’s ability to inhibit coronaviral transcription as monotherapy (viral RNA knock-down assay). Compounds demonstrating the greatest inhibition of FIPV RNA production were GC376 (a 3C-like coronavirus protease inhibitor), GS-441524, EIDD1931 and EIDD2801, the latter three all being nucleoside analogs (Figure 5). Those with the least inhibitory effect on viral RNA transcription include elbasvir, nelfinavir and ritonavir. Ritonavir – a protease inhibitor – is used in combination with lopinavir to treat HIV-1 infection (Kaletra; AbbVie). Lopinavir monotherapy has poor oral bioavailability in people; however, when used in combination, ritonavir has been demonstrated to markedly improve lopinavir’s plasma concentration. 26 Therefore, despite the relatively minimal FIPV inhibition identified with ritonavir as monotherapy, this compound was assessed for combined anticoronaviral efficacy with ritonavir.
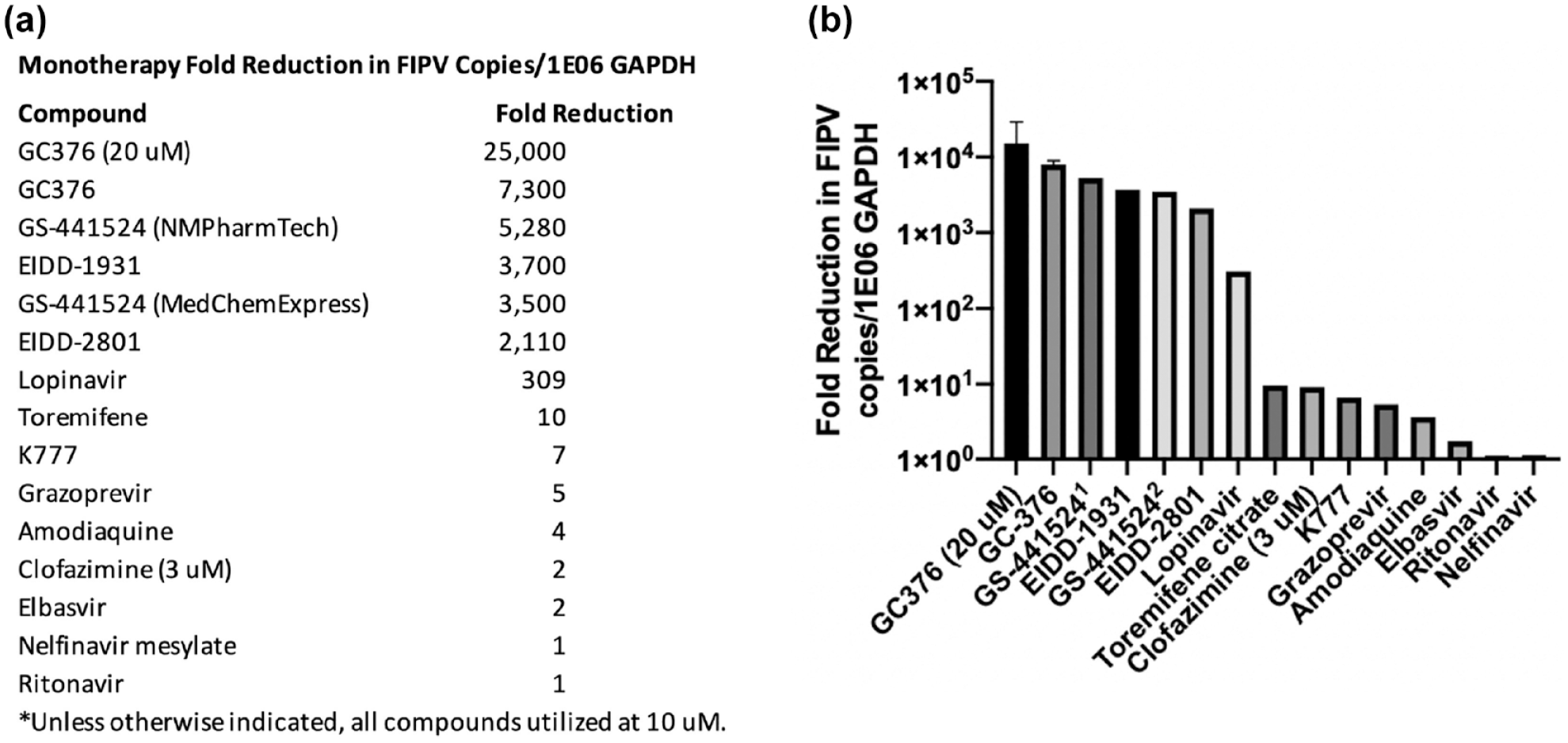
Fold decrease in feline infectious peritonitis virus (FIPV) RNA copy number using antiviral compounds as monotherapy. (a) FIPV-infected Crandell-Rees feline kidney cells were incubated for 24 h with various antiviral compounds. Viral copy number was subsequently determined via quantitative RT-PCR and normalized to feline GAPDH copy number, in order to determine the compound’s fold decrease effect. All compounds were tested at 10 µM unless otherwise specified. All experimental treatments were performed in triplicate wells and the fold decrease calculated by dividing the average experimental, normalized FIPV copy number by the average normalized FIPV copy number determined for untreated, FIPV-infected wells. (b) Graphical representation of fold decrease values in FIPV RNA copy number. Graph bars with SD error bars represent experiments that were repeated. 1 GS-441524 sourced from NMPharmTech (China); 2 GS-441524 sourced from MedChemExpress (China)
Quantification of compound inhibition of viral RNA production with cACT
To identify drug combinations with additive or foldover-additive (synergistic) antiviral activity over monotherapy, combinations of two or more drug compounds were selected based upon: (1) established combinations used for other viral infections like HIV-1 and HCV; (2) drugs with different mechanisms of action; (3) potential variations in systemic distribution of the compound (eg, predicted ability to penetrate the blood–brain or blood–ocular barriers); and (4) minimal cytotoxicity (based on the CSP). For each cACT, any resulting decrease in FIPV copy number over the calculated additive effect for each drug used as monotherapy was considered to be synergistic (Figure 6).

Additive and foldover-additive reduction in feline infectious peritonitis virus (FIPV) viral RNA copy number using combined anticoronaviral therapy. The expected additive effect reflects the sum of the fold reduction in viral RNA based on each agent used as monotherapy (from Figure 5). (a) List of anti-FIPV combinations with associated fold reduction and foldover-additive reduction in FIPV copies normalized to 1 × 106 GAPDH copies. (b) Bar graph demonstrating comparison between overall fold reduction in FIPV copies and foldover-additive values. 1 = GC376 (20 µM) + amodiaquine + toremifene; 2 = GC376 (20 µM) + K777; 3 = GC376 (20 µM) + toremifene; 4 = GC376 (20 µM) + nelfinavir mesylate; 5 = GC376 (20 µM) + clofazimine (3 µM); 6 = GS-441524 (NMPharmTech) + clofazimine (3 µM); 7 = elbasvir (5 µM) + lopinavir; 8 = elbasvir (5 µM) + GC376 (20 µM); 9 = GC376 (10 µM) + amodiaquine; 10 = GC376 (10 µM) + grazoprevir; 11 = GC376 (20 µM) + GS-441524 (NMPharmTech); 12 = GC376 (10 µM) + amodiaquine + elbasvir (5 µM); 13 = GC376 (10 µM) + GS-441524 (MCE); 14 = GC376 (20 µM) + ritonavir; 15 = K777 + lopinavir; 16 = GC376 (10 µM) + GS-441524 (NMPharmTech); 17 = GC376 (20 µM) + lopinavir; 18 = lopinavir + ritonavir; 19 = lopinavir + ritonavir + toremifene; 20 = GC376 (10 µM) + EIDD-2801; 21 = K777 + toremifene
Owing to the pronounced anti-FIPV activity of GC376, as well as its potential availability for moving forward into in vivo pharmacokinetic studies and clinical applications, this compound was focused on in a series of mono- and combined viral RNA knock-down assays. Overall, 20 µM of GC376 demonstrated superior anti-FIPV activity when used as monotherapy and in combinatorial therapies in vitro. Although the combination of elbasvir and lopinavir demonstrated the highest cACT synergy, this combination was ultimately less effective than either GC376 or GS-441524 used as monotherapy. Interestingly, cACT using GC376 or GS-441524 resulted in no or limited detectable synergy.
Discussion
As there is no currently effective vaccine for FIP, there is an immediate clinical need for effective, affordable and available antiviral treatment options for FIPV-infected cats. Here we describe the in vitro screening of 90 compounds, resulting in the identification of 26 antiviral agents with antiviral efficacy against the feline coronavirus, FIPV serotype II. Based on the results of both the plaquing and viral RNA inhibition (RT-qPCR) assays, the most effective antiviral compounds that were identified include GC376, GS-441524, EIDD2801 and EIDD1931. Importantly, the agents demonstrating the least cytotoxicity include GC376 and GS-441524, with no evidence of toxicity up to 100 µM.
We also documented antiviral efficacy for combinations of antiviral agents (cACT). A few of the examined drug combinations demonstrated evidence of limited synergistic antiviral activity. cACT synergism, determined using RT-qPCR, was defined as the fold reduction in viral copy number beyond the additive effect (combined reduction in viral copy number for each individual agent). The cACT with the greatest foldover-additive (synergistic) effect was determined to be elbasvir and lopinavir. However, all the cACTs proved to be ultimately less effective than GC376, GS-441524 and both EIDD compounds used as monotherapies. This may be the result of the relative effectiveness of these agents when used as monotherapies in our in vitro culture-based systems and an assay-limited inability to identify an antiviral effect beyond a certain level of viral inhibition. Attempts were made to further optimize the detection of compound synergy by modifying the experimental protocol without success. It remains possible that combinations of agents may manifest synergy in in vivo studies. In addition, combinatorial therapy may also serve as a method to prevent the development of antiviral resistance. However, this would require further investigation using long-term culture, viral sequencing and in vivo studies, and is beyond the scope of this study.
Repeated plaque assay data indicated that elbasvir prevented the formation of viral plaques with an impressive EC50 of 0.16 µM. However, there was essentially no detectable difference in the viral RNA copy number between infected cells treated with or without elbasvir. Subjective visual analysis of FIPV-infected CRFK cells that were treated with elbasvir revealed evidence of CPE. Interestingly, the treated cells with evidence of CPE did not appear to detach from the culture plate and, as a result, the absorbance values acquired in the plaque assay were comparable to uninfected control wells. This discordant result between the plaquing and viral RNA knock-down assay results suggests that the putative antiviral effect of elbasvir may be downstream of viral transcription (eg, translation, virion assembly and other) and, as a result, elbasvir may reduce overall viral replication but not protect cells from the accumulation of viral RNA and cellular injury.
Prior in vivo clinical successes using GS-441524 or GC376 in cats with experimental and naturally occurring FIP demonstrate that an effective antiviral cure for FIP is an achievable goal. However, therapeutic challenges in treating non-effusive (granulomatous), multisystemic neurologic and ocular FIP remain. A cACT utilizing a compound(s) with effective penetration into these anatomic reservoirs (brain and eye) may be required to achieve a cure in multisystemic FIP. The 3C protease inhibitor GC376 has been demonstrated to be effective in the treatment of experimental FIPV infection but appears to be less effective in treating and eradicating the more chronic neurologic or ocular forms of the disease. 13 Optimizing the dose and administration of GC376 to establish an effective tissue distribution in the brain and eye may improve the overall clinical success of this compound.
Conclusions
This study reports the screening of 90 putative anti-viral agents and the identification of 26 compounds with variable anti-FIPV activity. Further, we designed strategic combinations of efficacious anti-FIPV compounds based on differing antiviral mechanisms of action to determine the presence of additive or synergistic activity. Although evidence of compound synergy was identified, overall, the most effective antiviral compounds were determined to be specific monotherapies, mechanistically featuring either a nucleoside analog or protease inhibitor.
Supplemental Material
Figure S1
Complete list of compounds screened for anti-FIPV activity
Footnotes
Supplementary material
The following file is available online:
Figure S1: Complete list of compounds screened for anti-FIPV activity.
Conflict of interest
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
We are grateful for the generous funding provided by the EveryCat Health Foundation (formerly Winn Feline Foundation – W17-020, W19-026 and W20-003) and the University of California, Davis Center for Companion Animal Health (CCAH) through gifts specified for FIP research by multiple individual donors and organizations (SOCK FIP, Davis, CA, USA) and private foundations (Philip Raskin Fund, Kansas City, KS, USA).
Ethical approval
This work did not involve the use of animals and therefore ethical approval was not specifically required for publication in JFMS.
Informed consent
This work did not involve the use of animals (including cadavers) and therefore informed consent was not required. No animals or people are identifiable within this publication, and therefore additional informed consent for publication was not required.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.