
Abstract
Objectives
Hyperammonemia occurs in cats with hepatobiliary and nutritional (cobalamin and arginine deficiency) disorders, and has also been documented in four cats with renal azotemia. We hypothesized that in cats with renal azotemia, fasting hyperammonemia would correlate with indices of worsening kidney function, and would be independent of cobalamin, potassium, systemic inflammation or urinary tract infection (UTI) with urease-producing bacteria.
Methods
A fasted blood sample was prospectively collected for ammonia and cobalamin analysis from 18 client-owned cats with renal azotemia (creatinine [Cr] ⩾1.6 mg/dl, urine specific gravity <1.030 or documentation of historical chronic kidney disease [CKD]). Correlations between blood ammonia and selected biochemical parameters were analyzed using Pearson’s correlation coefficient.
Results
Seven castrated males and 11 spayed females with a median age of 12 years (range 4–19 years) were enrolled. Ten of 18 (56%) cats presented for acute kidney injury (AKI) or acute on chronic kidney disease (AoCKD), and 8/18 (44%) presented for progressive CKD. The median Cr was 5.9 mg/dl (range 1.9–24.7 mg/dl). Hyperammonemia was documented in 4/18 (22%) cats, with a median of 95 µmol/dl (range 85–98 µmol/dl), and all four of these cats were classified as AKI/AoCKD. Blood ammonia concentrations had a significant moderate positive correlation between blood urea nitrogen (BUN) (r = 0.645, P = 0.003), Cr (r = 0.578, P = 0.012) and serum phosphorus (r = 0.714, P = 0.0009) but not with cobalamin, potassium or white blood cell count. No cats had UTIs with urease-producing bacteria.
Conclusions and relevance
A correlation exists between blood ammonia and BUN, Cr and phosphorus in cats with renal azotemia. Future studies are warranted in a larger population of cats to determine the true prevalence, etiology and potential therapeutic effect of medical management of hyperammonemia on long-term prognosis in cats with kidney disease.
Keywords
Introduction
Blood ammonia is derived primarily from the action of the intestinal biome in breaking down protein and amino acids in the colon. Protein degradation results in urea production, which is acted upon by urease-producing bacteria to produce ammonia. Additionally, enterocytes use glutamine as an energy source, the breakdown of which also produces ammonia.1–3
To keep blood ammonia within the normal range, the body utilizes various organs, including the liver, kidneys, intestines, muscles and brain. The main mechanism of detoxification is the urea cycle in the liver, through which ammonia, primarily derived from the intestine, is converted into urea.1–3 Urea is water soluble and can be excreted by the kidneys into the urine. The liver, intestines, muscles and brain detoxify ammonia outside of the urea cycle through the conversion of ammonia and glutamate to glutamine via glutamine synthetase.1–3
If there is an increased ammonia load in the body, then blood ammonia levels rise. Ammonia is a gas that can freely traverse cellular membranes, including the blood–brain barrier. Elevated brain ammonia concentration causes astrocytes to swell and alters neurotransmission in the brain, leading to the development of neurologic signs.1–3 Effective therapies to treat hyperammonemia exist and include medications that decrease ammonia production in the gastrointestinal tract, increase ammonia detoxification in non-hepatic tissues or correct precipitating factors such as dehydration, hypokalemia, metabolic alkalosis or infection with ammonia-producing bacteria. 4
Hyperammonemia resulting from impaired detoxification can be primary or secondary. 3 Primary hyperammonemia accompanies urea cycle enzyme deficiencies or transporter defects, and is reported rarely in dogs and cats.5–7 Secondary hyperammonemia is more common and occurs when the urea cycle is indirectly affected by substrate deficiencies or systemic metabolic derangements, or bypassed by the shunting of portal blood directly to the systemic circulation. In cats, secondary hyperammonemia is most commonly due to hepatic disease (congenital or acquired portosystemic shunting), 8 but there are also reports of hyperammonemia secondary to cobalamin and arginine deficiency.9–12 Urinary tract infections (UTIs) with urease-producing bacteria have been reported to cause hyperammonemia in dogs, although a similar finding has not been reported in cats. 13 In humans, non-hepatic hyperammonemia has also been reported with hematologic neoplasms, the use of certain drugs (eg, valproic acid, carbamazepine, asparaginase) and increased protein loading (total parenteral nutrition, gastrointestinal bleeding, status epilepticus).1,14 Recently a retrospective study of hyperammonemia in the intensive care setting also identified acute kidney injury as a potential cause. 15
Fasting hyperammonemia was reported in four cats with renal azotemia, though an underlying mechanism was not identified. 16 We speculated that hyperammonemia may be an underreported phenomenon in cats with kidney disease. Thus, the objective of the current study was to document blood ammonia concentrations in cats that presented for evaluation of renal azotemia. We hypothesized that fasting blood ammonia will correlate with blood urea nitrogen (BUN), creatinine (Cr) and phosphorus, but will be independent of other risk factors for the development of hyperammonemia.
Materials and methods
Study population
Eighteen client-owned cats that presented to the Foster Hospital for Small Animals at Cummings School of Veterinary Medicine at Tufts University between April 2016 and September 2017 for assessment or monitoring of renal azotemia were enrolled in this prospective pilot study. The patient’s signalment, current and prior medical history, body condition score (BCS), diet and mentation (bright, quiet, dull, comatose) were routinely noted in the medical record upon admission. Inclusion criteria were historical or newly diagnosed kidney disease with confirmed presence of renal azotemia (Cr ⩾1.6 mg/dl; urine specific gravity <1.030). Cats were classified as having acute kidney disease (AKI)/acute on chronic kidney disease (AoCKD) based on a combination of history (acute presentation of clinical signs, known nephrotoxin exposure, UTI, ureteral obstruction), physical examination findings (pyrexia, renomegaly, pain on renal palpation), laboratory findings (pyuria, neutrophilic leukocystosis, newly documented or acutely progressive azotemia) and/or imaging findings suggestive of acute injury such as obstructive uropathy (hydronephrosis, hydroureter), renomegaly, pyelectasia, pyonephrosis or perinephric steatitis. Cats were classified as having CKD based on medical records of referral veterinarians when available with history of renal azotemia >3 months, consistent diagnostic imaging findings of decreased kidney size, irregular shape or parenchymal changes, and lack of historical, laboratory and imaging findings described previously that would be supportive of an acute component to their disease.
Exclusion criteria were congenital portosystemic shunts or other pre-existing liver disease (defined as serum alanine aminotransferase or alkaline phosphatase activity >1.5 upper limit of normal), cobalamin deficiency (cobalamin <150 ng/l), lactulose or probiotic administration and UTI with urease-producing bacteria. Owners signed an informed consent form and the Foster Hospital Clinical Science Review Committee approved the study.
Hematologic analysis
Following enrollment, a fasted (>12 h) sample of 3 ml of whole blood was collected by jugular venipuncture. Approximately 0.5 ml of whole blood was placed into a lithium heparin microtube for the initial ammonia level to be performed by the Foster Hospital Clinical Pathology Laboratory. The sample was placed on ice and brought immediately to the laboratory for centrifugation and in-house analysis (Roche Cobas 501c Analyzer; reference interval [RI] 30–65 µl). The remaining 2.5 ml of whole blood was placed into a serum separator tube and submitted for a cobalamin level (IDEXX 2013; RI 276–1425 ng/l).
A packed cell volume (PCV), total solids (TS), BUN, Cr, phosphorus and potassium were obtained at the Foster Hospital within 24 h of enrollment (Cobas C 501; RI for BUN 15–33 mg/dl, RI for Cr 0.9–21 mg/dl, RI for phosphorus 3.0–6.3 mg/dl and RI for potassium 3.4–5.2 mEq/l). A complete blood count, biochemical profile and urinalysis were performed either at the Foster Hospital Clinical Pathology Laboratory or at a referring veterinary hospital within 2 weeks of enrollment. Sixteen of 18 cats had a urine culture and sensitivity performed. The remaining two cats without cultures had inactive sediments.
Statistical analysis
Data were tested for normality by inspection of histograms and evaluation of kurtosis and skew. Owing to a mixture of normal and non-parametric data, all data were expressed as median ± range. Pearson’s correlation coefficient after log transformation of the data, if necessary, was used to determine the relationships between blood ammonia and BUN, Cr, phosphorus, cobalamin, potassium, PCV, TS and white blood cell (WBC) count. Statistical significance was defined as a P value <0.05.
Results
Seven castrated male cats and 11 spayed female cats were enrolled, with a median age of 12 years (range 4–19 years). There were 12/18 (66.7%) domestic shorthairs, 2/18 (11.1%) domestic mediumhairs, 2/18 (11.1%) domestic longhairs, 1/18 (5.5%) Siamese and 1/18 (5.5%) Ragdolls. Based on a review of the clinical history, laboratory parameters and imaging findings, 10/18 (55.5%) cats presented for AKI or AoCKD, and 8/18 (44.4%) presented for progressive CKD. Selected biochemistry values from this population of cats are included in Table 1, and complete baseline information for all cats is included in Table 1 in the supplementary material. All cats had increased BUN and Cr, with 9/18 (50%) also having increased phosphorus.
Selected clinical parameters in 18 cats with renal azotemia

RI = reference interval; PCV = packed cell volume; TS = total solids; WBCs = white blood cells; ALP = alkaline phosphatase; ALT = alanine aminotransferase; BUN = blood urea nitrogen; NH3 = ammonia
Hyperammonemia was documented in 4/18 (22.2%) cats, none of which showed overt neurologic signs or had a documented infection with urease-producing bacteria. Table 2 compares subgroups of cats with and without increased blood ammonia. Of the hyperammonemic cats, median ammonia was 95 µmol/l (range 85–98 µmol/l). The four hyperammonemic cats were profoundly azotemic, with a median Cr of 11 mg/dl (range 6.8–24.7 mg/dl) and a median BUN of 212 mg/dl (range 160–261 mg/dl).
Comparison of cats with and without increased blood ammonia concentration

P <0.005†P <0.05
NH3 = ammonia; F = female; M = male; BCS = body condition score; PCV = packed cell volume; TS = total solids; WBCs = white blood cells; ALP = alkaline phosphatase; ALT = alanine aminotransferase; BUN = blood urea nitrogen
As depicted in Figure 1, blood ammonia concentrations had a significant moderate positive correlation with BUN (r = 0.645, P = 0.003), Cr (r = 0.578, P = 0.012) and phosphorus (r = 0.714, P = 0.0009). There was no correlation between blood ammonia and potassium (r = 0.045, P = 0.786), PCV (r = 0.09, P = 0.876), TS (r = 0.091, P = 0.854), WBC count (r = −0.218, P = 0.31) or BCS (r = 0.409, P = 0.092).
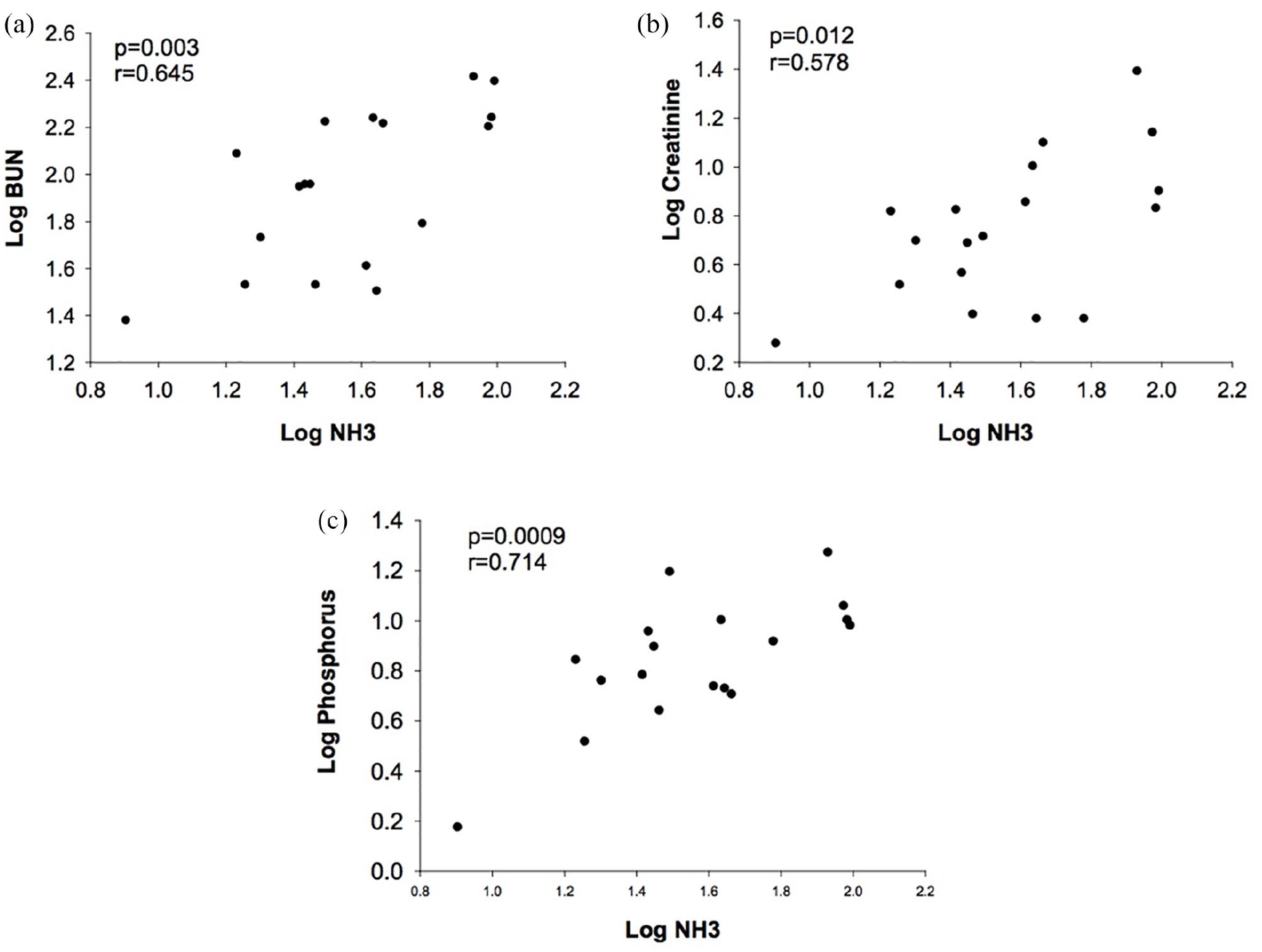
Correlations between blood ammonia concentration and indices of kidney function. Scatterplots showing the relationship between blood ammonia (NH3) concentration and (a) blood urea nitrogen (BUN), (b) creatinine (Cr) and (c) phosphorus. The data were log transformed and underwent testing for a significant correlation with Pearson’s correlation coefficient. A P value <0.05 was considered significant
Cats with AKI/AoCKD (n = 10) and those with progressive CKD (n = 8) are compared in Table 3. Cats with AKI/AoCKD had significantly higher blood ammonia, BUN, Cr and phosphorus than cats that presented with CKD. All four cats with high blood ammonia concentrations had AKI/AoCKD. There was no difference in cobalamin, PCV, TS or WBCs between the two groups; however, cats with AKI/AoCKD had higher BCSs (5.5, range 3–8) than cats with CKD (3.5, range 2–5).
Biochemical parameters in cats with with acute (AKI) or acute on chronic kidney disease (AoCKD) and chronic kidney disease (CKD)

Significantly different P <0.05
RI = reference interval; NH3 = ammonia; BUN = blood urea nitrogen; WBCs = white blood cells
Three cats had cobalamin below the RI (median 243 ng/ml, range 205–270 ng/l), but none had severe hypocobalaminemia previously reported to be associated with hyperammonemia in cats.9,10 There was no correlation between blood ammonia and cobalamin (r = 0.025, P = 0.92).
Discussion
In the current study, hyperammonemia was documented in 4/18 (22%) cats with renal azotemia and was positively correlated with BUN, Cr and phosphorus. Cats with an acute component of their kidney disease had the highest blood ammonia levels, likely reflecting more severe azotemia in this group. The strongest correlation identified was between blood ammonia and serum phosphorus. Increases in serum phosphorus predict progression of stable CKD in cats and are associated with decreased survival.17–20 Less is known about phosphorous in AKI/AoCKD, but recent studies in people and dogs suggest it may also serve as a biomarker of disease severity in this context as well.21–23 The pathophysiology for hyperammonemia in cats with kidney disease is unknown, but various potential contributing factors will be explored.
Hyperammonemia in cats with kidney disease could be due to decreased elimination owing to disruption of the hepatic urea cycle or increased ammonia load. In rats, argininosuccinate lyase, a key enzyme in the urea cycle, is inhibited by the presence of high urea.24,25 However, little is known about the regulation of the urea cycle enzymes in cats. Increased protein (ammonia) loading in cats with renal azotemia could occur with the feeding of high-protein diets, gastrointestinal bleeding, infections with urease-producing bacteria or excessive protein catabolism. Diet history was known for 14/18 cats in the current study, and all were on commercial cat foods, none of which had excessive protein levels. None of the cats in the current study had UTIs with urease-positive organisms or had clinical signs compatible with gastrointestinal bleeding such as melena, hematemesis or a recent drop in PCV. Although sarcopenia associated with muscle catabolism is common in cats with CKD, the BCS of the cats in this study was not correlated with blood ammonia.
Renal ammoniagenesis in response to acute or chronic metabolic acidosis or hypokalemia might contribute to hyperammonemia in this population. Renal ammoniagenesis plays an important role in maintaining systemic acid–base balance. Although the bulk of the ammonia produced is excreted in the urine, there is some spillover into the systemic circulation.26,27 In CKD, the kidneys progressively lose their ability to excrete acids, which leads to chronic metabolic acidosis. This acidosis triggers renal ammoniagenesis. 28 Similarly, with hypokalemia, the lack of potassium ions leads to a compensatory increase in intracellular hydrogen ions in the tubules, resulting in intracellular acidosis and renal ammoniagenesis. 27 The influence of these factors in this study cannot be determined, although at the time of evaluation none of the cats was hypokalemic. While some cats had clinical determination of their pH during hospitalization, this was not consistently performed at the same time of sampling for blood ammonia. Studies that measure acid–base status and urinary ammonia excretion of azotemic cats will be necessary to determine if renal ammoniagenesis contributes to blood hyperammonemia.
Hyperammonemia in azotemic cats could result from alterations in the gut microbiome that lead to increased gastrointestinal ammonia production and/or absorption. In humans with kidney disease, decreased renal excretion of urea leads to increased secretion of urea into the gastrointestinal tract, which is converted by urease-producing bacteria to ammonia.29–31 Studies in uremic humans with kidney disease show that there are changes in the gastrointestinal environment, which result in decreased microbiome diversity and dysbiosis.31–33 In this dybiosis, there is expansion of bacterial families that produce urease and other enzymes associated with the formation of uremic toxins (eg, uricase, indole and p-cresol-forming enzymes).31–33 Additionally, microbial dysbiosis can alter gut barrier function, which increases risk for translocation of uremic toxins, endotoxins and bacteria into the systemic circulation. 33 A recent study of the fecal microbiome of cats with CKD showed decreased microbiome diversity and increased serum indoxyl sulfate, 34 and a follow-up study by the same authors evaluating fecal fatty acid concentrations in cats with CKD showed evidence for protein malassimilation in this population. 35 As such, it is possible that a combination of protein malassimilation and increased delivery to the colon could be contributing to increased ammonia production. Additionally, although none of our cats presented with a history of overt tenesmus or fecal retention on imaging, it would be impossible to completely rule out a role for sublinical constipation.
Other known causes of hyperammonemia were unlikely in our population of cats. Congenital portosystemic shunting would be unusual given the advanced age of our cats and the lack of clinical signs earlier in life. Acquired shunting secondary to portal hypertension from hepatobiliary disease would be accompanied by some clinical (icterus, ascites) or biochemical evidence of synthetic liver failure (increased liver enzymes or hyperbilirubinemia). Although we did not measure plasma arginine and thus cannot rule out hypoargininemia 11 as a cause of hyperammonemia in our population, nutritional arginine deprivation is unlikely, as the cats in this study were fed commercial diets. Hypoargininemia secondary to azotemia alone is also unlikely, as a study has already demonstrated no difference between plasma arginine in azotemic and non-azotemic cats. 36 However, a recent case report did demonstrate acquired hypoargininemia in a cat with concurrent CKD and inflammatory bowel disease. 12
Hyperammonemia secondary to severe hypocobalaminemia has been reported in the dog 37 and cat.9,10 Cobalamin deficiency results in increased methylmalonic acid (MMA), which indirectly inhibits carbamoyl phosphate synthetase 1, the rate-limiting enzyme of the urea cycle. 11 Three cats in our population were found to have mild cobalamin deficiency, but there was no correlation between blood ammonia and cobalamin levels. However, MMA concentration, considered a more accurate marker of cobalamin deficiency at the cellular level,38 was not measured.
Hyperammonemia could have several consequences in cats with kidney disease. First, it can lead to varying degrees of hepatic encephalopathy (HE) owing to increased brain ammonia concentrations. This HE might be manifested clinically in many ways from mild decreases in mentation, altered behavior or inappetence to more severe signs such as altered mentation or seizures. Accumulating evidence also suggests that hyperammonemia/HE is a proinflammatory and procoagulant state,39–44 both of which could fuel progression of kidney disease. Hyperammonemia has also recently been linked to the generation of sarcopenia by various mechanisms, including increased expression of myostatin, increased phosphorylation of eukaryotic initiation factor 2a, mitochondrial dysfunction and increased autophagy-mediated proteolysis.45,46 Loss of muscle mass and/or strength is a frequent occurrence in cats with late-stage CKD. Treatments to address hyperammonemia, such as reduction in dietary protein, L-ornithine L-aspartate, lactulose, probiotics and antibiotics, could be used to enhance the cat’s quality of life, modulate loss of muscle mass and potentially slow the progression of disease.
Our study had several limitations, including a small sample size, lack of comprehensive diet histories, lack of muscle condition scoring and difficulty in absolutely distinguishing between CKD, AKI and AoCKD. Additionally, four cats were undergoing antibiotic treatment at time of enrollment, which could have decreased ammonia production in the gastrointestinal tract. We opted to permit antibiotic administration in this pilot study owing to concerns that withholding antibiotics until blood ammonia sample collection could be dangerous in our population,47,48 and because, besides portosystemic shunting, the majority of disorders associated with hyperammonemia in people are not particularly responsive to antibiotic therapy.49–52 Some individual cats did not have as high blood ammonia as would be expected for their level of azotemia, which we cannot explain, but assume could be related to the aforementioned limitations. To better understand this phenomenon, future studies should consider enrolling cats prior to antibiotic therapy, the length of azotemia, carefully assessing dietary histories, precise assessment of BCS by a single individual, objective use of ultrasonographic techniques to determine the degree of sarcopenia, determination of blood and urine pH, assessment of arginine and citrulline levels in the blood and diet and analysis of the gastrointestinal microbiome.
Conclusions
Hyperammonemia is present in some cats with renal azotemia and it appears to correlate with the degree of azotemia. Larger studies are needed to determine its true prevalence and etiology.
Supplemental Material
Supplementary Table 1
The following file is available online: Study population baseline data
Footnotes
Supplementary material
The following file is available online: Table 1: Study population baseline data.
Conflict of interest
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article
Funding
The study was funded by the intramural Companion Animal Health Fund at Cummings School of Veterinary Medicine at Tufts University.
Ethical approval
This work involved the use of nonexperimental animals only (including owned or unowned animals and data from prospective or retrospective studies). Established internationally recognized high standards (‘best practice’) of individual veterinary clinical patient care were followed. Ethical approval from a committee, while not specifically required for publication in JFMS, was nonetheless obtained, as stated in the manuscript
Informed consent
Informed consent (either verbal or written) was obtained from the owner or legal custodian of all animal(s) described in this work (either experimental or non-experimental animals) for the procedure(s) undertaken (either prospective or retrospective studies). No animals or humans are identifiable within this publication, and therefore additional informed consent for publication was not required.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.