
Abstract
Objectives
Papillomaviruses (PVs) are ubiquitous host- and site-specific viruses. PV infections in cats are associated with oral papillomas, viral plaques, Bowenoid in situ carcinomas (BISCs), squamous cell carcinomas and sarcoids; this association is primarily based on PCR detection of PV DNA within said lesions. PV DNA is frequently detectable on normal feline skin; thus, it is possible that some of the implicated DNA is commensal rather than associated with lesion formation. Therefore, the aim of the present study was to use fluorescence in situ hybridization (FISH) to localize PV DNA within feline BISCs, to provide additional evidence that PV infection may influence the development of these neoplasms.
Methods
FISH probes targeting Felis catus papillomavirus type 2 (FcaPV2) DNA were used to localize FcaPV2 DNA within 42 BISCs from which FcaPV2 DNA had previously been amplified via PCR.
Results
Fifteen of 42 BISC lesions (35.7%) demonstrated intralesional FcaPV2 using FISH. Probe annealing was predominantly located within the nuclei of koilocytes found in the upper strata of the epidermis. Probes were typically scattered multifocally within the lesions; most commonly this was near the periphery of the BISCs.
Conclusions and relevance
These results confirm that a proportion of BISCs contain FcaPV2 DNA. These results further support a causative association between FcaPV2 and BISCs in cats.
Introduction
Papillomavirus (PV) infection of cats was initially reported in 1990, though 10 years passed before the first Felis catus papillomavirus (FcaPV) was sequenced in its entirety.1,2 Today, FcaPVs have been detected worldwide with a total of five FcaPV genomes fully sequenced.2–6 At present, four feline skin lesions are recognized to have an association with the presence of PV: oral papillomas, viral plaques, Bowenoid in situ carcinomas (BISCs) and squamous cell carcinomas (SCCs). Of these, BISCs and SCCs are considered to have the most aggressive clinical behaviors. Cats can also be asymptomatically infected by FcaPVs.7,8
SCCs are the most common malignant cutaneous neoplasm of cats. 9 BISCs, so named from a human corollary, are an in situ SCC variant. Grossly, these lesions have similar appearances: single-to-multifocal crusting plaques of the epidermis. Microscopically, SCCs are characterized by islands and trabeculae of neoplastic keratinocytes invading into the dermis, whereas BISCs consist of foci of thickened dysplastic epidermis and lack invasion into or through the basement membrane. 10 BISCs may progress to SCCs. 11 In some instances, both lesion types have been reported in the same cat. 10
In humans, in situ hybridization (ISH) and ISH variants, including fluorescence in situ hybridization (FISH), are commonly used to detect human papillomaviruses within pre-neoplastic and neoplastic lesions. The main advantage of ISH over conventional PCR is that it can be used to provide a spatial location of specific nucleic acids via annealing of a labelled complementary probe. As PVs frequently asymptomatically infect feline skin, the demonstration of PV DNA within the neoplastic cells is an important finding that supports the PVs as a cause of the lesions. Previously, a study used ISH to detect PV RNA within a series of feline cutaneous SCCs; 12 however, to our knowledge, there have been no previous studies using ISH to detect PV DNA in feline BISCs.
Therefore, the purpose of the present study was to use FISH to localize FcaPV2 DNA within a series of BISCs that had previously been shown to contain FcaPV2 via PCR. The confirmation that the FcaPV2 DNA was present within the neoplastic cells would support a possible causative role of the virus in lesion formation.
Materials and methods
Sampling
Forty-two BISCs were included in this study. DNA had previously been extracted from formalin-fixed paraffin-embedded (FFPE) tissue scrolls from each lesion and the FcaPV2 DNA had been amplified via PCR using the JMPF/JMPR primers. 7
FcaPV2 detection and partial-L1 ORF amplification for FISH DNA probe generation
FISH FcaPV2 L1 DNA probes were generated from partial amplicons of 177 bps using primer set JMPF/JMPR. PCR reactions were performed on a Mastercycler thermocycler (Eppendorf) in 30 µl with the final concentrations of reagents described previously. 7 Amplification conditions were 94°C for 10 mins, 45 cycles of 94°C for 90 s, 50°C for 90 s, 72°C for 90 s and 72°C for 5 mins. Confirmation of amplification was performed in a 1% agarose gel containing 1:30,000 SYBR Safe (Invitrogen) subjected to electrophoresis at 90 V for 35 mins and viewed on a UV transilluminator (Fischer Biotech). Products were purified using the QIAquick PCR purification kit (Qiagen) and subsequent DNA concentration and purity determined using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies).
Negative control probe generation
Nested primer set DFA/ILK/KGI/TGV/IYG 13 was used to amplify equine herpesvirus DNA for use as negative control probes. PCR reactions were performed on the previously described thermocycler. Initial reactions totalled 50 µl with the following concentrations of reagents: 1× PCR reaction buffer, 2 mmol/l MgCl2, 200 µmol/l of each dNTP, 1 µmol/l of each primer (DFA, ILK, KGI), 1 U/reaction Taq DNA polymerase and 2 µl DNA template with PCR-grade water (AstraZeneca) added to equal the final reactant volume. The combined mixture was then cycled as previously described. 13 Confirmation of amplification, purification and determination of concentration were performed as described above.
Genomic feline DNA was extracted from feline hepatocytes for non-viral-positive control probes via a DNEasy Blood and Tissue Kit (Qiagen) from fresh-frozen feline liver (Murdoch University Pathology Service). Resultant products were subjected to restriction enzyme digestion in 20 µl containing the following: 1 µg extracted DNA, 2 µl 10 × Buffer E (Promega), 1 µl EcoRI (Promega), 1 µl HindIII (Promega) and 11.85 µl deionized PCR-grade water (AstraZeneca) in a water bath at 37°C for 10 h. Products were then checked to ensure sufficient digestion via agarose gel, and were purified and quantified as described above.
Sequencing of DNA amplicons and DIG-nick translation
DNA amplicons were sequenced prior to use. Sequencing reactions were carried out according to protocols described elsewhere, 3 with the following modified conditions: reactions were performed in a Mastercycler thermocycler (Eppendorf): 96°C for 2 mins, 25 cycles of 90°C for 10 s, annealing for 5 s (50°C for FcaPV2 or 46°C for herpesvirus) and 60°C for 4 mins.
Purified DNA was subjected to DIG-nick translation to facilitate probe visualization. The following reagents were added to a total volume of 20 µl: 1000 ng purified DNA and 4 µl DIG-nick translation mix (Roche), along with sterile and deionized water. This mixture was then placed in the thermocycler at 15°C for 2 h, where after the product size of the nick translation reaction was visualized on a 1% agarose gel as described above. After confirmation of DIG-nick translation, the reaction was stopped with the addition of 1 µl 0.5 mol/L EDTA pH 8.0 and denatured for 10 mins at 65°C. DIG-labelled DNA was then purified as described above.
Five-fold serial dilutions (1:5–1:15,625) of 1 µl aliquots of DIG-nick translated DNA were used to confirm probe labelling efficiency. Two microlitres of each aliquot were applied to a nylon membrane (Roche) and heated to 120°C for 30 mins in a Venticell oven (Medcenter Einnchtungen), then rinsed three times for 5 mins in buffer 1 (0.1 mol/l Tris pH 7.5, 0.1 mol/l NaCl, 2 mol/l MgCl2, 0.05% Triton-X 100 [v/v]), followed by soaking in buffer 2 (buffer 1 with 0.5% [w/v] commercial blocking reagent [Roche]) for 30 mins and a further two washes for 5 mins each in buffer 1. A 1:5000 dilution of alkaline phosphatase (ALKP)-conjugated anti-DIG Fab fragments (Roche) was then applied to cover the membrane and incubated at room temperature for 30 mins. The membrane was then rinsed twice in buffer 1 and soaked for 5 mins in buffer 3 (0.1 mol/l Tris pH 9.5, 0.1 mol/l NaCl, 50 mmol/l MgCl2) followed by overnight immersion in the ALKP substrate precipitating solution Permanent Red (Dako). Subsequently, the membrane was rinsed with water, dried and evaluated.
ISH mixture construction
Hybridization mixtures were prepared in a total volume of 1000 µl, which contained 10–20 µl DIG-nick translated DNA, 100 µl 20 × saline sodium citrate (SSC) (Sigma Aldrich), 500 µl formamide (Applied Biosystems), 200 µl 50% (w/v) dextran sulfate (Sigma Aldrich) and sterile, deionized water (AstraZeneca) to complete the total volume.
Determination of endogenous ALKP activity in FFPE tissue
FFPE sections comprised of feline integument were examined to determine the level of endogenous ALKP. Sections were deparaffinized in xylene for 1 min, followed by an additional 1 min deparaffinization in clean xylene, then rehydrated for 1 min each in 100% EtOH, 95% EtOH, 90% EtOH and 70% EtOH, immersed in tap water, and permeabilized in PBST (1 × phosphate buffered saline [PBS] with 0.05% [v/v] Tween-20). Sections were then developed in Permanent Red (Dako) for 20 mins at room temperature. Slides were rinsed with tap water, counter-stained with Brazilin hematoxylin (Murdoch University Histology Department) for 60 s, rinsed and allowed to air dry prior to permanent mounting in Faramount (Dako).
FISH protocol
A FISH protocol modified from a previously described ISH protocol was performed. 14 Briefly, FFPE tissue sections were deparaffinized, rehydrated and permeabilized as described above. Target site unmasking was performed using two rounds of 90% maximal power output (90 V; Kambrook Turntable Microwave oven KER-686LE) for 4 mins each in TE buffer pH 9.0 (1.21 g Tris, 0.37 g EDTA, dissolved in 1000 ml distilled water) followed by two rounds of 70% maximal power output for 4 mins each in TE buffer pH 9.0. Slides were cooled then rinsed with PBST. Fifty microliters of hybridization mixture was added to each slide, then slides were cover-slipped and cycled on a PHC-3 ISH-adapted thermocycler (Techne) at 95°C for 15 mins, and then cooled to room temperature. Slides were uncovered, rinsed with SSCT (2 × SSC with 0.05% [v/v] Tween-20), blocked in 100 µl blocking solution (1 ml TBST [0.05% Tween-20 in 1 × Tris-buffered saline] with approximately 20 bovine serum albumin flakes [Sigma Aldrich]) for 5 mins, and incubated in probe detection solution (1:500 anti-DIG Fab fragments dissolved in blocking solution) for 30 mins at room temperature. Each slide was rinsed with TBST and developed in Permanent Red for 20 mins at room temperature. Slides were then rinsed, stained and mounted as described above.
Determination of FISH results, image acquisition and adjustment
FISH probe binding was considered positive when signal was located intracellularly within the epidermis, including within keratinocytes and koilocytic keratinocytes (koilocytes) with eccentric nuclei displaced by a cytoplasmic vacuole. Hair shafts and erythrocytes were excluded from positive results, owing to endogenous ALKP activity. FISH slides were viewed using a fluorescently adapted microscope (Olympus BH2 with RFC attachment) and slides were catalogued according to results, with the observer blinded to the prior PCR results.
Results
FcaPV2 was detected in 15/42 BISCs (35.7%); no intralesional binding was visible in 25 BISCs (59.5%). Therefore, using PCR as the comparative gold standard, FISH was 35.7% sensitive and 100% specific for FcaPV2 detection. None of the samples contained detectible probe binding within any cells within the epidermis surrounding the BISCs or in any cell within the dermis. Positive and negative controls for the assay generated expected results.
In positive lesions, FcaPV2 DNA was detected in the stratum spinosum, stratum granulosum and stratum corneum in similar proportions. In contrast, no probes were observed in the stratum basale. FcaPV2 DNA was principally intranuclear; however, occasional intracytoplasmic probe binding was also present. Epidermal koilocytes (enlarged keratinocytes with eccentric, pyknotic nuclei surrounded by a large, clear, cytoplasmic halo) were the primary cells positive for probe binding, though signal intensity was variable (Figures 1 and 2). Epidermal keratinocytes and koilocytes had morphology consistent with prior descriptions of PV-associated infections.15,16 Not all koilocytes displayed FcaPV2 DNA probe binding.
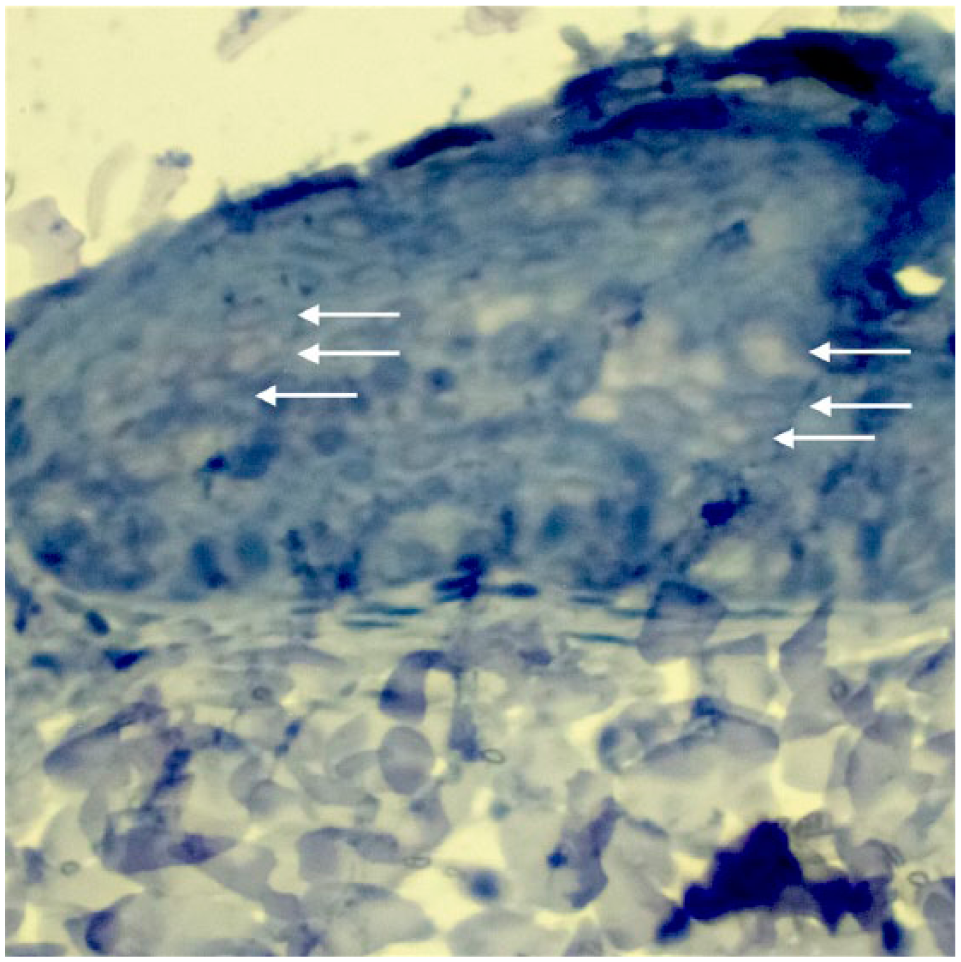
Bright-field conditions show koilocytes within the layers of the epidermis (arrows). Hematoxylin/permanent red

The same formalin-fixed paraffin-embedded tissue section imaged with a fluorescent filter. Focal uptake of FcaPV2 probe within the same koilocytes (arrows) is noted. Some background artefact is also present (star). Hematoxylin/permanent red
Within individual BISCs it was frequently observed that only a portion of the lesion contained probe binding, generally at the periphery of the neoplasm. Cell morphology in these regions was largely considered to display milder forms of cellular pathology (eg, hyperplasia). Conversely, more dysplastic or atypical regions, which also tended toward a central location in the lesion, often did not show probe binding. Hybridization ranged from the majority of cells in examined high-power-field sections (HPF; × 400 magnification) to <10 cells/HPF.
Discussion
Around a third of BISCs contained intralesional FcaPV2 DNA, as demonstrated by ISH. To our knowledge, this is the first study that has utilized ISH to locate the presence of PV DNA within a series of feline BISCs. The demonstration of FcaPV2 DNA within the BISCs supports the hypothesis that FcaPV2 could influence the development of these lesions in cats.
Prior to the present study, most data investigating the relationship between FcaPV2 and BISCs arose from conventional PCR and immunohistochemistry. Using PCR, detection rates of FcaPV2 in BISCs have ranged from 23.8–100%,17–20 with the wide range in detection rates most likely due to differences in the primer sets used in these studies. While such results suggested that FcaPV2 could be causative for BISCs, because FcaPV2 can also be detected in 52% of normal feline skin swabs, 8 such results could not exclude incidental PV infections in the surrounding epidermis. IHC has previously been used to detect PV antigen in 11% of BISCs, 21 which has the advantage of being able to locate the PV antigen within the lesion. However, IHC relies on the presence of PV L1 antigen and will therefore only detect PV when a lesion contains significant, active viral production of structural (late) proteins. Furthermore, the specific PV type cannot be determined using IHC. Thus, the main advantages of FISH are the ability to detect PV DNA in lesions in which productive infection is not present, the ability to localize the DNA within the lesion and the ability to determine the PV type present. 22
ISH was previously used to detect FcaPV2 DNA and RNA in a series of feline SCCs. 12 In this study, 5/18 (28%) SCCs contained hybridization signals. The similar rates of detection of intralesional PV DNA in the SCCs and in the present study of feline BISCs suggests that these lesions could both be influenced by PV infection. Therefore, these results support previous studies, suggesting FcaPV2 could be a significant cause of pre-neoplastic and neoplastic skin lesions of cats. 23
Surprisingly, the present study found that only 35.7% of tested BISCs contained demonstrable FcaPV2 DNA. This was lower than expected, as 100% of the lesions were FcaPV2 DNA-positive via PCR. PCR is inherently more sensitive than FISH, as it amplifies target DNA and can theoretically detect single viral DNA targets within a sample. At best, studies suggest FISH to have a detection threshold of 50 viral copies/cell, 24 though this figure is debated. 25 Additionally, when performing conventional PCR, DNA is typically extracted from a 10-μm-thick slice of tissue. Comparatively, ISH only detects DNA that is close to the surface of the lesion. Therefore, if only small amounts of DNA are scattered through the lesion, these small amounts may be undetectable by ISH, while still remaining amplifiable by conventional PCR. Sample processing is also known to affect FISH sensitivity. For instance, probe annealing can be impaired if the target tissue had been subjected to over-fixation prior to embedding in paraffin. 26 Finally, current probes were no longer than 177 bp in length, and probe sizes where the majority of the fragments are less than 200 bp have been shown to have lower sensitivity in target detection. 27 Redesigning the probe length, techniques to increase signal strength, or amplifying probe target, along with standardizing sample submission, may further help to increase FISH sensitivity.
Another factor that may have decreased the reported sensitivity of the present FISH assay is that the current study targets only a portion of FcaPV2 L1. This gene is important in capsid production, and is transcribed late in the normal viral life cycle. However, PV is a tumor virus, capable of oncogenic transformation in humans and other species.28,29 When transformation occurs, early viral genes may integrate into the host genome (E6 and E7), while other open reading frames are lost (eg, L1). 5 Therefore, neoplastic transformation via PV may occur, with a simultaneous decrease or lack of L1 production. This scenario would benefit a more sensitive testing modality such as PCR, which amplifies detected DNA, while possibly confounding ISH results. This is confirmed in other studies: for instance, in humans, L1 is more likely to be detected in pre-neoplastic lesions, than in neoplastic counterparts. 30 Thus, in the present report, some BISCs may have been transformed via PV infection but lacked sufficient quantities of L1 to enable FISH detection. Future investigations of feline BISCs may be better served to include a variety of FISH targets, such as E6 and/or E7 genes, or the use of RNA probes, which may better elucidate viral function.
FcaPV2 was most frequently detected within superficial strata of the feline epidermis within the BISCs. This correlated with reports of papillomavirus within the superficial epithelium in cats and other species,21,28 including other FISH-based L1 studies in humans. 31 Our results also confirm that koilocytes in feline BISCs, as noted in other species, often contain detectable PV DNA. Koilocytes in tested BISCs have similar morphology in FcaPV2 infections as those reported in other PV neoplasms in other species as evidenced by detection of L1, though it is uncertain why some koilocytes were not positive for FcaPV2 L1 in the present study.
PVs are currently associated with skin cancer in humans, 32 and are being investigated as possible causes of skin cancers in other species. 33 The detection of FcaPV2 DNA within the BISCs in the present report adds additional evidence supporting a role of this virus in causing feline BISCs.
Conclusions
In the present report, FcaPV2 was detected within dysplastic keratinocytes within approximately a third of feline BISCs. This is the first time that the presence of PV DNA has been demonstrated using FISH within a series of feline BISCs. The presence of intralesional viral DNA supports the hypothesis that FcaPV2 is associated with pre-neoplastic and neoplastic lesions of the skin of cats.
Footnotes
Acknowledgements
The authors wish to thank Dr Phil Nicholls, Mike Slaven, Gerard Spolestra, Dr Susan Kueh, Dr Tim Hyndman, Dr Andrea Ducki, Dr Claude Favrot and Dr Frances Brigg for sourcing materials and equipment associated with the project.
Conflict of interest
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.