
Abstract
During drug discovery, small molecules are typically assayed in vitro for secondary pharmacology effects, which include ion channels relevant to cardiac electrophysiology. Compound A was an irreversible inhibitor of myeloperoxidase investigated for the treatment of peripheral artery disease. Oral doses in dogs at ≥5 mg/kg resulted in cardiac arrhythmias in a dose-dependent manner (at Cmax, free ≥1.53 μM) that progressed in severity with time. Nevertheless, a panel of 13 different cardiac ion channel (K, Na, and Ca) assays, including hERG, failed to identify pharmacologic risks of the molecule. Compound A and a related Compound B were evaluated for electrophysiological effects in the isolated rabbit ventricular wedge assay. Compounds A and B prolonged QT and Tp-e intervals at ≥1 and ≥.3 μM, respectively, and both prolonged QRS at ≥5 μM. Compound A produced early after depolarizations and premature ventricular complexes at ≥5 μM. These data indicate both compounds may be modulating hERG (Ikr) and Nav1.5 ion channels. In human IPSC cardiomyocytes, Compounds A and B prolonged field potential duration at ≥3 μM and induced cellular dysrhythmia at ≥10 and ≥3 μM, respectively. In a rat toxicology study, heart tissue: plasma concentration ratios for Compound A were ≥19X at 24 hours post-dose, indicating significant tissue distribution. In conclusion, in vitro ion channel assays may not always identify cardiovascular electrophysiological risks observed in vivo, which can be affected by tissue drug distribution. Risk for arrhythmia may increase with a “trappable” ion channel inhibitor, particularly if cardiac tissue drug levels achieve a critical threshold for pharmacologic effects.
Introduction
During the process of drug discovery, numerous in vitro assays are incorporated into screening paradigms to identify and select low molecular weight compounds for further study. Screening paradigms are designed to require relatively low amounts of compound (i.e., mg quantities) which allows for numerous molecules to be tested in an efficient manner. Attributes related to physical-chemical properties, pharmacologic potency and selectivity, safety, and pharmacokinetics/metabolism are evaluated during the selection process with the objective of increasing the probability for identifying a compound that may progress through drug development.1-5 A battery of in vitro assays that includes ion channels, enzymes, membrane and nuclear receptors, and transporters are typically utilized to evaluate the potential of a compound to act as an agonist, antagonist, or inhibitor and thereby elicit potential pharmacologic effects unrelated to the desired primary pharmacologic target.6,7 These assays may be comprised of binding inhibition and functional assessments with results oftentimes referred to as secondary pharmacology or “off-target” effects. Reference to secondary pharmacology assessments is made in the ICH Guideline: S7A Safety Pharmacology Studies for Human Pharmaceuticals, with the results typically submitted to health authorities prior to initiation of human clinical studies. 7
The life-threatening ventricular arrhythmia, torsades de pointes, can arise with use of various low molecular weight drugs of diverse therapeutic class that cause delayed ventricular repolarization, as depicted by prolongation of the QTc interval of the electrocardiogram. 8 Significant concern lies with the potential for drugs to block voltage-dependent potassium channels, in particular the delayed rectifier current Ikr, which plays a key role in ventricular repolarization of the action potential in humans and other non-rodent animals typically utilized in preclinical investigations. The pore-forming subunit of this potassium channel is coded by the human ether-a-go-go-related gene (hERG). 9 A cellular in vitro electrophysiology test system incorporating this potassium channel, commonly referred to as the hERG assay, has become a standard but important assay in the battery of safety pharmacology tests conducted prior to first-in-human clinical studies. 10 The expectations of global health authorities for assessing cardiovascular safety of experimental drugs to support initial clinical trials are described in ICH Guideline: S7B Nonclinical evaluation of the potential for delayed ventricular repolarization (QT interval prolongation) by human pharmaceuticals.11,12 Furthermore, inhibition of other cardiac ion channels, such as the sodium current, can lead to other forms of arrhythmia associated with slow conduction.13,14 What has become evident is that inhibition of multiple cardiac ion channels can arise with a particular drug (and in relation to exposure levels) that can impact cardiac electrophysiologic function. Therefore, ion channels (K, Na, and Ca) relevant to cardiac electrophysiology represent a core component of secondary pharmacology evaluations.
Myeloperoxidase (MPO) is a heme protein synthesized during myeloid differentiation that constitutes the major component of neutrophil azurophilic granules. MPO produces hypochlorous acid (HOCl) from H2O2 as a microbicidal agent. 15 Prior research suggested that MPO is involved in various inflammatory pathologies including cardiovascular disease such as atherosclerosis.16-18 MPO activity is believed to result in increased lipid peroxides, including low density lipoproteins (LDL). Oxidized LDL is thought to be phagocytized by macrophages, leading to increased foam cells in atherosclerotic plaques. 19 The presence of active MPO in the vasculature and atherosclerotic plaques may further exacerbate arterial disease and lead to diminished regional blood flow. Peripheral artery disease (PAD) is a manifestation of systemic atherosclerosis in peripheral arteries, and it was hypothesized that MPO is involved with PAD pathology. 20 A drug discovery program was previously active at Novartis with the goal of developing a low molecular weight inhibitor of extracellular MPO to improve PAD vascular biology. 21 Medicinal chemistry efforts identified a novel indole pharmacophore as an irreversible inhibitor of MPO 22 with additional research leading to the identification of a pyrrolidinone indole series of irreversible MPO inhibitors. 23
Compound A (Figure 1) was identified as a potential drug candidate from the pyrrolidinone indole series.
23
Following initial drug discovery evaluations, Compound A was evaluated for safety in a dog dose range-finding toxicity study. In this study, severe cardiovascular changes were observed, which were not predicted by initial in vitro ion channel assays, including the hERG assay. Therefore, an investigation was undertaken to identify a potential mechanism of action and to better understand the lack of predictivity of the ion channel assays that were conducted. The rabbit ventricular wedge assay, which demonstrated highly accurate prediction of drug-induced proarrhythmic liability in a recent blinded study with drugs listed in the Comprehensive in vitro Proarrhythmia Assay (CiPA) initiative,
24
and human-induced pluripotent stem-cell derived (IPSC) cardiomyocytes were utilized for this investigation. Chemical structures of Compound A and Compound B.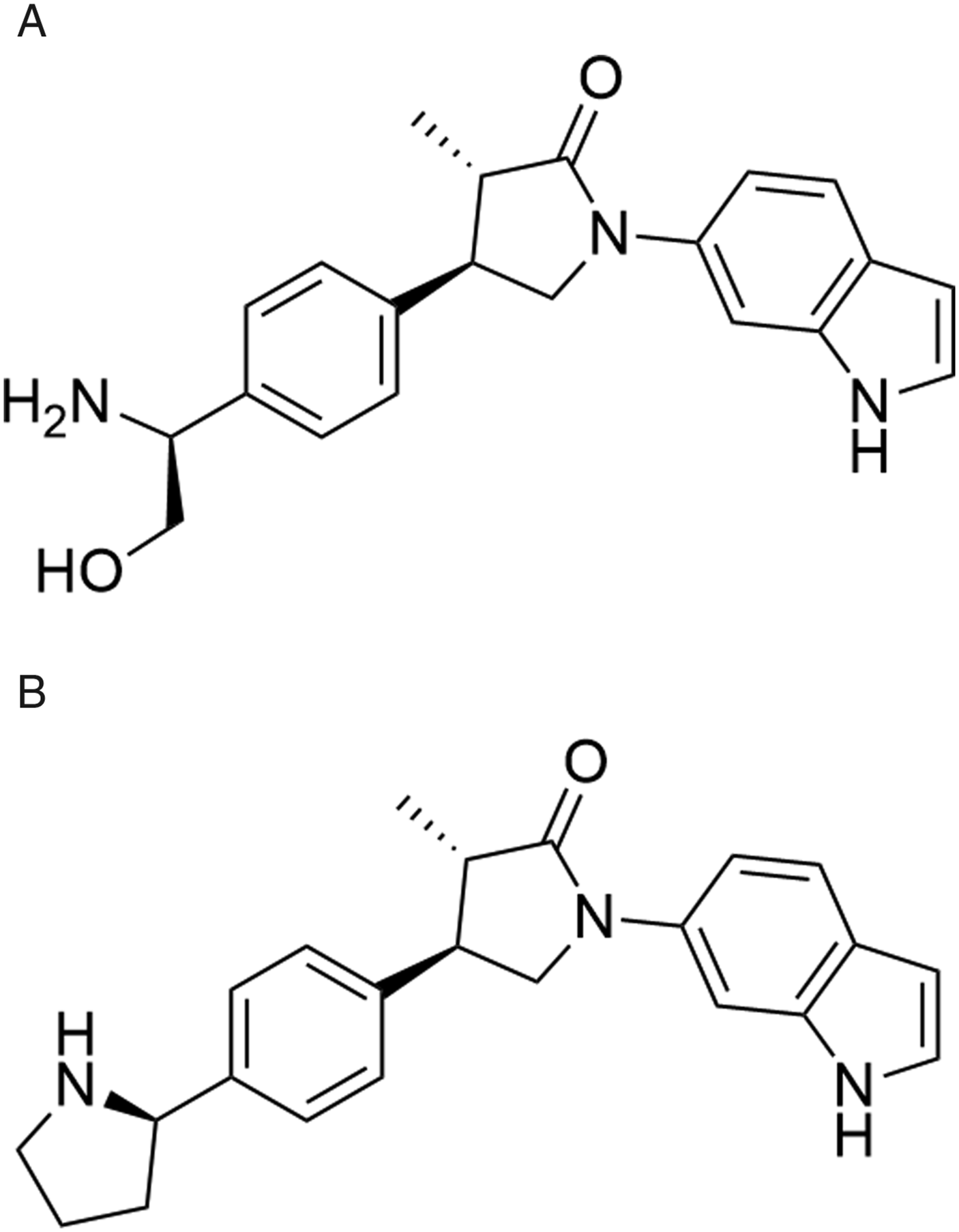
Portions of this paper were presented as a poster at the 2022 American College of Toxicology annual meeting in Denver, CO. 25
Materials and Methods
Compounds Tested
Compounds A (molecular weight of 349.4 g/mol) and B (molecular weight of 359.5 g/mol) were synthesized by the Novartis Institutes for Biomedical Research (NIBR), Cambridge, MA, USA (see Figure 1). The malate salt of Compound A (salt:base ratio of 1.384) was administered to dogs whereas the free base was administered to rats. Compound B was previously described (as IN-4) by Regard et al. 24
In Vitro Cardiac Ion Channel Assays
Compound A was tested for inhibition of Ikr in the hERG assay using the QPatch electrophysiology system 26 over the concentration range of .37–300 μM, along with the IonWorks Quattro electrophysiology system 27 at concentrations of 100 and 200 μM. Both assays were conducted at NIBR, Cambridge, MA.
In vitro electrophysiological assays were conducted on Compound A using either an 8-point concentration curve from .05 μM or 7-point concentration curve from .14 μM up to a top concentration of 100 μM using the IonWorks Quattro (K channels) or IonFlux HT electrophysiological platform (Na channels; KATP) (assays performed by Eurofins Pharma Bioanalytics Services US Inc, St Charles, MO). The ion channels included the voltage-gated K channels Kv4.3/KChIP2, Kv1.5, and KCNQ1/mink, the inward-rectifying voltage-gated K channel Kir2.1, the hyperpolarization-activated cyclic nucleotide-gated K channel HCN4, the voltage-gated Na channels Nav1.1 and Nav1.7, and the ATP-sensitive K channel KATP (agonist and antagonist assessments). Compound A inhibition data were normalized to vehicle negative control data (.3% DMSO) and ion channel-specific inhibitors were included as positive reference controls.
Compound A was tested for inhibition of Na/K-ATPase (protein from porcine cerebral cortex) in an in vitro assay using an 8-point concentration range of .03–100 μM (performed by Eurofins Pharma Bioanalytics Services US Inc, St Charles, MO).
The potential for Compound A to inhibit Nav1.5, Cav1.2, and KCNQ ion channels were conducted at NIBR using the IonWorks Quattro electrophysiology system 26 over a concentration range of .09–300 μM.
Dog Dose Range-Finding Toxicity Study
Male beagle dogs were obtained from Marshall BioResources, North Rose, NY, and at the initiation of dosing were approximately 2 to 4 years old and weighed 6.8 to 12.1 kg. Animals were pair/group-housed in runs except while wearing telemetry jackets when they were single-housed. Housing conditions included a 12-hour light/12-hour dark cycle at 68–76°F and 30%–70% relative humidity. Animals were fed Certified Canine Diet 5007 (PMI Feeds, Richmond, IN, USA) at approximately 250g daily and were fasted prior to scheduled blood collections for clinical laboratory tests and necropsy. Water was provided ad libitum. Compound A was administered by oral gavage (5 mL/kg dose volume) at doses of 0 (control), 5, 15, and 40 mg/kg/day (N = 3 dogs/group) for up to 14 days. Control animals received the vehicle alone, which was .5% (w/v) methylcellulose, type 1500cPs, in water. Clinical observations (at least twice daily) and body weight and food consumption were recorded throughout the study. Electrocardiography (ECG) measurements were collected using jacketed ECG telemetry equipment (emka ECG system) pretest and from 2 hours prior to dosing to approximately 24 hours post-dose on days 1, 7, and 13. Telemetry ECG recordings were made with the animal in its home cage. Heart rate and ECG intervals (RR, PR, QRS, and QT) were measured from Lead II using the ECG auto computer software and the last (fourth) 15-minute period of every hour reported. QT was corrected for heart rate using the Van de Water QT correction formula: QTc = QT − .087 (RR-1000). Change and percentage change from pre-dose baseline and group means were calculated. Difference (double delta) in baseline-adjusted change and percentage change between pretest and dose groups was also calculated. Blood was collected with EDTA anticoagulant at .5, 1, 3, 7, and 14 hours after dosing on days 1 and 13, and plasma was isolated and stored frozen at approximately −70⸰C. Plasma was analyzed for Compound A concentrations using a suitable liquid chromatography-mass spectrometer (MS)/MS method using electrospray ionization as an interface in positive ion mode with the lower limit of quantitation of 1 ng/mL. Blood collection for routine hematology, serum chemistry, and coagulation parameters were collected pretest (all animals) and on days 9 (15 and 40 mg/kg/day groups) and 14 (control and 5 mg/kg/day groups). Animals in the 15 and 40 mg/kg/day groups were euthanized early on day 9 due to humane reasons whereas animals in the 5 mg/kg/day group were euthanized on day 15. Necropsies and gross examinations were conducted for all drug-treated animals, and microscopic examinations made from a limited list of tissues from the 5 and 40 mg/kg/day groups, along with heart and gross lesions from animals in the 15 mg/kg/day group. Control animals were returned to the animal colony.
Isolated Rabbit Ventricular Wedge Assay
Methods and validation for the isolated ventricular wedge assay were previously published.24,28-30 New Zealand White rabbits (n = 4 per compound), age 2 to 3 months and weighing 2.0 to 2.8 kg, were anticoagulated with heparin (800 U/kg) via an ear vein and anesthetized by intramuscular injection of xylazine (5 mg/kg) and intravenous administration of ketamine HCl (30 to 35 mg/kg). The chest was opened via a left thoracotomy, and the heart was excised and placed in a cardioplegic solution consisting of cold (4°C) normal Tyrode’s solution. The tissue was cannulated via the left circumflex branch of the coronary artery and perfused with cardioplegic solution. Non-perfused areas of the left ventricle, identified from its reddish appearance because of the existence of unwashed erythrocytes, were removed. The preparation was then placed in a small tissue bath and arterially perfused with Tyrode’s solution at a temperature of 35.7 ± .1°C and perfusion pressure of 35 to 45 mmHg. Tyrode’s solution composition (in mM) was: NaCl, 129; KCl, 4; NaH2PO4, .9; NaHCO3, 20; CaCl2, 1.8; MgSO4, .5; glucose, 5.5; insulin 1 unit/liter; and buffered with 95% O2 and 5% CO2. The ventricular wedge was allowed to equilibrate in the tissue bath until electrically stable for 1 hour. The preparations were stimulated at basic cycle lengths (BCL) of 500, 1000, and 2000 ms using bipolar silver electrodes insulated except at the tips and applied to the endocardial surface.
A transmural electrocardiogram or “ECG” signal was recorded via a HP ECG amplifier (model 8811A) using extracellular silver/silver chloride electrodes placed in the Tyrode’s solution, bathing the preparation 1.0 to 1.5 cm from the epicardial and endocardial surfaces, along the same vector as the transmembrane recordings (Epi: “+” pole). The QT interval was defined as the time from the onset of the QRS to the point at which the final downslope of the T wave crosses the isoelectric line. Transmembrane action potential from the endocardium (Endo) was recorded for identification of early after depolarizations (EADs) only when QT prolongation was >30% compared with the value at baseline via a custom-made amplifier. The Tp-e interval, an index of transmural dispersion of repolarization, was defined as the interval between the end and the peak of T wave.
All measured biological signals including ECG and transmembrane action potentials were sampled via a D/A converter (Cambridge Electronic Design, CED 1401, England) and stored in electronic media (CD) and external hard drives. The raw signals of ECG and transmembrane action potentials were analyzed using Spike 2 software (CED, England).
Compounds A and B were prepared in DMSO and tested at the following concentrations: .3, 1, 5, 15, and 30 μM. These concentrations bracketed the minimal Cmax, free (1.53 μM) for Compound A that was associated with arrhythmia in dogs. The maximal DMSO concentration in the final perfusion solution was .1% (v/v). Each wedge preparation was exposed to each concentration, in an increasing fashion, for approximately 25 minutes. Control data comprised the pre-dose baseline perfusion period. Data recordings were made for 30 to 60 seconds at the end of perfusion for each concentration. Following a BCL of 2000 ms, the wedge preparation was paced at BCL of 1000 ms, followed by a burst pacing (15 to 20 seconds) at BCL of 500 ms to assess the effect of the test compound on the staircase response of contractility and rate-dependent change in QRS duration. Data collected at BCL of 2000 ms were used for QT and Tp-e analyses and at 500 and 2000 ms for QRS. Arrhythmic phenomena were recorded if observed. A TdP score was calculated (potential range of −2 to 14) which provides a risk score for torsades de pointes and was previously described by Liu et al.28,29
Human IPSC Cardiomyocyte Assays
Electrophysiological endpoints of beating human-induced pluripotent stem cell (IPSC) derived cardiomyocytes (IPSC-CM) monolayers were recorded using the Multiwell-MEA system (Multichannel Systems, Harvard Biosciences Inc.). IPSC-CM (FujiFilm, Cellular Dynamics) were thawed according to the Cellular Dynamics user guide, using Cellular Dynamics thawing medium and plated at a density of at least 28500 cells per well on the electrode area of 96-well MEA plates (Multichannel Systems, MCS) and cultured for 1 week using Cellular Dynamics maintenance medium. Media was changed every 2 days. The day prior to any measurement, the complete media was removed and the volume was set to 200 μL. The test items and dofetilide were dissolved in 100% DMSO and diluted 100 times in maintenance media (DMSO concentration: 1%). The 96-well MEA plates were placed in the Multiwell-MEA system (MCS Multiwell System) and acclimated for 5 minutes. A 2-min control phase was recorded before adding 22.2 μL of corresponding concentration using an 8-channel multi-pipette (final DMSO concentration: .1%). Measurements were performed using Multiwell Screen Software (MCS). The effect was assessed in 5 wells per concentration after a 2-minute wash in period at 2, 15, 30, and 60 minutes for a duration of 2 minutes each (10000 Hz sampling rate). The effect of the vehicle was measured in 10 wells. The effect of the positive control dofetilide (3 nM; source of MedChem Express) was measured in 6 wells. All measurements were performed at 37°C. If arrhythmia occurred, the field potential duration (FPD) was not able to be evaluated at these timepoints. All effects were normalized with pretreatment values of the same wells and the results were corrected with the corresponding time-matched vehicle effect. Analysis was performed using Multiwell Analyzer Software (MCS) and MS Excel.
Drug Tissue Distribution in Rats
Male IGS Wistar Hannover Rats; Crl: WI(Han) were obtained from Charles River Laboratories, Raleigh, NC, USA, and at initiation of dosing were approximately 9 to 10 weeks of age and weighed 293.5 to 346.8 grams. Animals were housed in pairs or groups in solid bottom caging in conditions that included a 12-hour light/12-hour dark cycle at 68–76°F and 30%–70% relative humidity. Certified Rodent Diet 18% #5LG3 Pellets (PMI Feeds, Richmond, IN, USA) was provided ad libitum, except when animals were fasted 12 to 20 hours prior to scheduled necropsy. Water was provided ad libitum. Compound A was dosed orally by gavage (dose volume 10 mL/kg) at 0 (control; vehicle alone), 100, 300, and 1000 mg/kg/day (N = 5/group) for 14 days. Compound A was formulated in .5% (w/v) methyl cellulose, Type cPs, .5% (v/v) polysorbate 80, and NF in water. The 1000 mg/kg/day dose was poorly tolerated and animals were euthanized by day 4. Remaining animals were euthanized on day 15 at approximately 24 hours after the last dose, and plasma and heart tissue collected and measured for Compound A concentrations using a suitable liquid chromatography-MS/MS method using electrospray ionization as an interface in positive ion mode with the lower limit of quantitation of 1 ng/mL.
Animal Welfare Statement
Both the dog dose range-finding toxicity study and rat study were conducted at the Novartis Pharmaceuticals Corporation East Hanover, NJ, USA, facility and the study protocols were approved by the Novartis Animal Care and Use Committee. The rabbit ventricular wedge assay was conducted at the Lankenau Institute for Medical Research, Wynnewood, PA, USA. Both the Novartis and the Lankenau animal facilities are AAALAC accredited.
Statistical and Data Analyses
Dog plasma toxicokinetic parameters were calculated using Watson LIMS software. Dog ECG data were recorded and analyzed using the emka ECG system (Study Designer, IOX, and ECG-Auto). Results of the rabbit ventricular wedge assay are presented as mean ± standard error of the mean (SEM) and statistical analysis was performed using the Student’s t test. A P < .05 was considered as statistically significant when compared to the values in the control perfusion.
Results
In Vitro Cardiac Ion Channel Assays
Compound A: In Vitro Cardiac Ion Channel Assays.

ahERG assay conducted using the QPatch platform.
bIon channel assay conducted using the IonWorks Quattro platform.
cTwo separate assays conducted on the IonWorks Quattro platform.
Dog Toxicology Study
A 2-week dose-range finding toxicity study of Compound A in dogs was conducted to obtain initial toxicology and toxicokinetic data, prior to planning of future studies. Male beagle dogs (N = 3/group) received oral (gavage) doses of Compound A at 0 (vehicle alone), 5, 15, and 45 mg/kg/day for up to 15 days. Electrocardiography (ECG) data were collected using jacketed telemetry devices on days 1, 7, and 13 from 2 hours prior to dosing through 24 hours post-dose. Arrhythmias were observed in all animals on day 1 at ≥15 mg/kg/day and were characterized by premature ventricular complexes (PVC) and bigeminy, with time of onset from approximately .5 to 2 hours post-dose. On day 7, severe and complex arrhythmias were seen that included PVC and bigeminy at ≥15 mg/kg/day and non-sustained ventricular tachycardia as well as second-degree AV-block at 40 mg/kg/day. The arrhythmias were sustained for approximately 18–24 hours at the high dose. An increase in QTc interval was observed on day 7 at ≥15 mg/kg/day of up to +95 msec (+38%) from baseline, which was sustained through the 24-hour recording period. In addition, the PR interval of the ECG was increased in one animal at 40 mg/kg/day for approximately 8 hours, with a maximal increase of 31 msec (+35%) from baseline at approximately 3 hours post-dose. Upon a veterinarian auscultation exam on day 9, arrhythmia, occasional irregular/skipped beats, pulse deficits, and/or delayed capillary refill time were noted in animals at 15 and 40 mg/kg/day. Due to the declining clinical condition and severity of the cardiovascular changes, all animals in the 15 and 40 mg/kg/day groups were euthanized on day 9 at approximately 5 hours post-dose. One animal at 5 mg/kg/day exhibited PVC on days 1, 5, and 13. However, clinical signs of toxicity were not evident in this animal at this dose, and the cardiovascular changes did not worsen over time. There were no test article-related effects on heart rate and ECG intervals at 5 mg/kg/day on days 1, 7, and 13 or on day 1 at ≥15 mg/kg/day. Other than the cardiovascular changes described above, no other target organ effects were identified. Microscopic evaluation of various tissues, including the heart, did not identify any treatment-related changes at all dose levels. Serum chemistry results did not indicate the presence of altered electrolyte levels in the animals.
Mean Toxicokinetic Parameters of Compound A in Dog Plasma.
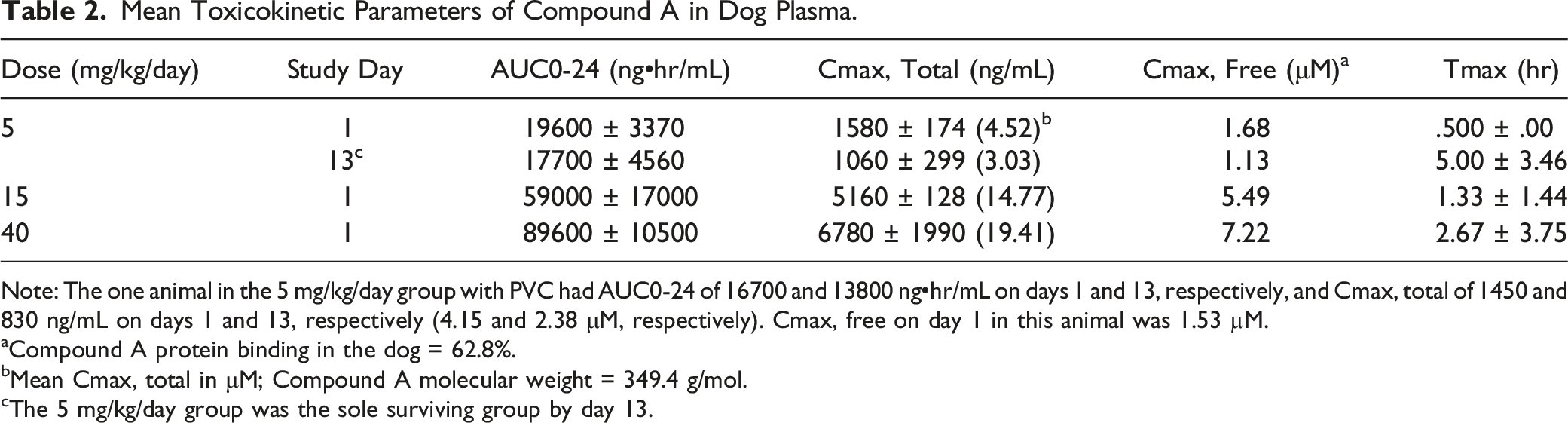
Note: The one animal in the 5 mg/kg/day group with PVC had AUC0-24 of 16700 and 13800 ng•hr/mL on days 1 and 13, respectively, and Cmax, total of 1450 and 830 ng/mL on days 1 and 13, respectively (4.15 and 2.38 μM, respectively). Cmax, free on day 1 in this animal was 1.53 µM.
aCompound A protein binding in the dog = 62.8%.
bMean Cmax, total in μM; Compound A molecular weight = 349.4 g/mol.
cThe 5 mg/kg/day group was the sole surviving group by day 13.
Rabbit Ventricular Wedge Assay
Rabbit Ventricular Wedge Results for Compound A.
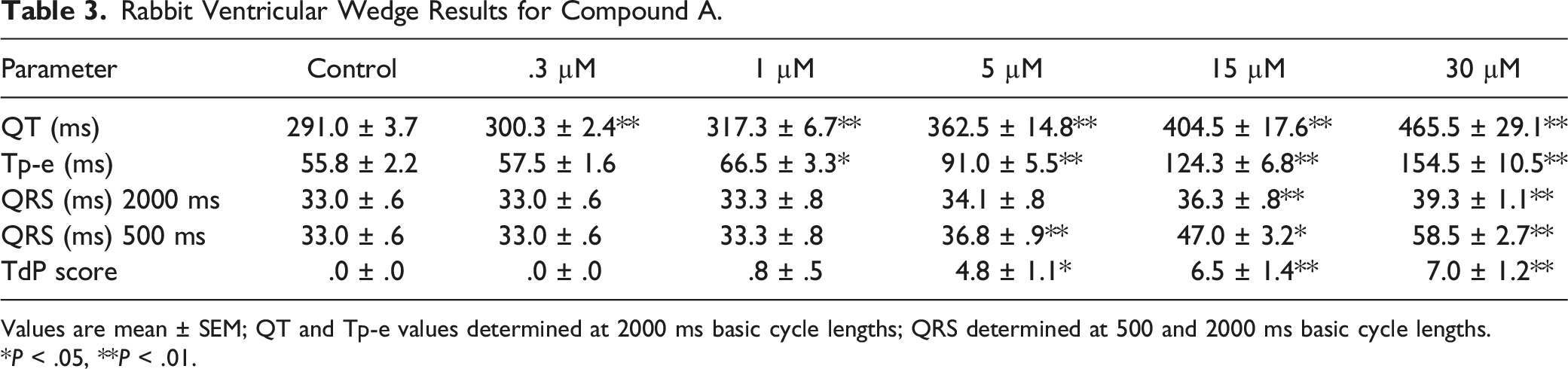
Values are mean ± SEM; QT and Tp-e values determined at 2000 ms basic cycle lengths; QRS determined at 500 and 2000 ms basic cycle lengths.
*P < .05, **P < .01.
Rabbit Ventricular Wedge Results for Compound B.
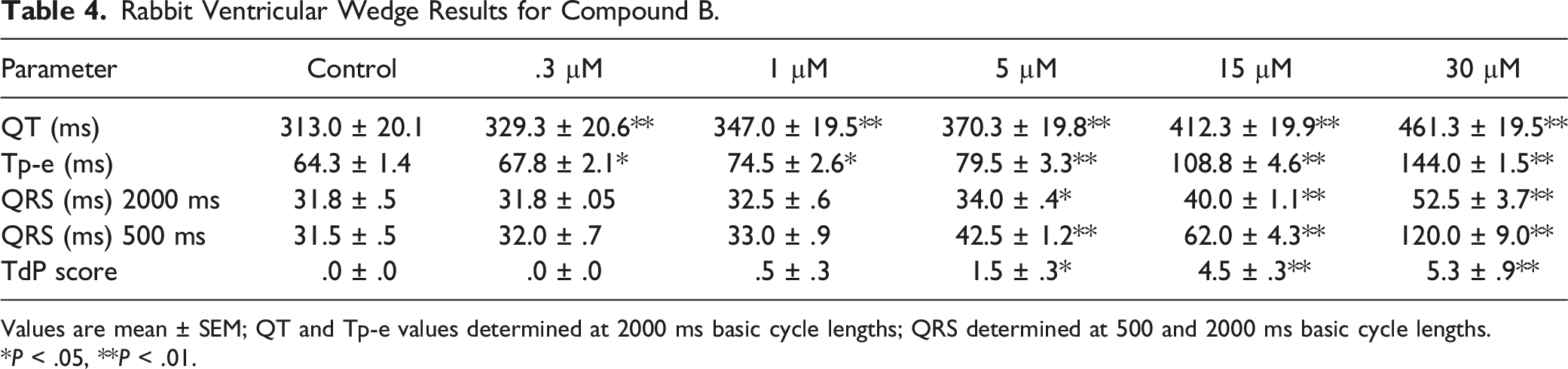
Values are mean ± SEM; QT and Tp-e values determined at 2000 ms basic cycle lengths; QRS determined at 500 and 2000 ms basic cycle lengths.
*P < .05, **P < .01.
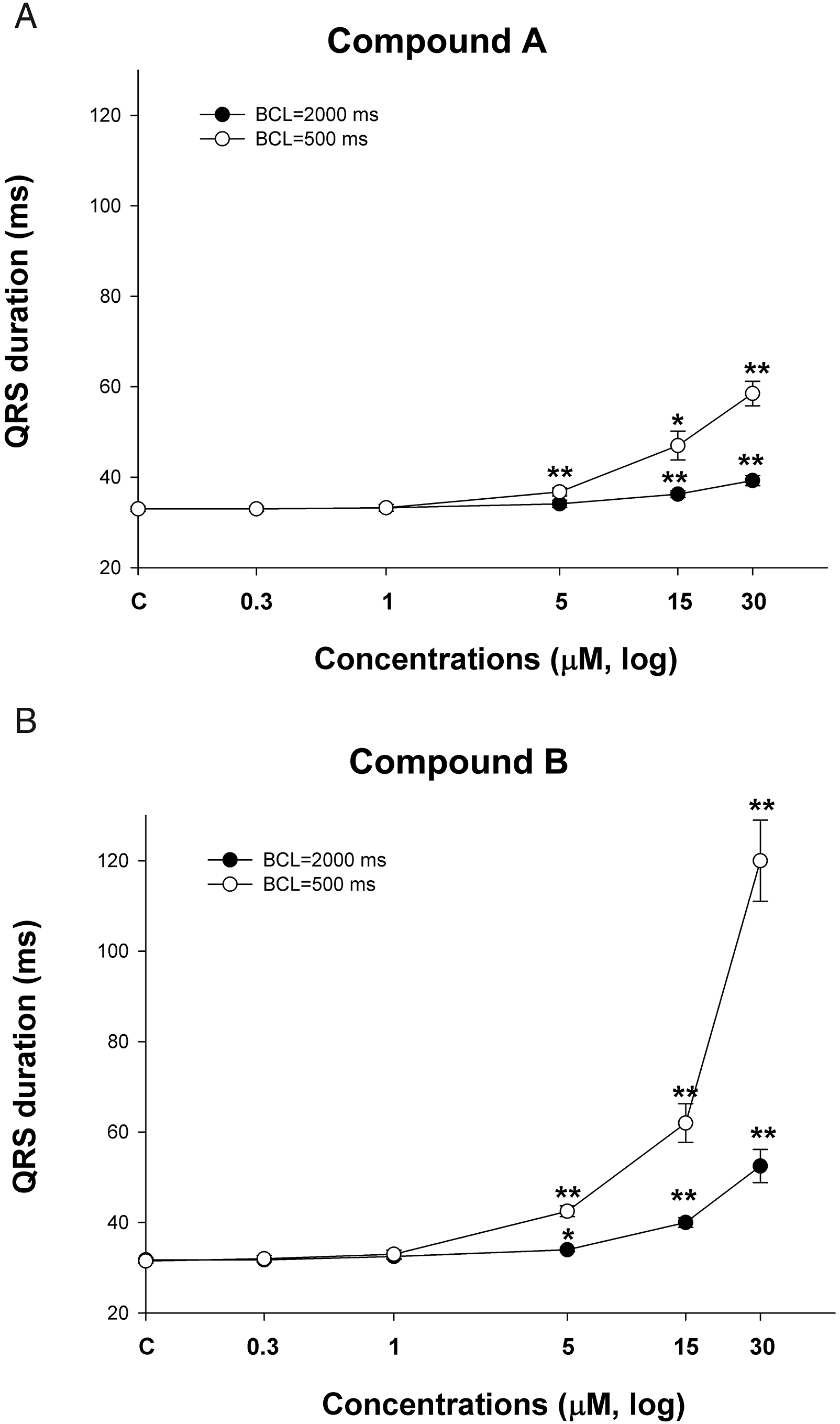
Measurement of QRS in the rabbit ventricular wedge assay. Compound A (Figure 2A) and Compound B (Figure 2B) were tested in the rabbit ventricular wedge assay at concentrations of 0 (control), .3, 1, 5, 15, and 30 μM at BCL of 500 or 2000 ms. QRS duration (ms) is plotted against drug concentration in log scale. *P < .05, **P < .01 versus control. Values are mean ± SEM.
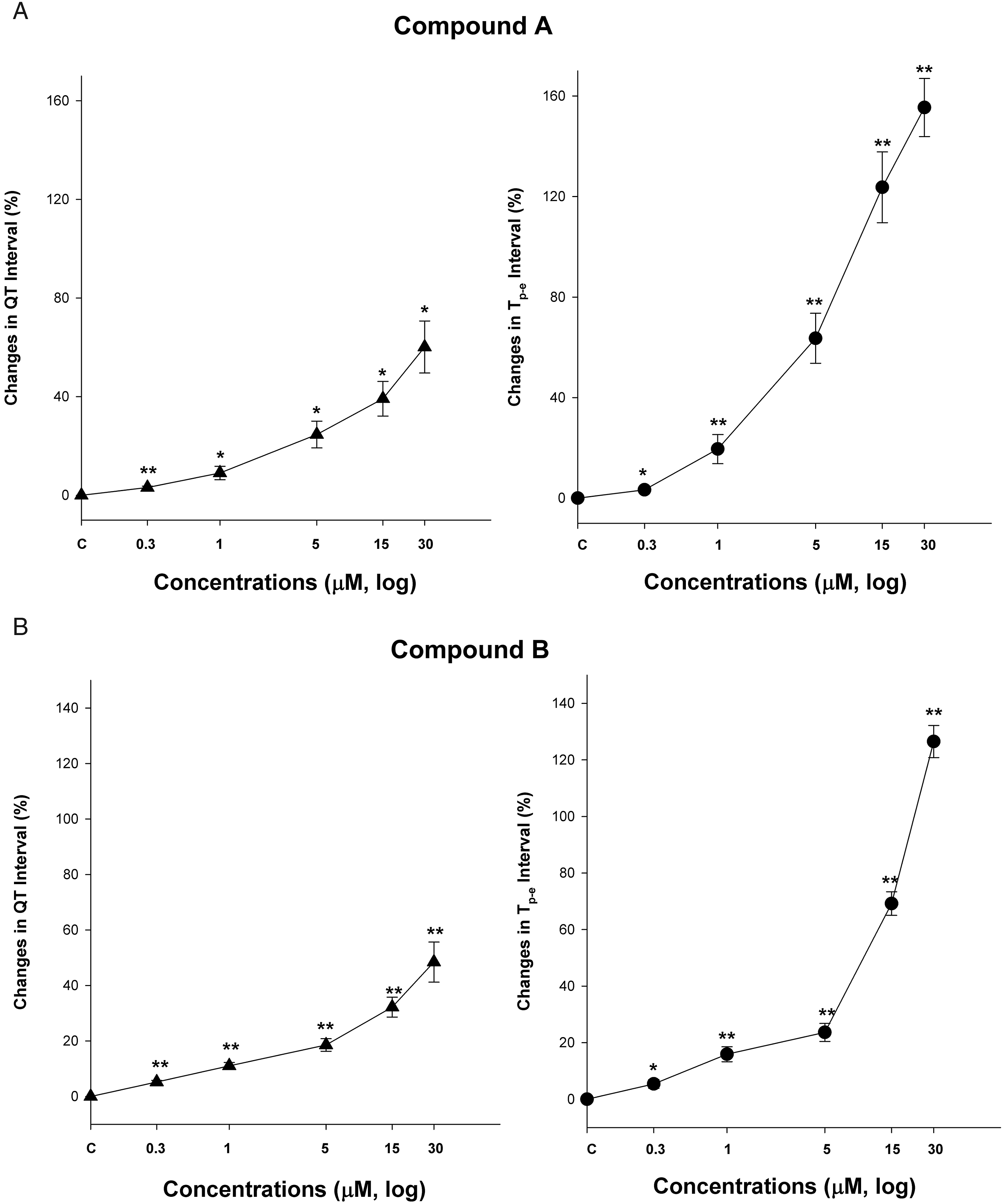
Measurement of QT and Tp-e in the rabbit ventricular wedge assay. Compound A (Figure 3A) and Compound B (Figure 3B) were tested in the rabbit ventricular wedge assay at 0 (control), .3, 1, 5, 15, and 30 μM. Percentage change in QT or Tp-e (time from peak to end of the T wave) intervals from control are plotted against drug concentration in log scale. *P < .05, **P < .01 versus control. Values are mean ± SEM.
Human IPSC Cardiomyocyte Assay
Human IPSC cardiomyocytes have recently become a recognized in vitro model system for testing for proarrhythmic potential of small molecules. 32 Compounds A and B were both tested over a range of concentrations (from 1 to 30 μM), with measurement of field potential duration (FPD) as the primary endpoint. These concentrations bracket the Compound A Cmax, free associated with arrhythmia in dogs. Compounds A and B both prolonged FPD at ≥3 μM and produced cellular dysrhythmia at ≥10 μM and ≥3 μM, respectively. Compound B suppressed electrical activity at 30 μM in which FPD could not be measured. Prolongation of FPD, followed by dysrhythmia, can occur with block of Ikr and/or Nav1.5 current. 33
Cardiac Tissue Distribution
Mean Heart and Plasma Concentrations of Compound A, and Heart:Plasma Ratio at 24 Hours Post-Dose in Rats.

Male rats (N = 5/group) were orally dosed with Compound A for 2 weeks and plasma and heart tissue were collected at approximately 24 hours after the last dose.
aProtein binding of Compound A in the rat is 64.3%.
Discussion
During the early phases of drug discovery, compounds are routinely tested in a battery of in vitro assays to evaluate the potential of the molecule to elicit pharmacologic activity, including intended (on-target) and unintended (off-target) pharmacology. Pharmacologic potency and selectivity are assessed, and decisions made to stop or progress those molecules demonstrating desirable features.5,34 Cardiovascular safety of pharmaceuticals is of particular interest due to the potential for severe or life-threatening adverse events to occur and the diversity of compounds known to affect the cardiovascular system. A major reason for attrition of drugs from development, or withdrawal from the market, is due to concerns regarding cardiovascular safety.35-38 In a retrospective analysis, cardiovascular safety liabilities were responsible for >20% of the attrition of candidate drugs during development and 45% of drugs removed from the market. 37 In addition to effects on hemodynamics and heart rate, drugs can inhibit cardiac contractility or be proarrhythmic. 37 This includes pharmaceuticals designed for cardiovascular indications along with those intended for non-cardiovascular pharmacologic activity. An example is the life-threatening and fatal ventricular arrhythmia, torsades de pointes, which can arise with use of various low molecular weight drugs of diverse therapeutic and chemical class and is associated with the prolongation of ventricular repolarization. 8 Additionally, inhibition of cardiac Na channels with drugs like flecainide can result in conduction block, leading to ventricular arrhythmias and reduced contractility and cardiac output.14,39 To mitigate against these potential risks, in vitro ion channel assays are typically incorporated into early screening paradigms for selecting candidate molecules for further evaluation. At Novartis, compounds early in drug discovery are routinely tested for inhibition of potassium, sodium, and calcium ion channels pivotal to the cardiac action potential and electrophysiology of the heart (Ikr or hERG; Nav1.5 and Cav1.2, respectively). 5
A drug discovery program was previously active at Novartis with the goal of developing a low molecular weight inhibitor of extracellular MPO for the treatment of peripheral artery disease.21,22 Medicinal chemistry, pharmacology, pharmacokinetic and early safety work lead to the identification of a novel pyrrolidinone indole series of irreversible MPO inhibitors. 23 Compound A (Figure 1) was identified as a potential drug candidate from this series and subsequently evaluated for safety in a dog dose range-finding study. In this study, severe cardiovascular changes were observed that included PVC and bigeminy (ectopic beat), which occurred following a single dose at ≥15 mg/kg/day and progressed in severity with daily dosing. Non-sustained ventricular tachycardia, second-degree AV-block, and increased PR intervals of the ECG were seen at 40 mg/kg/day. Increased QTc intervals were also observed after repeat dosing at ≥15 mg/kg/day. The minimal dose for producing cardiovascular changes in the dogs was 5 mg/kg/day, which lead to an increased occurrence of PVC.
The results of the dog study indicated that Compound A produced arrhythmias on day 1 in a dose-dependent fashion at ≥5 mg/kg (at Cmax, free ≥1.53 μM), which increased in severity with daily dosing at ≥15 mg/kg/day. The cardiovascular effects were not expected and resulted in termination of Compound A from further development. An investigation was initiated to identify a potential mechanism for the changes seen and to provide insight with respect to de-risking future molecules for cardiovascular safety. An additional objective of the research was to explore the potential for inclusion of other model systems for early cardiovascular safety evaluation.
Compound A was assayed in a panel of sodium, potassium, and calcium ion channels relevant for cardiac electrophysiology. Minimal to no activity was observed in the assays except for hERG and Nav1.5 which had IC50 values of 128 μM and 100 μM, respectively. These IC50 values are quite high and typically would not result in a notable concern as clinical exposures would be expected to be well below these values. However, integration of in vitro IC50 values and in vivo drug exposure data, such as Cmax, free, should be done when such data are available. Severe arrhythmias and QTc interval prolongation were seen in the dog study at ≥15 mg/kg/day. Protein binding in the dog is 62.8%, which is low to moderate, and can result in a significant amount of free drug that can interact with pharmacologic targets. The free (unbound to protein) drug hypothesis is that the free drug concentration at the site of action exerts its biological effect. 40 Mean Cmax, free at 15 mg/kg/day was 5.49 μM, which results in a margin of 23x and 18x versus the IC50 values for hERG and Nav1.5, respectively. Drug exposure at the 40 mg/kg/day dose (mean Cmax, free of 7.22 μM) results in further reductions in these margins. Increased PVC was seen at a Cmax, free of 1.53 μM (one animal at 5 mg/kg/day), which results in a margin of 84x for hERG and 65x for Nav1.5. The smaller the ratio between the IC50 for inhibition of an in vitro target to plasma drug levels (such as Cmax, free), the greater the potential for a pharmacologic response. Furthermore, consideration of the local tissue drug concentrations at the site of action, which could be significantly higher than levels in plasma is relevant.40,41 Although cardiac concentrations of Compound A are not available from the dog study, data in rats demonstrates that Compound A distributes to a high degree in cardiac tissue. This indicates that the local tissue levels of Compound A in the heart could have easily exceeded those in plasma, resulting in a critical concentration capable of inhibiting various cardiac ion channels. Confirmation of this hypothesis was demonstrated in the rabbit ventricular wedge and human IPSC cardiomyocyte assays, as discussed below.
The results obtained for Compound A using various in vitro ion channel assays did not provide sufficient information to generate a hypothesis for the arrhythmia observed in vivo. However, significant limitations exist with the utility of cell culture-based assays designed to test pharmacologic activity towards a single target. Such limitations include assay duration and voltage protocols, the use of non-human cell lines, data collection and analyses methods and the role that poly-pharmacology plays in complex biological systems. Drugs can inhibit multiple cardiac ion channels, with drug concentration impacting the net effect.31,42 Furthermore, high tissue distribution of a drug in myocardium (e.g., Compound A) is a factor that is not readily incorporated into cell-based ion channel assays. Therefore, alternate in vitro methods were utilized to investigate the proarrhythmic potential of Compound A.
Compound A and structurally related Compound B were studied for electrophysiological effects in the rabbit ventricular wedge assay. Compounds A and B prolonged QT at ≥.3 μM and Tp-e at ≥1 and ≥.3 μM, respectively, demonstrating that both drugs can inhibit Ikr (i.e., the hERG channel).24,29 Compound A also produced early after depolarizations and premature ventricular complexes at ≥5 μM. Compounds A and B both prolonged QRS at ≥5 μM, suggesting that both may inhibit the fast Na current (Nav1.5). The concentration-response data suggest that inhibition of Ikr can occur at lower concentrations than Nav1.5. QRS prolongation is associated with reduced left ventricle contractility and negative inotropy, as seen with the Class Ic antiarrhythmic flecainide. 43 The ion channel properties for both compounds were similar to one another, and both demonstrated the potential to elicit ventricular arrhythmia and conduction block. The concentrations of Compound A that elicited these changes (≥.3 μM) were less than the Cmax, free at the lowest dose in the dog study, supporting a pharmacodynamic–pharmacokinetic relationship for adverse cardiovascular effects.
Human IPSC cardiomyocytes have recently become a recognized in vitro model system for testing for proarrhythmic potential of small molecules and considered for use as a component of the Comprehensive in vitro Proarrhythmia Assay (CiPA).32,42 Human IPSC cardiomyocytes are also promoted as an in vitro system for evaluating drug-induced effects on cardiomyocyte repolarization by the ICH (see ICH Guidance E14/S7B Implementation Working Group: Clinical and nonclinical evaluation of QT/QTc interval prolongation and proarrhythmic potential Q&A, 21 February 2022). Compounds A and B both prolonged field potential duration (FPD) at ≥3 μM and produced cellular dysrhythmia at ≥10 μM and ≥3 μM, respectively, in this human cellular assay. Prolongation of FPD, followed by arrhythmia, can occur with block of Ikr alone or in combination with inhibition of Nav1.5 current, 33 which corresponds with the results obtained in the rabbit ventricular wedge assay although Compounds A and B prolonged QT at a 10-fold less concentration (i.e., at ≥.3 μM). The concentrations in which Compound A prolonged FPD in the human IPSC cardiomyocytes were in the same range of free plasma drug levels in dogs with arrhythmia.
The results of the studies conducted in the rabbit ventricular wedge and human IPSC cardiomyocytes demonstrated the potential for Compound A to block Ikr (hERG) and Nav1.5 currents providing a mechanism for the development of arrhythmia in vivo. The concentration ranges in which the ion channel effects were observed overlapped the free plasma drug levels in dogs providing a means for developing pharmacodynamic–pharmacokinetic relationships and support the translatability of data across in vitro and in vivo model systems. The ability for Compound A to distribute to a high extent in cardiac tissue provides an additional mechanism by which ion channel blockade could arise. Low molecular weight drugs that are sterically compatible with closed hERG channels can become trapped, whereas those that are incompatible with the closed/closing state become expelled during deactivation. Trappable blocker occupancy accumulates to steady-state levels over time, whereas that of non-trappable blockers vascillate during each action potential cycle (in which case, fast on-rates are essential for achieving proarrhythmic occupancy of the hERG channel). 44 Occupancy by a drug in the hERG channel at therapeutic levels may be tolerated for non-trappable blockers, but not for trappable blockers capable of building to a proarrhythmic occupancy level. 44 In the dog study, the cardiovascular changes observed with Compound A were sustained and increased in severity during the study, rather than peaking with Cmax and reversing by Cmin. These results are consistent with trappable ion channel blockers.
In 2013, the Comprehensive In Vitro Assay (CiPA) was proposed as a mechanistic, model-informed approach for predicting drug-induced torsades de pointes. The intent is to utilize mechanistic in silico cardiac electrophysiology models parameterized by in vitro data of drug action on various cardiac ion channels, with potential utilization of human stem cell-derived cardiomyocytes to assess the risk for proarrhythmia as an alternative to reliance upon the hERG assay or minor changes in QTc.45,46 While development of in silico models is ongoing, a white paper outlining general principles for validation of such models is available. 47 As with any model system, experimentation and validation will be necessary before widespread utilization occurs. This is particularly relevant for in silico-based approaches which can vary due software, data sets utilized for development, and parameters selected. In silico modeling was not performed for our investigation; however, structure-activity relationships with respect to hERG blockade and proarrhythmic risk is an area of active research. 48 Thoughtful consideration for the use and integration of in silico, in vitro, and in vivo cardiovascular data, along with pharmacokinetics, will be needed for evaluating the potential for proarrhythmia risk.12,49
In conclusion, in vitro ion channel assays may not identify cardiovascular risks observed in vivo, which can be affected by complex pharmacology such as multichannel effects, pharmacokinetics, and tissue drug distribution. The risk for arrhythmia may increase with a “trappable” ion channel inhibitor, particularly if cardiac tissue drug levels achieve a critical threshold for effect. A suggested strategy for derisking compounds for cardiovascular liabilities early in drug discovery is the following. Initially, standard in vitro ion channel assays (e.g., hERG, Nav1.5, and Cav1.2) should be conducted, and then on a case-by-case basis, testing in an ex vivo cardiac tissue assay (e.g., rabbit ventricular wedge) or in human IPSC cardiomyocytes should follow, prior to in vivo evaluation. Design of the testing strategy should consider (1) ion channel results of concern, (2) pharmacokinetic properties (especially high tissue or volume of distribution), (3) physical-chemical properties of the molecule, (4) primary pharmacology, and (5) intended indication.
Footnotes
Acknowledgments
The authors acknowledge the following individuals who lead the MPO inhibitors research program at the Novartis Institutes for Biomedical Research: Martin Marro and Andrew Patterson. The authors also acknowledge the assistance of Duncan Armstrong, Anatoly Lvov, Nargis Nasrin, and Richard Brochu for the conduct of the in vitro ion channel assays, and Robert Pearlstein for his insight regarding trappable and non-trappable hERG blockers.
Author Contributions
Brown, Alan P. contributed to conception and design, contributed to analysis and interpretation, drafted manuscript, and critically revised manuscript; Friedrichs, Gregory S. contributed to conception, contributed to interpretation, and critically revised manuscript; Tang, Hai-Ming contributed to design, contributed to acquisition, analysis, and interpretation, and drafted manuscript; Traebert, Martin contributed to design, contributed to acquisition, analysis, and interpretation, and drafted manuscript; Weber, Valerie contributed to acquisition and analysis and drafted manuscript; Yao, Nancy contributed to design, contributed to acquisition and analysis, and drafted manuscript. Yan, Gan-Xin contributed to design, contributed to acquisition, analysis, and interpretation, and drafted manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The authors Alan P. Brown, Greg Friedrichs, Hai-Ming Tang, Martin Traebert, Valerie Weber, and Nancy Yao declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: When these data were generated, these authors were employed by the Novartis Institutes for Biomedical Research. Gan-Xin Yan is employed by the Lankenau Institute and received funding from the Novartis Institutes for Biomedical Research to conduct the rabbit ventricular wedge assay.
Funding
The author(s) disclose receipt of the following financial support for the research, authorship and/or publication of this article: This work was supported by the Novartis Institutes for Biomedical Research.