
Abstract
Embryofetal toxicity studies are conducted to support inclusion of women of childbearing potential in clinical trials and to support labeling for the marketed pharmaceutical product. For biopharmaceuticals, which frequently lack activity in the rodent or rabbit, the nonhuman primate is the standard model to evaluate embryofetal toxicity. These studies have become increasingly challenging to conduct due to the small number of facilities capable of performing them and a shortage of sexually mature monkeys. The low number of animals per group and the high rate of spontaneous abortion in cynomolgus monkeys further complicate interpretation of the data. Recent FDA guidance has proposed a weight of evidence (WoE) approach to support product labeling for reproductive toxicity of products intended to be used for the treatment of cancer (Oncology Pharmaceuticals: Reproductive Toxicity Testing and Labeling Recommendations), an approach that has also supported the approval of biotherapeutics for non-cancer indications. Considerations to determine the appropriateness and content of a WoE approach to support product labeling for embryofetal risk include known class effects in humans; findings from genetically modified animals with or without drug administration; information from surrogate compounds; literature-based assessments about the developmental role of the pharmaceutical target; and the anticipated exposure during embryofetal development. This paper summarizes the content of a session presented at the 42nd annual meeting at the American College of Toxicology, which explored the conditions under which alternative approaches may be appropriate to support product labeling for reproductive risk, and how sponsors can best justify the use of this approach.
Keywords
Introduction
The expectations for developmental and reproductive toxicity (DART) testing of pharmaceutical agents are outlined in the ICH S5(R3) guideline. 1 This guideline describes the expectations for nonclinical testing to assess potential risks to fertility, embryofetal development (EFD), and effects on individuals who may be exposed in utero and/or during lactation (pre- and postnatal development, PPND). In general, most compounds are developed in accordance with the recommendations put forth in ICH S5(R3) 1 unless they are otherwise exempt (eg, for use in a patient population for which a particular test is not pertinent, such as embryofetal development studies in drugs for prostate cancer) or unless these tests are otherwise technically not feasible or justified.
According to the ICH S5(R3) guideline, 1 testing of embryofetal development for most drugs is expected to be conducted in two species, one rodent and one nonrodent. This expectation for a multi-species approach to DART testing arose in part as a result of the false negative result observed in rodents tested with thalidomide, 2 which formed the basis for its marketing approval in pregnant women in many parts of the world. While rodents failed to model the congenital malformations observed in humans, these effects were replicated in rabbits and NHPs. Although thalidomide is a small molecule, the thalidomide experience emphasized one of the key considerations for the design and conduct of predictive reproductive toxicity studies: that inter-species differences in the manifestation of embryofetal toxicity necessitate careful scientific justification when selecting species use to assess effects on embryofetal development in humans. 2
Given their exquisite specificity and high potency, most toxicity observed with biotherapeutics derives from extended or exaggerated pharmacology. The key criterion, therefore, for the selection of species used for safety evaluation of a biotherapeutic is pharmacological relevance. Although most toxicities associated with biotherapeutics are pharmacologically mediated, the extent to which animal studies predict outcomes in humans is in part a function of how well-matched the target-related processes are between humans and animals. Factors contributing to the predictive value of the preclinical model include similarities in target sequence homology (presumably reflective of binding affinity); physiological similarities between animals and humans for the intended pharmaceutical target; differences in pharmacokinetic properties of the drug, etc. In many cases, unless spontaneous human deficiency syndromes have been characterized in humans, the role of the intended target in embryofetal development may be better characterized in animals than in humans.
For drugs directed toward highly conserved targets, sufficient binding may be achievable to permit toxicological evaluation in a rodent model. For most biotherapeutics, however, binding and/or pharmacological activity is inadequately conserved between humans and rodents, and the only suitable toxicology species is the nonhuman primate. Monkeys pose difficulties for evaluation of many reproductive effects. Fertility, for example, cannot be readily evaluated in the monkey. It is, for example, generally agreed that it is impractical to conduct mating studies in nonhuman primates. 3 This is in part due to the low rate of pregnancy (estimated to be around 36% for successful male breeders). 4 A similarly low rate of pregnancy has been observed in female nonhuman primates. Per the ICH S5(R3) guideline, 1 inference about potential effects on fertility can typically be gleaned from histopathological evaluation of reproductive tissues taken from male and female animals in general toxicology studies of at least 3 months duration. In males, histopathology is considered the gold standard for detection of effects on spermatogenesis and most findings that result in decreased or abnormal sperm production will be visible histologically. A determination about the mechanistic underpinning of the effects and potential for reversibility can often be made by a pathologist with specialized expertise in gonadal histopathology. In males, effects on sperm maturation or capacity of sperm to successfully fertilize an ovum, however, are more difficult to detect histologically and may require an analysis of sperm motility and morphology, which requires specialized equipment and staff trained to perform such evaluations. Because semen collection can be performed non-terminally in the context of general toxicology studies, serial observations can be conducted within individual animals.
Administration of exogenous proteins in the monkey often leads to formation of anti-drug antibodies (ADA) leading to increased clearance and/or neutralization of the pharmacodynamic properties of the drug in pregnant dams. Concerns about the impact of ADA on the interpretation of data are more pronounced in studies of longer duration. In rats and mice, for example, ADA formation may not occur at levels sufficient to impact overall study interpretation because the duration of dosing and exposure are relatively short. In monkeys, however, in which the duration of dosing and exposure are much longer, the likelihood that maternal ADA formation may impact fetal exposure to the drug is higher. In some cases, ADA formation has precluded meaningful evaluation of biotherapeutics in reproductive studies in NHPs and such an occurrence may be adequate to justify the use of an alternative model. In addition, placental pathology secondary to immune complex deposition (either secondary to autoimmune disease, or in theory, to ADA-related immune complexes) may increase the risk of pregnancy loss5-7 and may confound overall study interpretation regarding the risk of embryolethality.
In the past few years (2020-2021) FDA has approved biologic products in neoplastic and non-neoplastic indications using alternative approaches for fulfilling the reproductive and developmental toxicity requirements. These alternative approaches have included utilizing surrogate therapeutics in mice, as well as supporting the decision not to conduct studies based on the known mechanism of action, literature and/or reported clinical findings of the product or products in the class. In the US Package Insert (USPI) for Rybrevant® (amivantamab-vmjw), a bispecific epidermal growth factor receptor (EGFR) and mesenchymal-epithelial transition (MET) receptor antibody approved in 2021, the reproductive and developmental toxicology labeling references literature in transgenic mice, so no studies were conducted with Rybrevant. Jemperli® (dostarlimab-gxly), a programmed death receptor-1 (PD-1)–blocking monoclonal antibody, includes labeling based on the class of PD-1/PD-L1–blocking antibodies which are known to have the potential to cause fetal death. The USPI for Spevigo® (spesolimab-sbzo), an interleukin-36 receptor antagonist antibody (anti-IL36R), references embryofetal and fertility studies in mice with a surrogate antibody. Similarly, Opdualag®, a combination product of nivolumab and relatlimab-rmbw, an anti-LAG3 (lymphocyte activation gene-3) antibody, conducted mouse reproductive and developmental studies with a surrogate anti-LAG3. This is consistent with current regulatory guidance1,8 which highlights the potential use of alternative models as well as waiving in vivo studies when scientifically justified with other relevant data (mechanism of action, literature, clinical safety, and drug class information).
Another consideration for the conduct of reproductive toxicity studies with biotherapeutics is the difference between animals and humans in the kinetics of compound distribution across the placenta. Unlike small molecules, which are typically capable of crossing the placenta, distribution for many biotherapeutics is often tightly controlled and may change throughout gestation. Although some biotherapeutics with teratogenic properties have been identified in animals, 9 in most species, placental transfer of the biotherapeutic to the fetus is low during the period of organogenesis, and the mechanism by which antibodies and other biotherapeutics is transferred across the placenta often differs between humans and other species. This results in a pattern of exposure in animals that may not fully replicate the anticipated effects in humans. In mice, rats, rabbits, and guinea pigs, antibody transfer across the placenta occurs via the inverted yolk sac splanchnopleure. In humans and NHPs, FcRNs that can transport mAbs are expressed in the placenta sometime during the second trimester which is why embryofetal mAb exposure is limited early in gestation. 6
Despite these differences, for the testing of most monoclonal antibodies, demonstrating maintenance of adequate maternal exposure has been considered sufficient to support product labeling when evaluated in rodents or rabbits (when pharmacologically relevant) and in nonhuman primates.
For products with unique structural properties for which placental transfer mechanisms and/or kinetics may be unknown, additional data beyond standard binding and pharmacodynamics may be needed to support selection of a suitable toxicology species on a case-by-case basis. Similarly, if considering a model not widely used to characterize the reproductive effects of biotherapeutics, a study of the kinetics and placental transfer may be needed to adequately justify the model. These concepts and scientific rationale are consistent with ICH guidance.
The most common design for evaluation of embryofetal and postnatal development in the nonhuman primate is the enhanced pre- and postnatal development study. These studies are typically designed to administer the drug in pregnant dams from the confirmation of pregnancy (GD20) to parturition, and offspring are subjected to a limited assessment of relevant developmental parameters (eg, neurodevelopmental and immunological). For the studies to be interpretable, they should also be designed to include a large enough number of pregnancies and conceptuses that it is feasible to differentiate spontaneous, low-frequency events from drug-induced changes. The design of these studies should be sufficient to allow a meaningful interpretation of the data. Commonly, 16 pregnant dams are used per group and the studies often explore dose-related effects through inclusion of two or more active dose levels. As a result, these studies often contain more than 60 pregnant dams, although as described below, single dose-level evaluations have been performed and found to be acceptable in some contexts. Given the high rate of spontaneous abortion in macaques, the number of resulting offspring per group may be considerably less than 16. 10 One study estimated that a vehicle control group with a group size of 16-20 pregnant dams had an 80% probability of having 13-16 pregnancies on GD 100 to evaluate, indicating that the rate of spontaneous abortion was approximately 20%. 10 A more recent study 11 found that group sizes of 14-24 maternal animals resulted in more than 6-8 infants, supporting a reduction in group size to 14; this is slightly lower than the current recommendation in ICH S5(R3) 1 of 16 maternal animals per group.
Standard embryofetal studies in the rodent use 20 litters per group, each with multiple offspring per litter. These group sizes were statistically powered to differentiate spontaneous events from treatment-related effects. With only 1 infant per monkey, the number of evaluable offspring per group is relatively low for NHP studies. The ICH S5(R3) guidance (2021) 1 recommends at least 16 maternal cynomolgus monkeys per group and 2 dose groups which appears to reflect flexibility for the required animal numbers for the ePPND study. CROs conducting the NHP ePPND study have different rates of spontaneous abortion which may change over time and should be evaluated prior to conducting a study.
For these studies to be interpretable, highly trained, and knowledgeable staff are needed and the facility needs to have sufficient experience and historical data to reliably distinguish treatment-related findings from spontaneous background events; thus, relatively few facilities perform ePPND studies in NHPs. These studies are also extremely long in duration given the low rate of pregnancy in female monkeys and the long duration of gestation. Because of the low rate of pregnancy, females are enrolled onto the study one at a time; therefore, accrual can take several months to complete. In addition, to evaluate effects on developmental endpoints, infants are often retained for up to 6 months postpartum. During times of extreme shortages of sexually mature animals, such as occurred during and immediately after the peak of the COVID-19 pandemic, the times to schedule and run these studies were much longer, which can impact the overall timelines for the development and approval of drugs, particularly in non-oncology indications. Some laboratories schedule studies 2 years in advance and in many cases, there is a waiting list for those placements, so the actual time to initiate a study may be considerably longer than 2 years.
Overview of DART Testing: Scientific Considerations for the Use of Alternative Models to Assess Reproductive Risk With a Biotherapeutic
Traditional Testing Methodology for Small Molecules
The ICH S5 R3 (2021) guidance 1 for detection of toxicity to reproduction for human pharmaceuticals and the ICH S11(2020) guidance, Non-Clinical Safety Testing in support of Development of Pediatric Pharmaceuticals 12 provides the guidance for testing any new drug (small or large molecule) or vaccines. These guidance documents specifically exclude gene and cell therapy products.
The stages of reproductive life that are covered are noted in the reproductive life cycle Figure 1 with the actual testing guidance to cover the life cycle divided into segments that provide a mechanism for collecting data on the hazard of exposure to a part of the life cycle, thereby providing data on when to and when not to use a drug. These segmented designs include: three types of studies covered in the ICH S5(R3) guidance
1
and juvenile toxicity studies covered in ICH S11.
12
Stages of the reproductive life cycle and the phases of reproductive toxicology studies over which drug-related effects on reproductive parameters are commonly tested.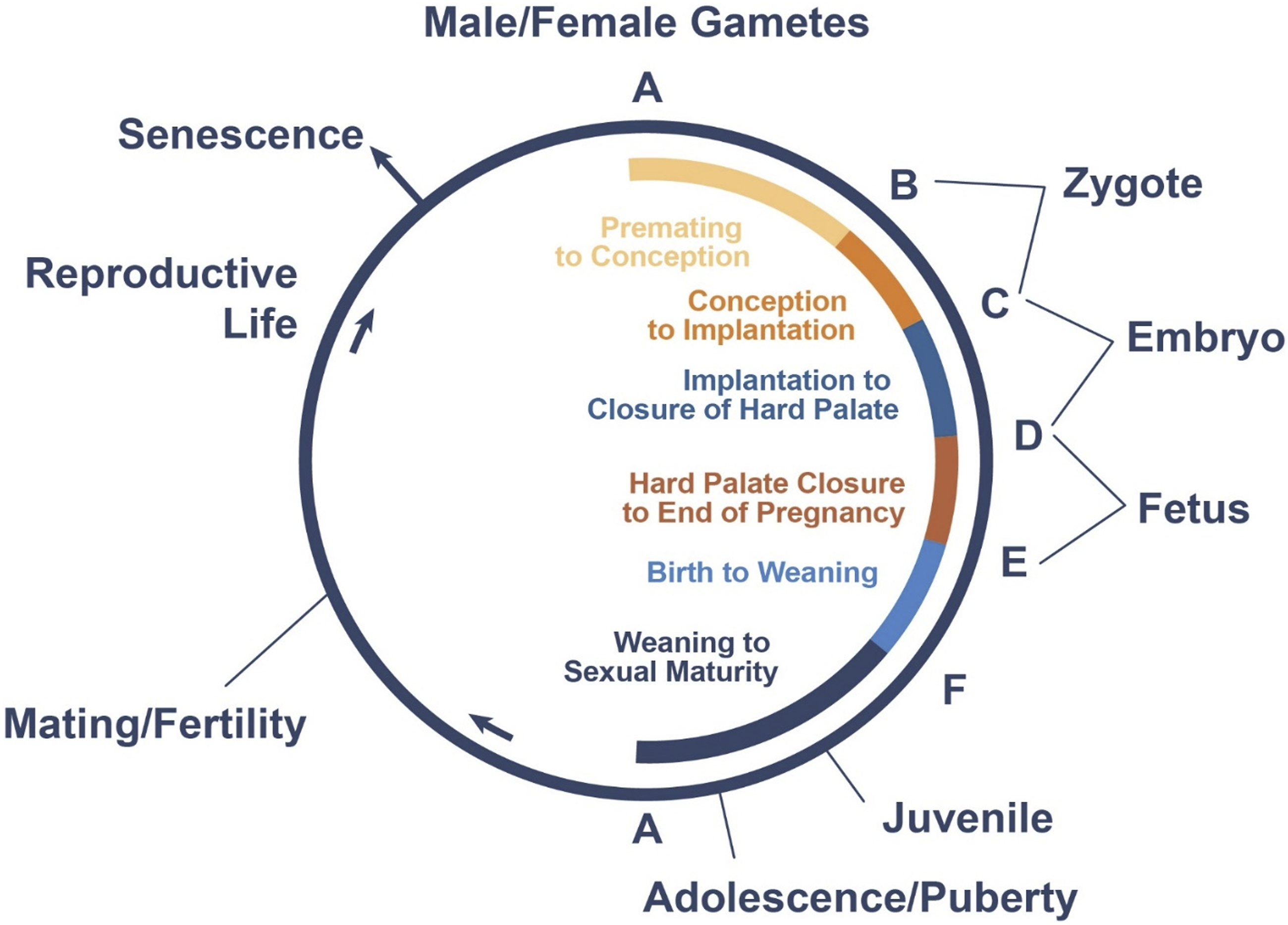
The studies are: • Fertility and Early Embryonic Development (FEED) Study: (previously known as Segment I, covering ICH Stages A and B) and used to assess the hazard of the test article to functional male and female fertility including adverse effects on male and female gametes, mating performance, and development of the fertilized ova through implantation. • Embryo-Fetal Development (EFD) (previously known as a Segment II, Teratology or Organogenesis Study; covering ICH Stages C to D): designed to assess the hazard to the embryo for three of the four classic endpoints of developmental toxicity (fetal death, malformation, growth). • Perinatal/Postnatal Development Evaluation: (PPND) (previously known as Segment III; covering ICH Stages C Through F): designed to assess the hazard of the test article to the embryo, fetus, gestation length, duration of parturition, maternal nursing behavior, offspring morbidity, growth, and development through production of an F1 generation; following the selection of at least one male and female from each litter in a group at weaning, the F1 generation is monitored for sexual maturation and functional assessments (the fourth endpoint of developmental toxicity) including behavioral changes prior to production of an F2 generation. • Juvenile Toxicity Study: Designed to assess the hazard to offspring not exposed in utero or in the milk, but from direct exposure to the test article during critical periods of postnatal development of most organ systems.
Figure 2 provides a schematic of the relationship of the three evaluations for reproduction and developmental toxicity. While most of the DART and juvenile studies are conducted in rodents and rabbits, other rodents including hamsters and guinea pigs and non-rodents including mini-pigs and dogs can be used especially when the metabolism more closely matches humans for small molecules or when homology of proteins (biopharmaceutical) is closer to humans in one of these species. Schematic depicting the designs of the standard reproductive toxicology studies for pharmaceuticals. Illustrated are the designs of the FEED (fertility/FEED); EFD, embryofetal; and PPNDI, pre- and postnatal development studies.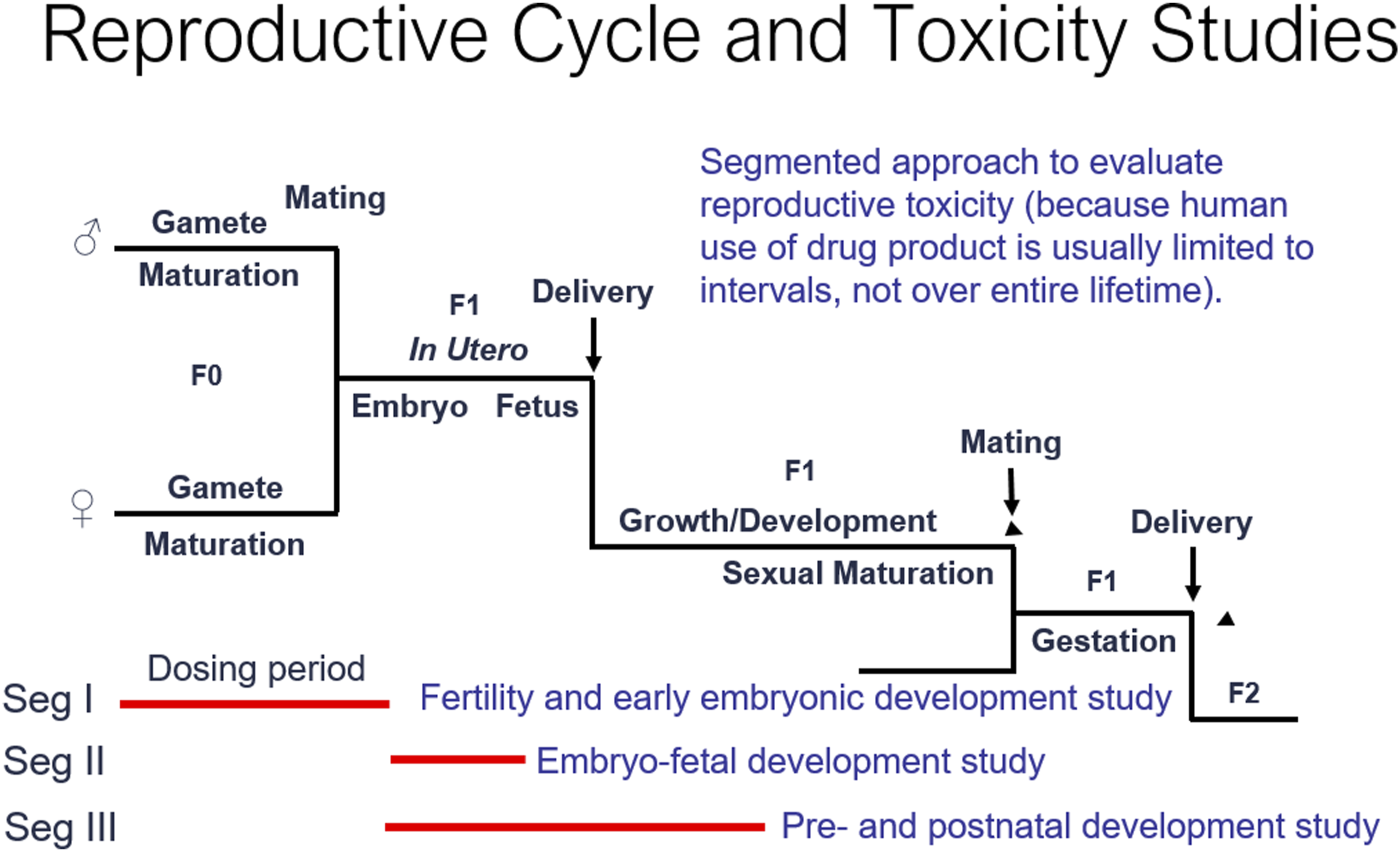
Group size for rodents is typically 20 pregnancies (minimum 16) per group. Similar group sizes are used for male rodents as mating should be one male to one female.
Modifications of Traditional Methodology
Antigenicity can often occur if a human protein is administered to another animal species. The antigenicity can be neutralizing for the drug resulting in no or minimal exposure occurring in the test species. The presence of anti-drug antibodies (ADA) does not necessarily disqualify an animal species if exposures are not markedly altered. However, if ADA does not develop for two weeks or even after only a few days, a rodent and/or rabbit model may be an appropriate species for the EFD study. The number of doses and timing of the doses will depend on the extent of ADA and the timing of the development. TK and the effect of ADAs are generally used to determine the frequency of dosing for molecules that have long biological half-lives, such as mAbs. With mAbs, a once-weekly dosing regimen is often acceptable since large concentrations of the drug are often still circulating in plasma 7 days after dosing.
When possible, the traditional models (rodents and rabbits) should be considered as regulators understand these models and large historical control databases exist. Daily dosing of a biopharmaceutical in rodents and especially rabbits can also be limited because of toxicity (usually renal toxicity) due to protein overloading; however, due to their long plasma half-lives, dosing every other day or even less frequently may further still ensure exposure without causing protein overload. When NHPs are the only appropriate model, the FEED study will almost never be conducted due to the low success rate of mating NHPs in the laboratory environment. As described in ICH S6(R1) 3 fertility-related endpoints in NHPs are only evaluated in a general toxicity study in sexually mature animals by monitoring functional endpoints, if data from the drug’s mechanism, or repeat-dose toxicity findings warrant it. Such monitoring often includes: hormonal assessments throughout the female cycle and/or observational evaluation of normal cycling in females, the monitoring of sperm production in males and histology of the reproductive organs, including a stage aware analysis of testicular sperm production and ensuring all stages of follicle assessment in females. When using NHPs for evaluation of the effects on embryo/fetal development, a combined EFD and PPND study [enhanced PPND (ePPND) study] design is used as all four endpoints of developmental toxicity can be monitored while using approximately one-half of the NHPs that would be needed if separate studies are conducted. Only an EFD study might be considered when teratogenicity is expected and/or postnatal survival is expected to be compromised.
Advantages of Using Rodents for Embryofetal Assessments of Biotherapeutics.

Alternative Models
Surrogate molecules that mimic the human pharmacology in a species that is not responsive to the clinical therapeutic may provide an opportunity to evaluate DART and juvenile toxicity, with the caveat that any surrogate molecule will need to be manufactured and characterized just like the clinical product even though it will only be used for non-clinical studies in support of use in humans. Although there are no established criteria or regulatory guidance for studying the pharmacology and toxicology of a surrogate, characterization should include a comparison of the dose of the surrogate relative to the clinical product. All this work for a molecule that will never be in the clinic can be time consuming and expensive.
Transgenic models (usually transgenic mice), either knock in (humanized) or knock out models, can provide an appropriate species for testing. When a human gene is inserted into the mouse model to allow the clinical candidate to be tested it must be understood that only one human gene has been inserted and the rest of the genes are still from the mouse. Data, such as the use of a functional biomarker, should be collected to justify the ability of these models to replicate anticipated effects in humans.
Alternatively, a knock out model with the deletion of a gene whose product is the protein being suppressed by a monoclonal antibody, can also provide useful information for hazard assessment, understanding that the total elimination of a protein is different from pharmacologic suppression of a protein. Studies in knock-out mice can be straightforward as the entire reproductive life cycle can be covered in one study; combined FEED, EFD, and PPND with only one set of knock-out mice being compared to the wild-type mouse used to make the knock-out model.
Conclusions
• Do not forget traditional methods and adaptations of these methods • Three main options are used as alternate strategies for the evaluation of reproductive toxicity of therapeutic proteins • Surrogate molecules • Knock out/transgenic mouse • Knock in/transgenic mouse with administration of surrogate • The reasons for needing an alternate strategy could be due to species specificity, and the need to reduce the use of NHPs • Per ICH S6(R1)—if there is not species specificity, the lower order animals are preferred for a mAb or protein even if it also is active in NHPs. • All options have caveats, as well as resource considerations, and must be evaluated as part of a “weight of evidence” approach. • Consider your needs early in the program!
Use of WOE in Assessing Embryofetal Developmental Toxicity of Therapeutic Proteins: A CDER Perspective
Introduction
As noted above, there are multiple approaches that can be taken for assessing the potential of a biopharmaceutical to cause embryofetal developmental (EFD) toxicity. Which approach is most appropriate is going to be defined by program-specific parameters, such as (1) the indication, (2) the patient population, (3) knowledge of the target’s role in supporting embryofetal development, (4) availability of a pharmacologically relevant species, and (5) the ability of the biopharmaceutical to affect endogenous molecules. This program-dependent flexibility is captured in multiple guidances, as described above.
In light of the on-going, COVID-19-related disruption in the supply of NHPs—especially sexually mature NHPs, we wanted to understand to what extent alternatives to studies conducted in the NHP have been used to inform on EFD risk for biopharmaceuticals approved over the last few years, and whether there were opportunities to further reduce NHP use in assessing the potential for EFD toxicity. To approach this question, the labels for CDER-regulated BLAs (excluding biosimilars) approved over the 6-year period from 2015 to 2020 were analyzed for the source of nonclinical information that supported the risk summary statement in section 8.1 of the approved product label. If either EFD or PPND endpoints were evaluated in the NHP, the label was considered to be informed by the NHP, even if other models were also used in the developmental toxicity risk assessment.
Results
Of the non-biosimilar BLAs that were identified as being approved by CDER between 2015 and 2020, inclusive, we found that, across all indications, 67% of the risk summary statements appearing in section 8.1 of the labels did not use data from the NHP to inform on EFD risk Figure 3 (Panel A). 29% relied on data from studies of the active pharmacological ingredient (API) conducted in the rodent and rabbit; 21% relied on a WoE assessment wherein the mechanism of action (MoA) was sufficient to characterize and communicate risk; 10% relied on a WoE assessment of negligible risk based on mode of action (eg, exogenous target) or patient population (these are captured as “no assessment” in Figure 1), 4% relied on studies in the rodent using a rodent-active surrogate, while 3% relied on genetically modified animals—either modified to respond to the API, or knock-out intended to model the effect of the API. Percentage of Labels for BLAs approved by CDER between 2015 and 2020, inclusive, in which the risk summary appearing in Section 8.1 of the label is informed by the data source indicated. (A) All indications, B) non-oncology indications, and C) oncology indication.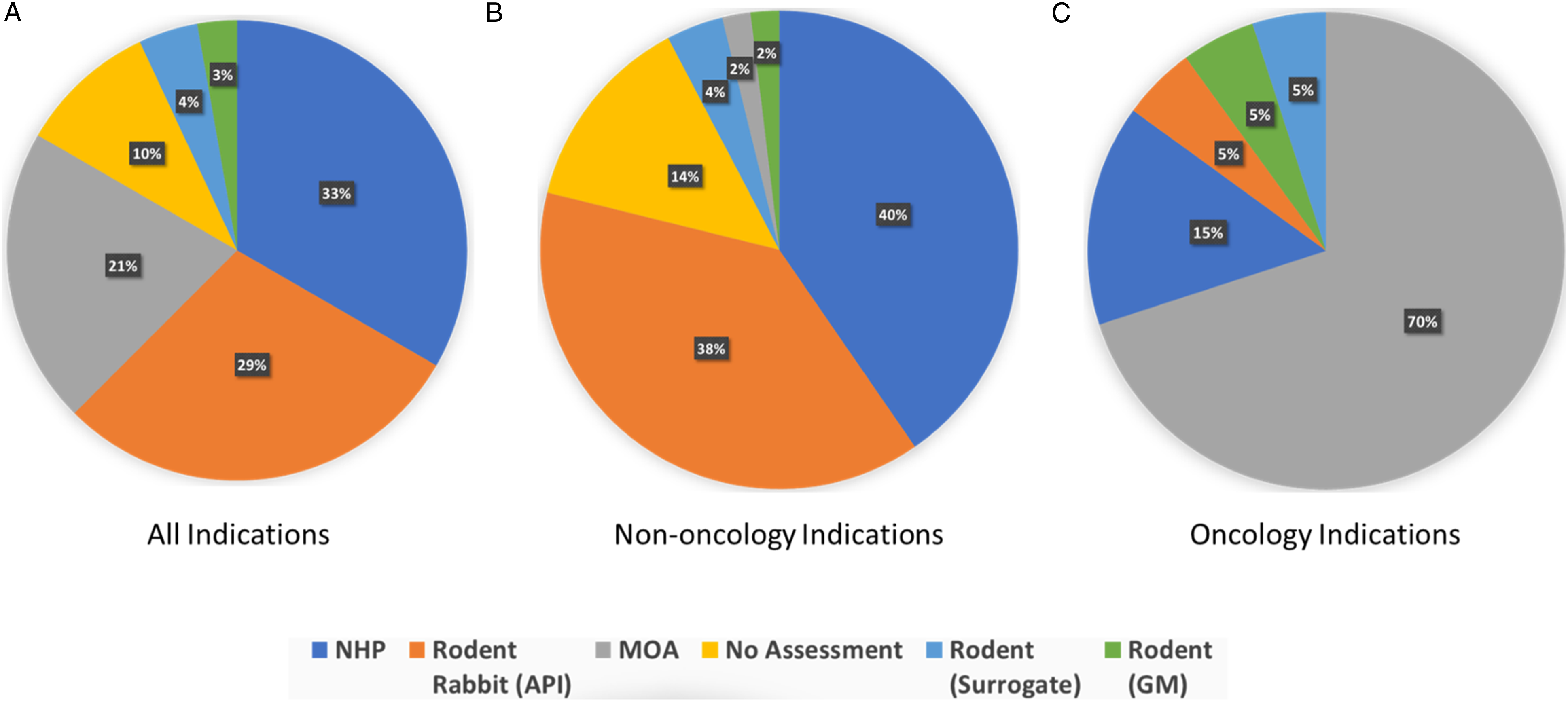
When analyzing only the BLAs for non-oncology indications Figure 3 (Panel B), we found that 40% relied on data from studies conducted with the API in the NHP, 38% relied on data from studies of the API conducted in the rodent and rabbit, 14% relied on a WoE assessment indicating negligible risk (captured as “no assessment”), 4% relied on studies in the rodent using a rodent-active surrogate, 2% relied on an a WoE assessment wherein the mechanism of action (MoA) was sufficient to characterize and communicate risk, and 2% relied on genetically modified animals (modified to respond to the API).
Looking at only the oncology BLAs Figure 3 (Panel C), we found that fully 70% of labels relied on the MoA for assessing and communicating risk, 15% relied on data generated in the NHP, 5% relied on studies of the API conducted in the rodent and rabbit, 5% relied upon studies of a rodent-active surrogate, and 5% relied upon genetically modified animals (knock-out animals).
Oncology Products With Labels Supported by WoE Assessment.
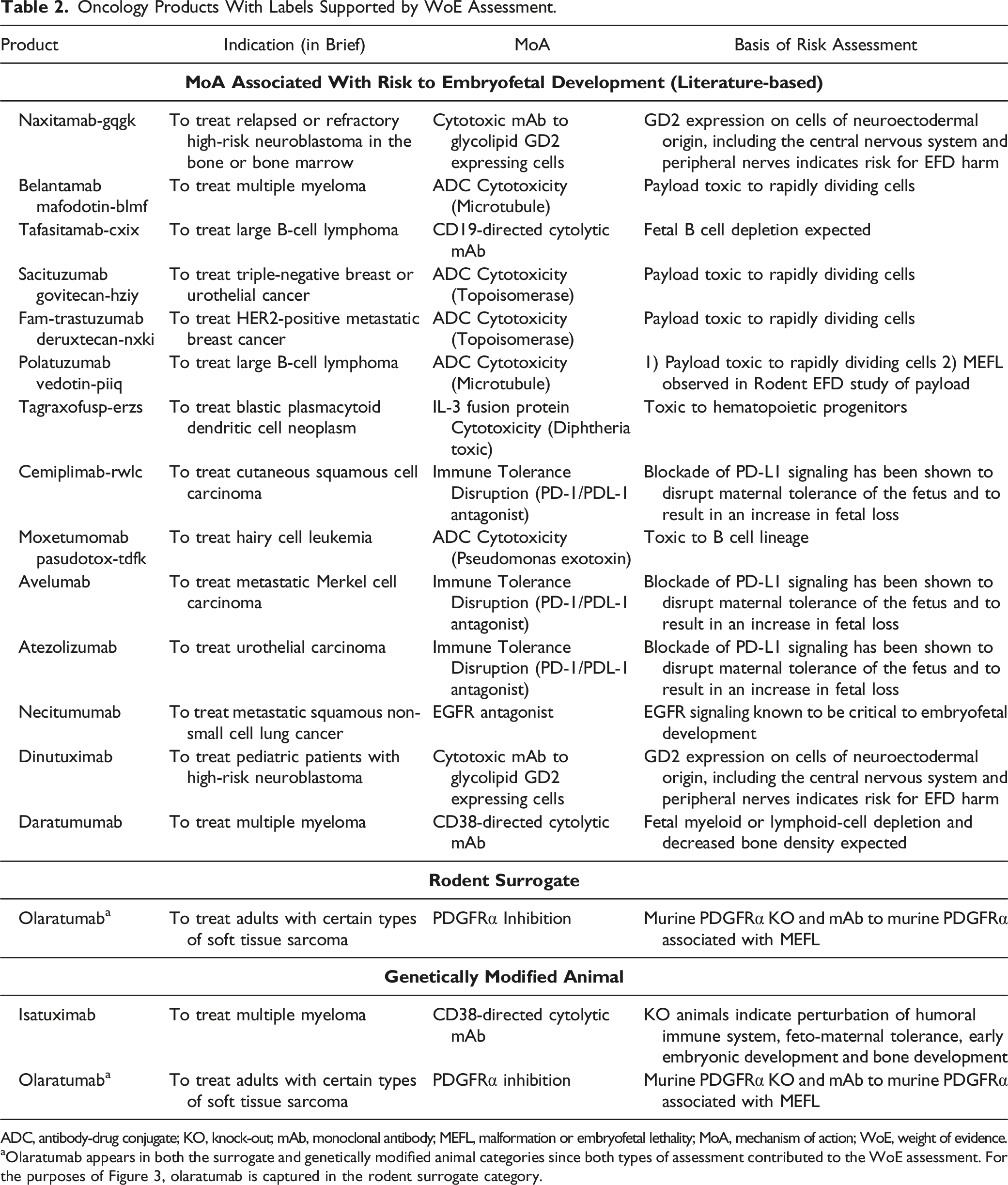
ADC, antibody-drug conjugate; KO, knock-out; mAb, monoclonal antibody; MEFL, malformation or embryofetal lethality; MoA, mechanism of action; WoE, weight of evidence.
aOlaratumab appears in both the surrogate and genetically modified animal categories since both types of assessment contributed to the WoE assessment. For the purposes of Figure 3, olaratumab is captured in the rodent surrogate category.
Non-Oncology Products With Labels Supported by WoE Assessment.
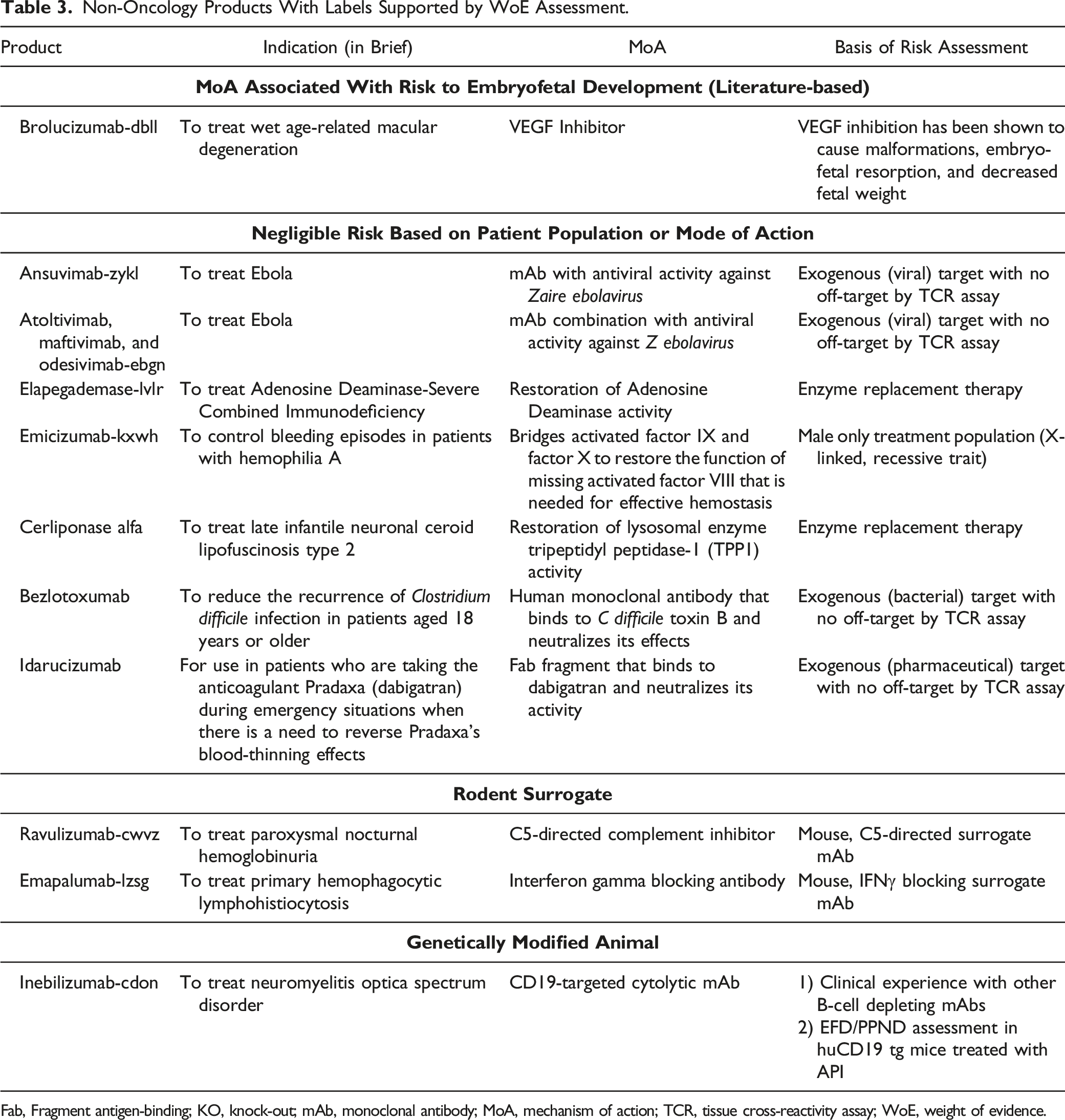
Fab, Fragment antigen-binding; KO, knock-out; mAb, monoclonal antibody; MoA, mechanism of action; TCR, tissue cross-reactivity assay; WoE, weight of evidence.
So, in practice, CDER has supported WoE labeling for developmental toxicity risk when there is clearly evident risk associated with the mechanism of action, or when there is a clearly evident absence of risk (or negligible risk) based on the patient population or mode of action.
Considerations
Clearly, sponsors are having some success in licensing BLAs in the United States using data sources other than the NHP for assessing EFD toxicity risk. To the extent that this is scientifically appropriate for their particular biopharmaceutical development program, sponsors should consider whether there are adequate data that already exist (or that can be generated) that would allow for adequate characterization and communication of risk for EFD toxicity, rather than defaulting to conducting a developmental toxicity assessment in the NHP.
In 2022, FDA published guidance intended to help sponsors mitigate the effects of the COVID-19 pandemic on the NHP supply 13 This guidance (which was allowed to lapse in May of 2023 with the termination of the COVID-19 public health emergency) restated and emphasized the flexibilities provided in other FDA guidances that support approaches to EFD assessment that do not rely on testing in NHPs, such as WoE assessments, including studies in the rodent with a rodent-active surrogate or using genetically modified animals. 14
The FDA also recognizes that there can be legal challenges in the United States to relying on data not owned by the sponsor, when seeking to license a 351(a) (non-biosimilar) biologic, especially if these data are owned by competitors. FDA encourages industry to consider whether there may be opportunities to make EFD data generated in the NHP by innovator products available by right-of-reference in order to avoid sponsors having to conduct scientifically unnecessary and wasteful studies in the NHP.
A Pharmaceuticals and Medical Devices Agency Perspective on the Necessity for Developmental and Reproductive Toxicity Studies Using Non-Human Primates in Biopharmaceuticals
Introduction
Developmental and Reproductive toxicity (DART) of biopharmaceuticals should be evaluated according to the indication and target patient population in clinical practice. In addition, DART studies should be considered based on the ICH S5(R3) 1 and ICH S6(R1) 3 guidelines; the ICH S6(R1) guideline 3 recommends that for biopharmaceuticals that are pharmacologically active only in NHPs, consideration should be given to conducting DART studies using NHPs. However, given the limitations of using NHPs for DART evaluation and the recent increase in the adoption of animal welfare principles of replacement, reduction, and refinement (3Rs), as well as the shortage of NHP supply due to the COVID-19 pandemic, the need for animal studies using NHPs should be more carefully considered. Here, we would like to present our alternative approach to DART risk assessment of biopharmaceuticals when NHPs are the only relevant animal species.
DART Risk Assessment When the NHP Is the Only Relevant Species
DART studies in NHPs are considered to have advantages in terms of similarity to humans such as phylogenetic and physiological characteristics and placental transfer of antibodies. On the other hand, from the standpoint of study feasibility and animal welfare, the DART studies using NHPs have limitations, including difficulty in quantitative risk assessment because it is usually conducted in a single dose group. Considering these limitations in DART studies using NHPs, we believe that it is possible to evaluate not only necessarily by conducting the studies, but also by utilizing a weight of evidence (WOE) approach and surrogate-based studies, Figure 4. DART risk assessment when the NHP is the only relevant species.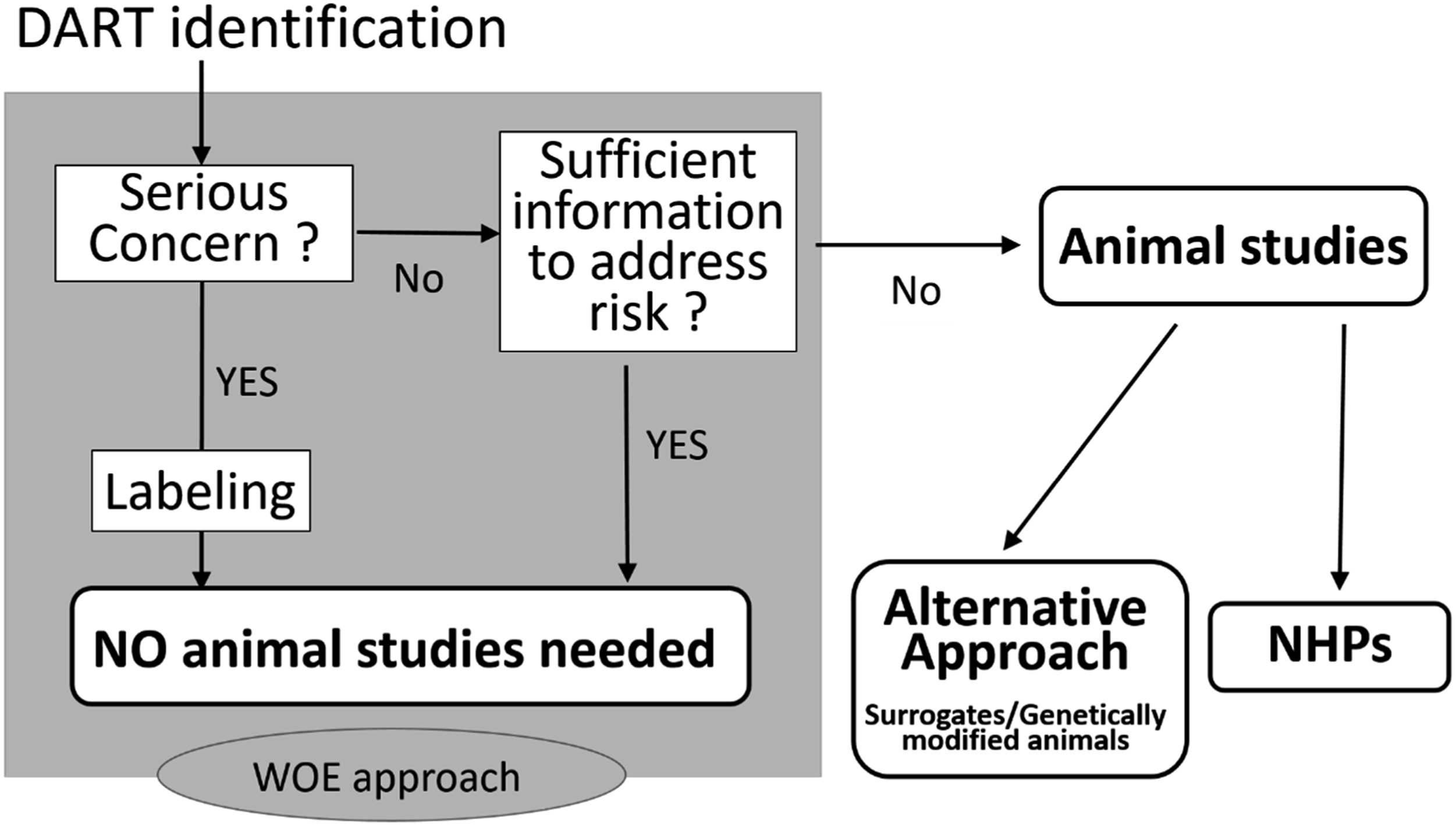
(1) WOE Approach
Biopharmaceuticals are considered to have high specificity to their targets and have a lower potential to cause DART due to off-target toxicity compared to small chemical entities. Therefore, an approach, including review of scientifically relevant information from various sources can be useful. By comprehensively reviewing publicly available information (eg, MOA of the biopharmaceuticals, the detailed biological properties of the target molecule, information of genetically modified animals or human disease, and information on class effects), it would be possible to collect information on the potential hazard of biopharmaceuticals without conducting in the DART studies in NHPs.
In cases where relevant information from literature and databases indicates that the MOA raises serious concerns for embryo-fetal development due to the exaggerated pharmacology of a biopharmaceutical, it would be difficult to deny the hazard even if a DART study is conducted and no fetal toxicity is detected. For example, biopharmaceuticals that are expected to cause serious concerns about embryo-fetal development based on their MOA include anti-VEGF antibodies (embryo-fetal lethality or malformation due to inhibition of angiogenesis) or anti-PD-1 and anti-PD-L1 antibodies (embryo-fetal lethality due to destruction of immune tolerance during pregnancy). For such cases, as stated in ICH S6(R1), 3 we believe that information on adverse effects on embryo/fetal development should be provided without animal studies. When administered to women of childbearing potential, subjects should be nonpregnant and use highly effective contraception.
On the other hand, where the biopharmaceutical properties of the target molecules of biopharmaceuticals are well characterized by existing information (eg, phenotype of genetically modified animals and information on class effects), it may be possible to judge from this information that the risk of DART is low and avoid another DART study using NHPs.
In addition, as DART studies for biopharmaceuticals can be conducted during Phase III, it may be possible to determine the necessity of DART studies based on PK data from clinical trials, in accordance with ICH M3 guidance. 15 In case of locally administered biopharmaceuticals, such as intravitreal preparations, systemic exposure of the drug can be very low. If the systemic exposure in humans is less than the minimum anticipated biological effect level (MABEL), DART associated with the pharmacological effect is not expected to occur. In such cases, the value of conducting DART studies using animals could be low.
As described above, we believe that the WOE approach based on MOA, detailed information on biological properties of target molecules, information on class effects, and information on systemic clinical exposure level can make it possible to determine the necessity of DART studies using NHPs. However, in the WOE approach, the sufficiency of the obtained data, consistency and reliability of the results should be considered, and the necessity of a DART study using animals should be carefully considered.
(2) Use of Homologous Protein/Genetically Modified Animals
When sufficient information on DART cannot be obtained by the WOE approach, it is necessary to consider conducting a DART study. In such cases, evaluation by an alternative approach using homologous proteins or genetically modified animals expressing human target proteins can be considered instead of NHP studies. As shown in ICH S6 guideline, 3 studies with the alternative approach are generally not useful for quantitative risk assessment because the biological activity of homologous proteins and clinical candidates is not identical and the expression levels and functions of human target molecules in genetically modified animals are not necessarily equivalent to those in humans. However, similar limitations exist in DART studies using NHPs; thus, it is considered possible to avoid NHP studies by using the alternative approach. When the sponsor chooses the alternative approach, it is necessary to explain its appropriateness in terms of the pharmacological effects caused by the test substance, the target specificity of the homologous protein, and the biodistribution of the target molecule in genetically modified animals.
Conclusions
Considering the limitations of DART risk-assessment, and ethical issues of NHP studies, there may be room for the active use of WOE assessment and alternative approaches with homologous proteins/genetically modified animals when the clinical candidate is pharmacologically active only in NHPs, utilizing accumulated information on the MOA of biopharmaceuticals, literature information of target molecules, and phenotypes of genetically modified animals. When these alternatives to NHPs studies are scientifically appropriate, we believe it would be possible to avoid the NHP studies. In addition, the necessity and study design of DART studies vary depending on the clinical indication, target patient population, and treatment options in Japan. It is recommended that these approaches be discussed and agreed upon with the regulators at the appropriate time.
When developing a weight of evidence assessment, a thorough evaluation of the available mechanistic data gleaning the role of the target protein in human and/or animal development; data from studies in genetically-modified or -deficient animals; data from spontaneous human deficiencies; toxicology data defining the known toxicities of the drug and the feasible or tolerable dose-ranges evaluable in NHPs; and data from clinical pharmacology evaluations in patients should be synthesized in the request. The weight of evidence evaluation should address both the known or suspected developmental effects defined by proprietary and/or published scientific literature, as well as any residual uncertainty. Because a refusal to grant a WoE assessment can have a significant impact on the timeline of a development program, early feedback is recommended.
Workshop Summary and Future Perspectives
• A growing number of biotherapeutics have been approved using alternative approaches. • Although the European Medicines Agency (EMA) was not represented in this session, representatives from the other founding ICH regulatory jurisdictions indicate a willingness to consider a flexible approach to evaluating the reproductive toxicity of biotherapeutics, including a weight of evidence assessment; the use of transgenic models; and the use of species-specific surrogate molecules in alternative animal species instead of conducting an NHP DART study. That EMA has stated that it will provide special support
16
for developers who are intending to use alternative approaches to animal testing suggests that they would, as warranted and on a case-by-case basis, consider the use of appropriately justified alternative strategies for reproductive toxicity testing of biotherapeutics. • Surrogate molecules may represent a significant investment in time and manufacturing cost because compounds used in reproductive toxicology studies need to be adequately characterized to meet the expectations for GLP compliance. • The use of an alternative species, particularly a genetically manipulated model (eg, transgenics and KO mice) should be scientifically justified to support biological relevance to humans. • Considering the limited availability of monkeys, the long lead times needed to initiate a study in monkeys and the long duration of testing, sponsors should seek advice on the acceptability of an alternative reproductive assessment plan as early as practicable in case a standard approach in monkeys is needed. It is recommended that sponsors seek input from global health authorities to ensure that all regions agree with the proposed alternative model. • When seeking input from regional health authorities, sponsors may wish to clarify whether a study in the monkey is needed if the results obtained in the alternative model deviate from expectation (particularly if an unexpectedly negative result is obtained).
Footnotes
Acknowledgments
The authors wish to acknowledge the contributions of the anonymous reviewers who provided many thought-provoking and insightful comments that improved our summary of this session.
Author Contributions
Hoberman, A contributed to acquisition. Misaki, N, Fumito, M, and Kazushige, M contributed to conception and design and contributed to acquisition, analysis, and interpretation. Lansita, J, and Weis, S contributed to conception and design and contributed to acquisition. Wange, R contributed to design and contributed to acquisition and interpretation. All authors drafted manuscript, critically revised manuscript, gave final approval, and agrees to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.