
Abstract
An important safety consideration for developing new therapeutics is assessing the potential that the therapy will increase the risk of cancer. For biotherapeutics, traditional two-year rodent bioassays are often not scientifically applicable or feasible. This paper is a collaborative effort of industry toxicologists to review past and current practice regarding carcinogenicity assessments of biotherapeutics and to provide recommendations. Publicly available information on eighty marketed protein biotherapeutics was reviewed. In this review, no assessments related to carcinogenicity or tumor growth promotion were identified for fifty-one of the eighty molecules. For the twenty-nine biotherapeutics in which assessments related to carcinogenicity were identified, various experimental approaches were employed. This review also discusses several key principles to aid in the assessment of carcinogenic potential, including (1) careful consideration of mechanism of action to identify theoretical risks, (2) careful investigation of existing data for indications of proliferative or immunosuppressive potential, and (3) characterization of any proliferative or immunosuppressive signals detected. Traditional two-year carcinogenicity assays should not be considered as the default method for assessing the carcinogenicity potential of biotherapeutics. If experimentation is considered warranted, it should be hypothesis driven and may include a variety of experimental models. Ultimately, it is important that preclinical data provide useful guidance in product labeling.
Introduction
For potential new human therapeutics, particularly those that will be used continuously for periods of greater than six months, an important safety assessment question is determining whether the new therapy will increase the risk of cancer in patients. For traditional small-molecule therapeutics, this concern has primarily been addressed by two-year carcinogenicity studies in both rats and mice (International Conference on Harmonization [ICH] S1A 1995; Jacobs and Jacobson-Kram 2004), with increasing use in recent years of six-month studies in transgenic mice as an alternative. These studies, commonly referred to as bioassays, have long been used in the evaluation of the carcinogenic potential of environmental chemicals (Huff 1988). Despite the extensive use of these models, there remains considerable debate regarding the ability of these studies to predict human risk (Alison, Capen, and Prentice 1994; Cohen 2004; Jacobs 2005). For biotherapeutics, traditional rodent bioassays are often not scientifically applicable owing to a lack of relevant pharmacology or technically feasible owing to immunogenicity consequences, thereby making this assessment even more challenging. Because biotherapeutics do not induce direct genotoxicity, do not form active or genotoxic metabolites, and have effects that are primarily on target, the concern is not that the therapeutic will act as a complete carcinogen or general tumor promoter, but rather that it may increase the incidence or growth rate of a specific neoplasm or group of neoplasms.
The primary regulatory guidance related to nonclinical evaluation of biotherapeutics is the International Conference on Harmonization (ICH; 1997b) S6 Document, “Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals.” This guidance recognizes the unique challenges involved in the nonclinical assessment of biotherapeutics, and a common theme in the document is the adoption of a flexible, case by case, science-based approach. Specific to carcinogenicity assessments, the guidance states the following: Standard carcinogenicity bioassays are generally inappropriate for biotechnology-derived pharmaceuticals. However, product-specific assessment of carcinogenic potential may still be needed depending upon duration of clinical dosing, patient population and/or biological activity of the product (e.g., growth factors, immunosuppressive agents, etc.) When there is a concern about carcinogenic potential a variety of approaches may be considered to evaluate risk.
Also of relevance is the ICH (1995) S1A guidance, “Guideline on the Need for Carcinogenicity Studies of Pharmaceuticals.” Although the guidance primarily focuses on small-molecule therapeutics, the guidance states the following relevant to biotherapeutics: Although not usually necessary, long-term carcinogenicity studies in rodent species should be considered for the other biotechnology products noted above, if indicated by the treatment duration, clinical indication, or patient population (providing neutralising antibodies are not elicited to such an extent in repeated dose studies as to invalidate the results). Conduct of carcinogenicity studies may be important in the following circumstances: (1) For products where there are significant differences in biological effects to the natural counterpart(s); (2) for products where modifications lead to significant changes in structure compared to the natural counterpart; and (3) for products resulting in humans in a significant increase over the existing local or systemic concentration (i.e., pharmacological levels). an increasingly diverse range of therapeutic targets that have varying levels of concern regarding carcinogenic potential. For most biotherapeutics, the theoretical concern is not that the therapeutic will act as a complete carcinogen, but rather through epigenetic mechanisms such as enhancement of cellular proliferation, mechanism-based influence on specific neoplasms, or altered immune function. lack of relevant biology (receptors, signaling pathways, etc.) and/or the presence of immunogenicity in rodent species that makes traditional carcinogenicity bioassays in rodents technically unfeasible or of limited value for assessing risk to humans. carcinogenicity studies that are not feasible in nonhuman primates, which are often the only pharmacologically relevant toxicology species for biotherapeutics. the ability of nonclinical studies to dissect the complex relationship between altered immune function and increased carcinogenic risk, either through decreased immune surveillance or emergence of oncogenic viruses. limited knowledge of the clinical relevance and inexperience using alternative assessments of carcinogenic potential including rodent homologous molecules, target-specific transgenic animals, in vitro and in vivo cell proliferation data, and in vitro or in vivo measurements of tumor growth promotion.
Owing to these uncertainties in this area, the BioSafe organization formed an ad hoc group to study past and current practice regarding carcinogenicity assessments of biotherapeutics. This paper presents the results of this effort and specifically provides a review of publicly available data for approved biotechnology-derived pharmaceuticals and corresponding carcinogenicity assessments, and recommendations for assessing carcinogenic potential of new biotherapeutics.
Materials and Methods
Nonclinical Information on U.S. Food and Drug Administration (FDA; http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm) and European Medicines Agency (EMA; http://www.ema.europa.eu/htms/human/epar/eparintro.htm) Web sites were reviewed for information on carcinogenicity assessment of approved biotherapeutics. Molecules included in the assessment included monoclonal antibodies, fusion proteins, hormones, growth factors, recombinant plasma factors, and cytokines. Antisense oligonucleotides, plasma-derived proteins, enzyme-replacement therapies, and growth factors incorporated into devices for local administration were not included in this review.
From publicly available information, each biotherapeutic was evaluated to determine whether any assessments of carcinogenic potential or tumor growth enhancement were conducted. Each molecule was evaluated for the following information: Conduct of traditional two-year carcinogenicity bioassays in rodent species Conduct of one of the alternative mouse carcinogenicity bioassays (p53+/− knockout, rasH2 transgenic, TgAC transgenic, homozygous XPA knockout) Modified versions of the traditional or alternative carcinogenicity bioassays mentioned above Conduct of other alternative in vivo studies, including studies with duration of less than two years or inclusion of comparator arms Conduct of in vitro or in vivo studies to assess mitogenicity, cell proliferation, or promotion of tumor growth Precautions or warnings regarding animal studies or clinical data on carcinogenicity or tumor promotion in the drug label or prescribing information
The review did not include a listing of genotoxicity assessments that may have been conducted. It is now understood that mechanisms like modulation of the immune system or cell division may be more relevant mechanisms for biotherapeutic-induced promotion or progression of tumors rather than direct genetic damage.
The materials reviewed included product labels or leaflets (United States Package Insert [USPI] and Medication Guides for FDA-approved products, Summary of Product Characteristics [SmPC] for EMA-approved products), and approval documents (FDA Summary Basis of Approval [SBA] Documents, EMA Public Assessment Reports [EPAR]). In some cases, only documents for a single country were available owing to approval in only one geographic region or lack of toxicology data in the approval documents.
The results of the review were summarized and tabulated (Tables 1 –6). A subset of molecules was selected for additional discussion, as they illustrated important scientific or regulatory principles. For these cases, additional information from the peer-reviewed literature was reviewed. These details are provided in case example narratives in the Results section.
Peptide hormones and growth factors.
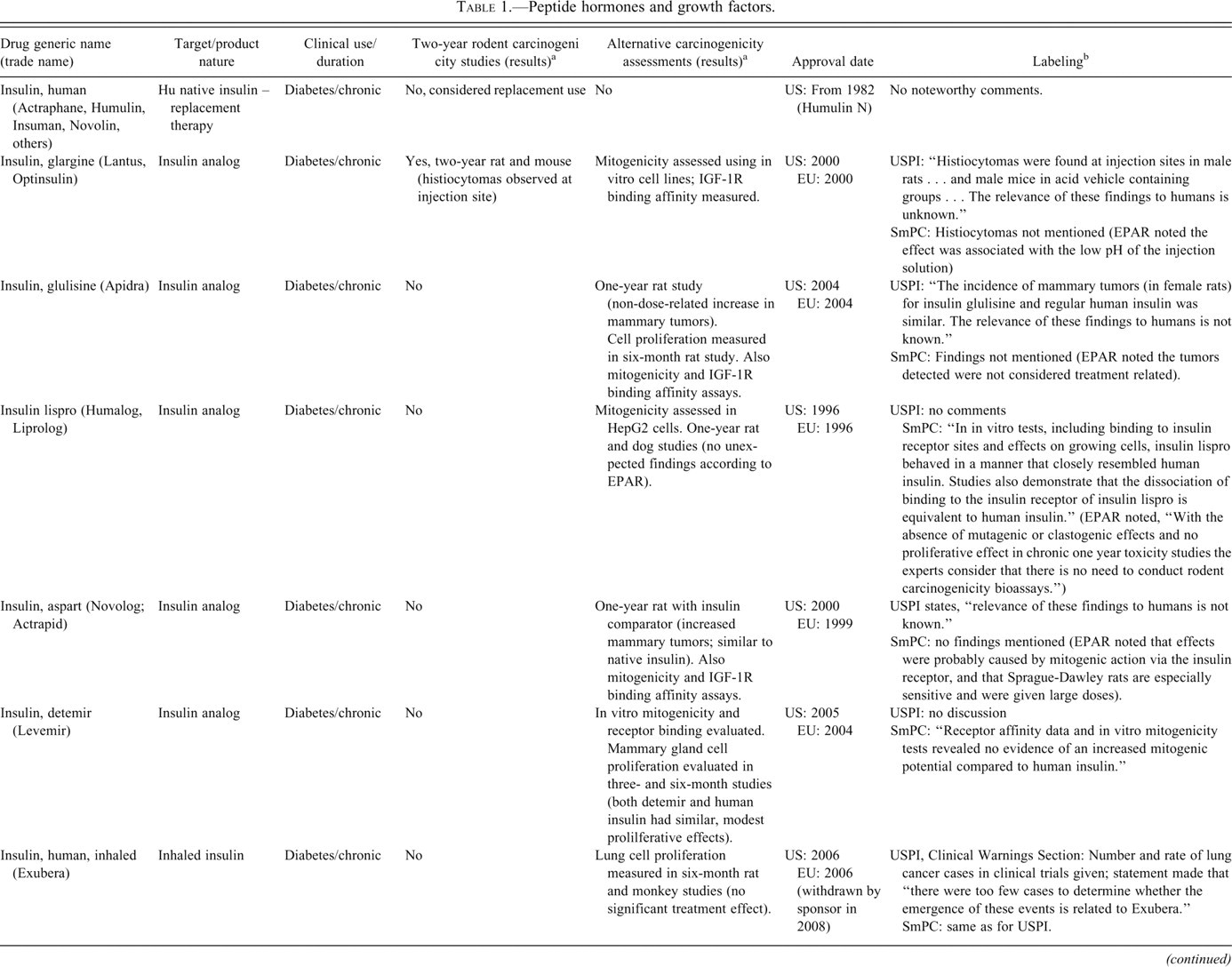
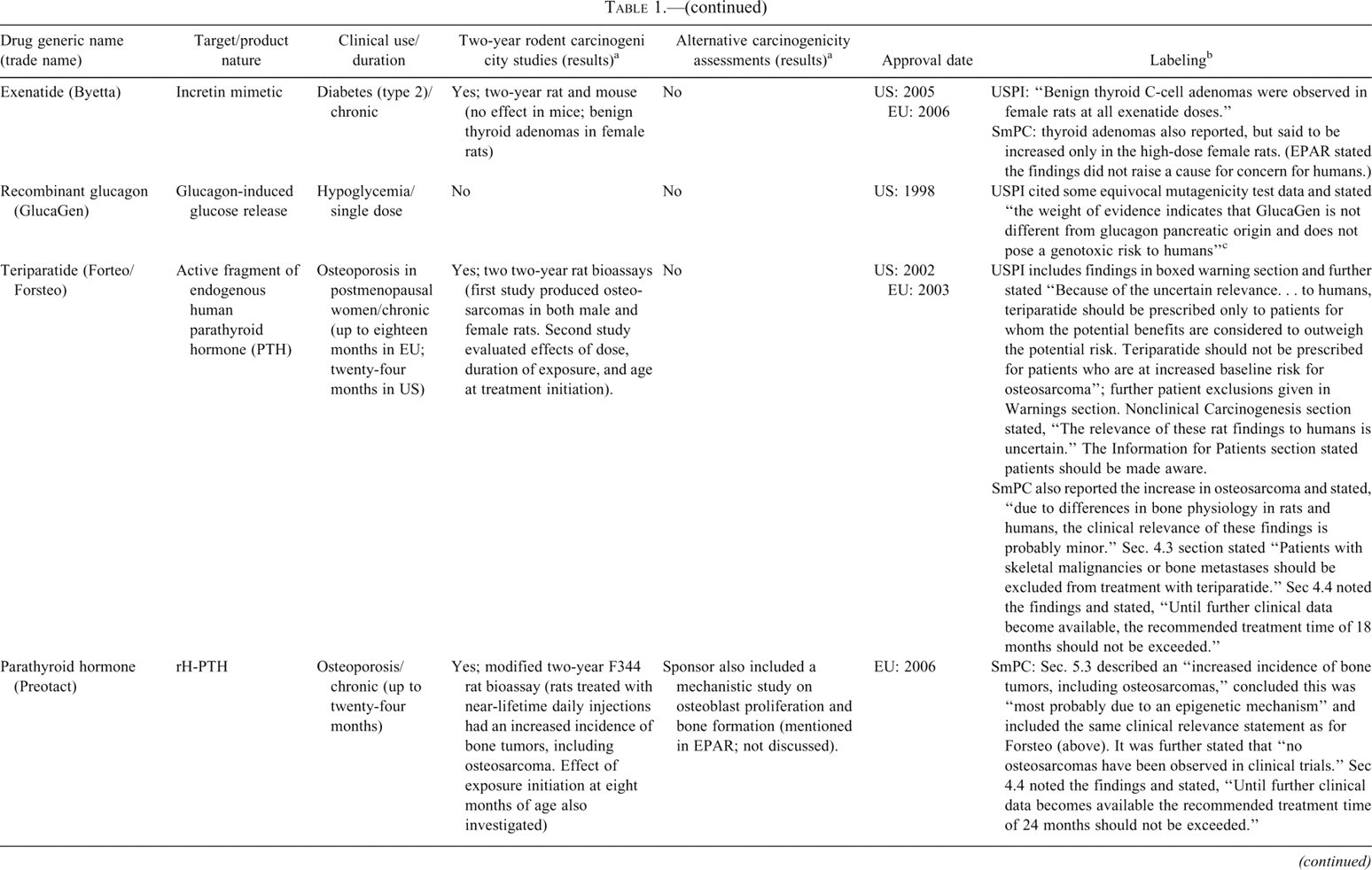
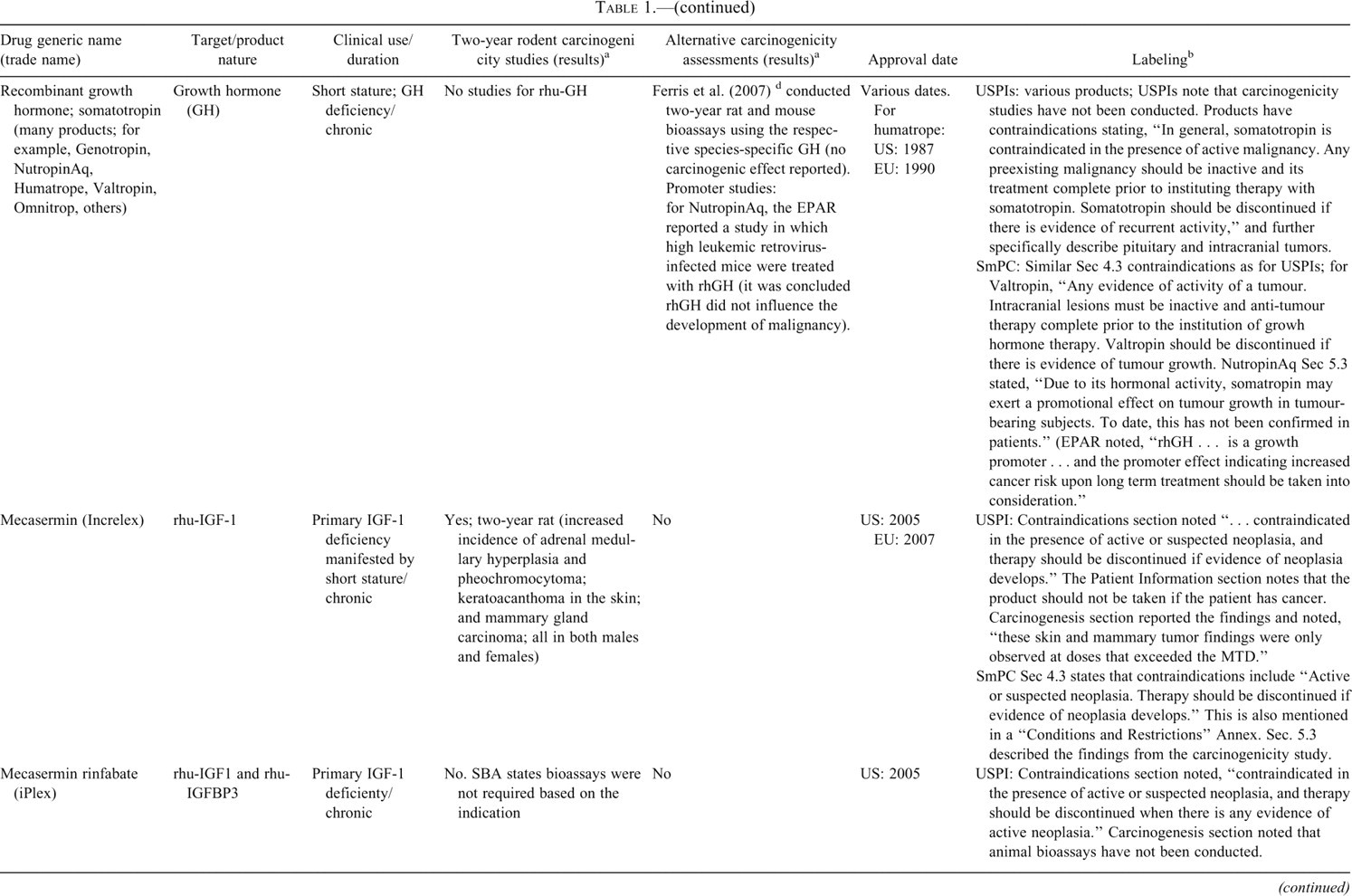

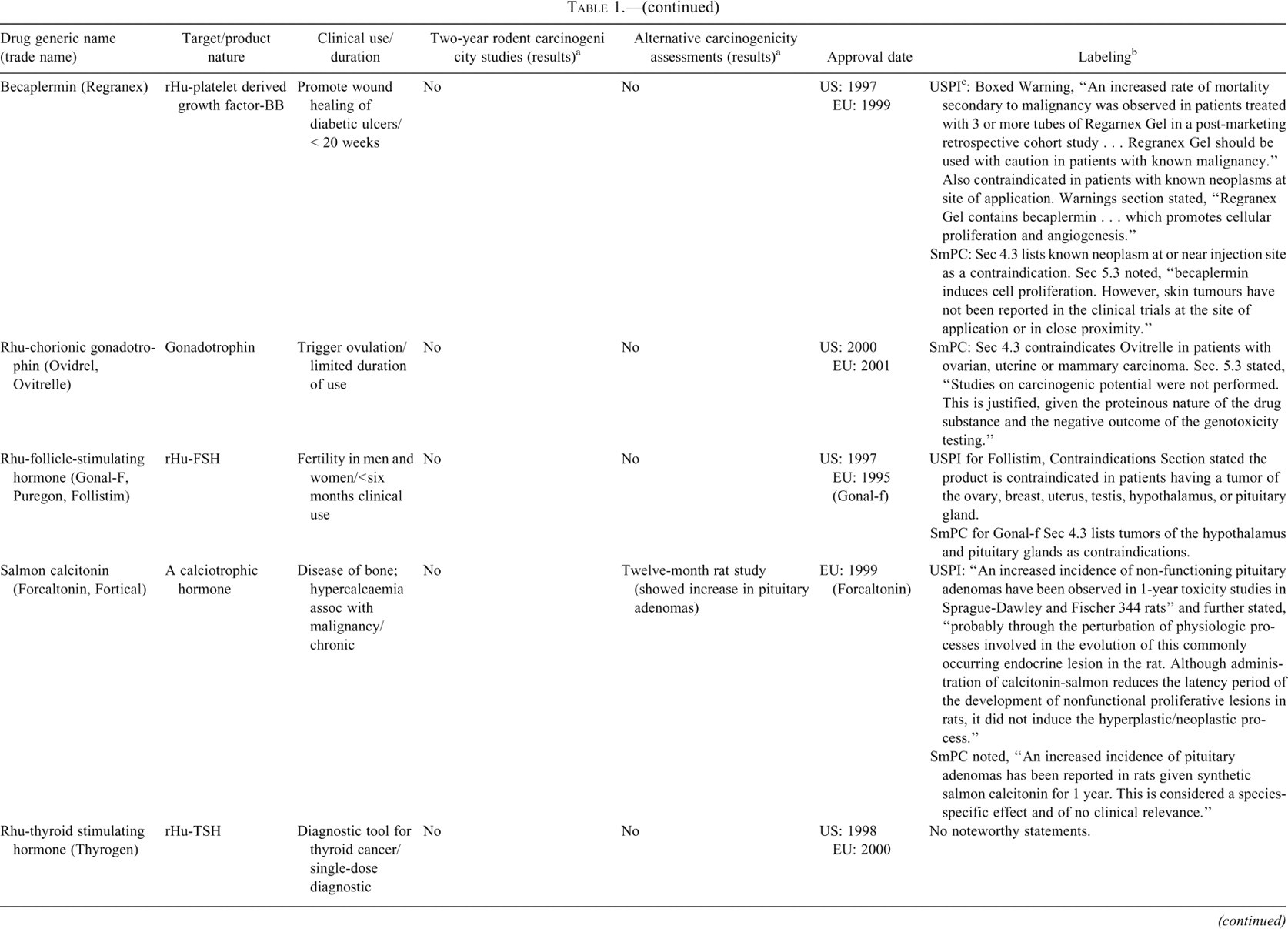
a Unless otherwise noted, data were obtained from U.S. FDA Summary Basis of Approval (SBA) documents. (http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm) or the EMA European Public Assessment Report (EPAR) (http://www.ema.europa.eu/htms/human/epar/eparintro.htm).
b Unless otherwise noted, labeling statements were obtained from nonclinical section(s) of U.S. Package Inserts (USPI) or Summary of Product Characteristics (SmPC); most recent version available up through December 2008 accessed.
c Label obtained from company Web site.
d Farris et al. 2007.
Monoclonal antibodies.
a Unless otherwise noted, data were obtained from U.S. FDA Summary Basis of Approval documents (http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm) or the EMEA European Public Assessment Report (http://www.ema.europa.eu/htms/human/epar/eparintro.htm).
b Unless otherwise noted, labeling statements were obtained from nonclinical section(s) of U.S. Package Inserts (USPI) or Summary of Product Characteristics (SmPC); most recent version available up through December 2008 accessed.
Fusion proteins; other protein immunomodulators.


a Unless otherwise noted, data were obtained from the U.S. FDA Summary Basis of Approval Documents (http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm) or the EMA European Public Assessment Report (EPAR) (http://www.ema.europa.eu/htms/human/epar/eparintro.htm).
b Unless otherwise noted, labeling statements were obtained from nonclinical section(s) of U.S. Package Inserts (USPI) or Summary of Product Characteristics (SmPC); most recent version available up through December 2008 accessed.
Hematology factors.
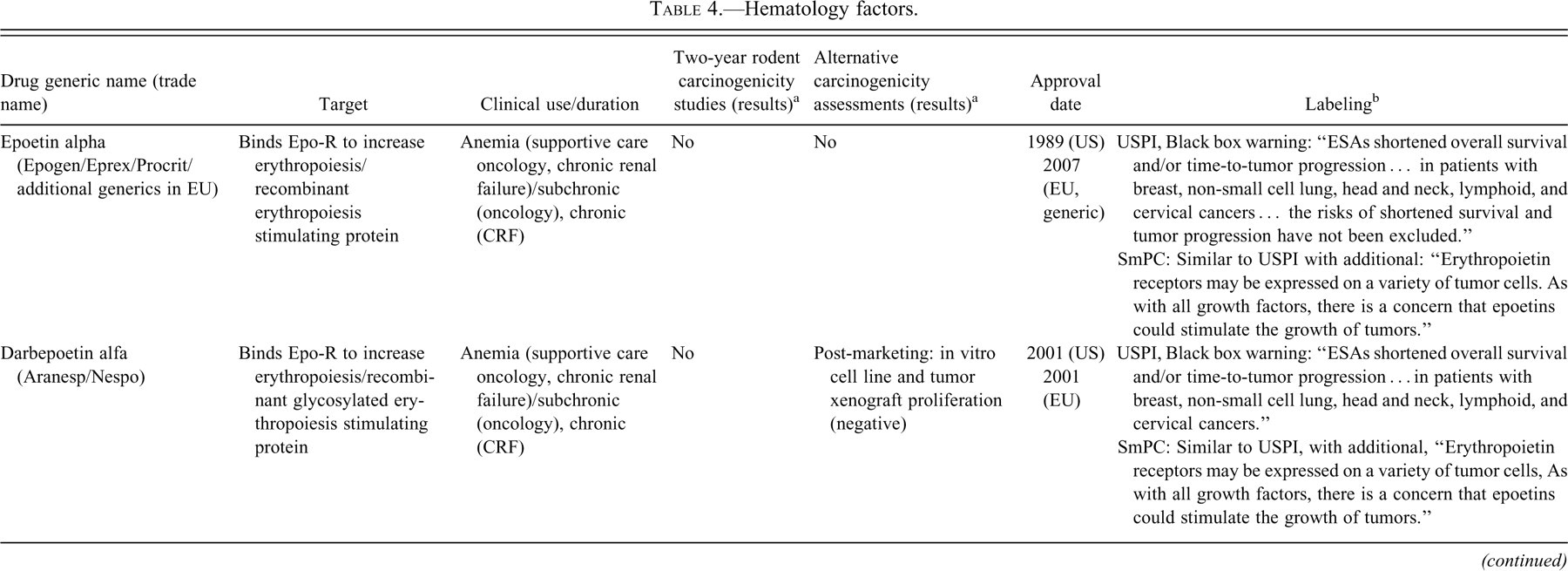
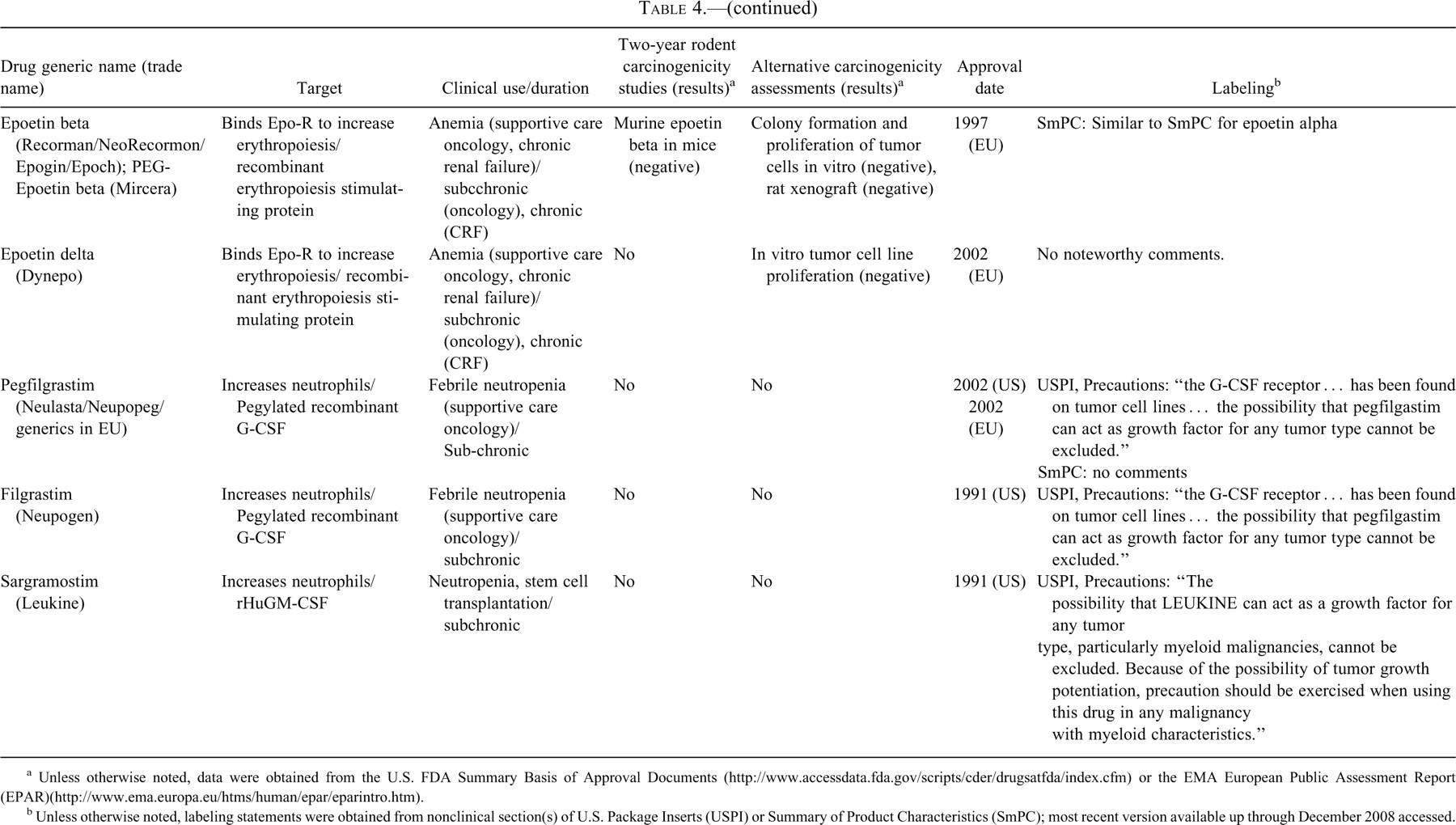
a Unless otherwise noted, data were obtained from the U.S. FDA Summary Basis of Approval Documents (http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm) or the EMA European Public Assessment Report (EPAR)(http://www.ema.europa.eu/htms/human/epar/eparintro.htm).
b Unless otherwise noted, labeling statements were obtained from nonclinical section(s) of U.S. Package Inserts (USPI) or Summary of Product Characteristics (SmPC); most recent version available up through December 2008 accessed.
Coagulation factors.
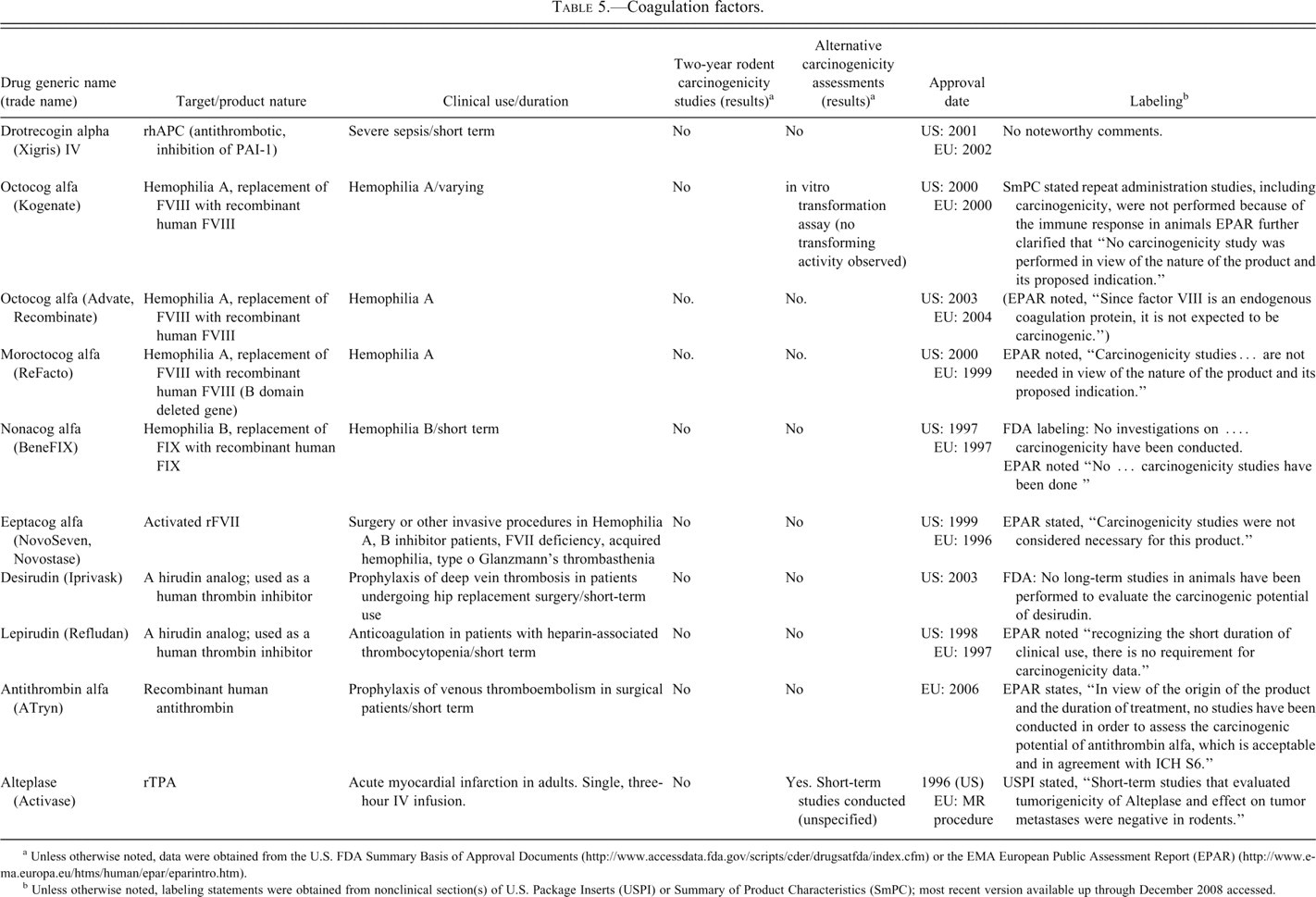
a Unless otherwise noted, data were obtained from the U.S. FDA Summary Basis of Approval Documents (http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm) or the EMA European Public Assessment Report (EPAR) (http://www.ema.europa.eu/htms/human/epar/eparintro.htm).
b Unless otherwise noted, labeling statements were obtained from nonclinical section(s) of U.S. Package Inserts (USPI) or Summary of Product Characteristics (SmPC); most recent version available up through December 2008 accessed.
Interferons and others.
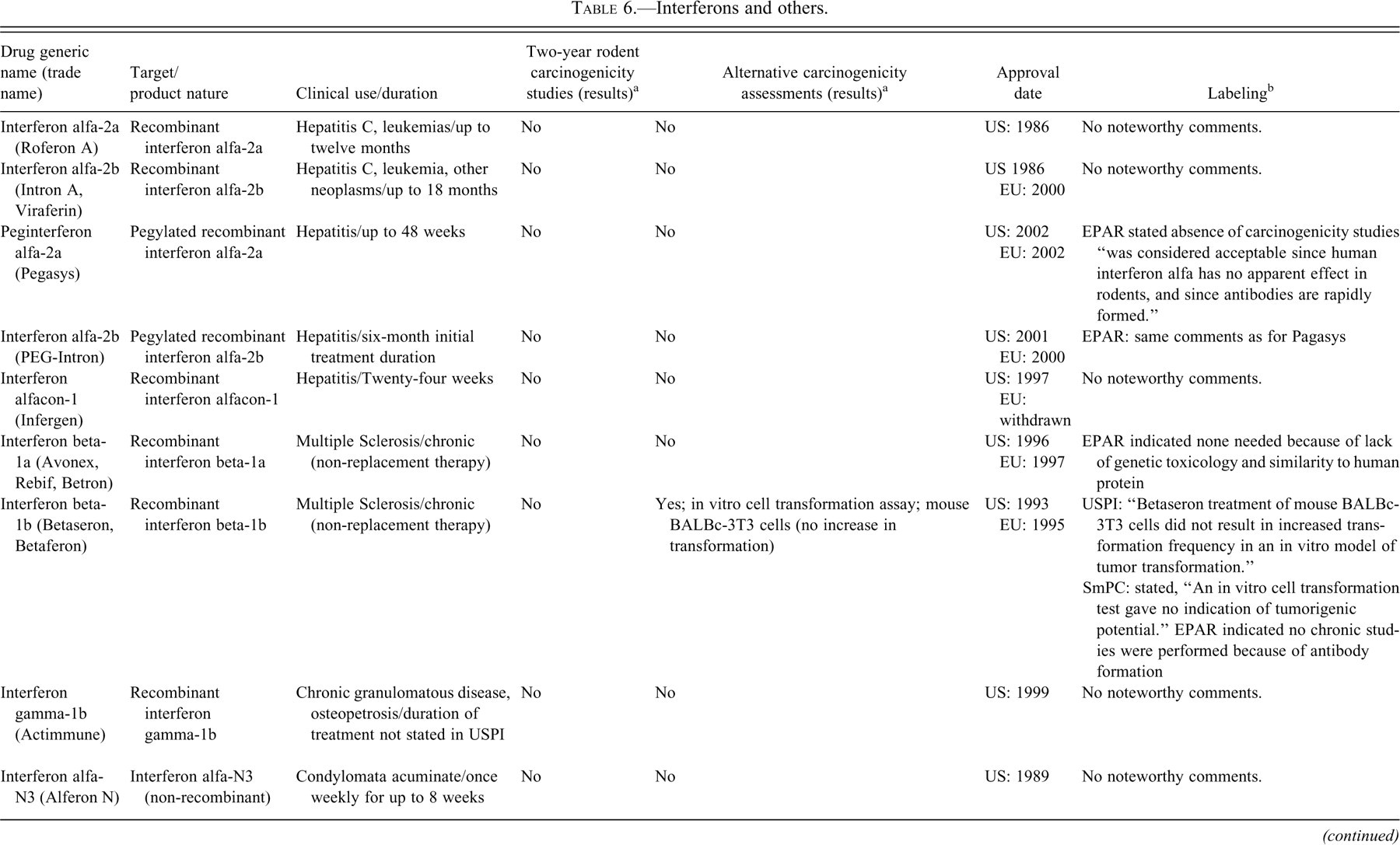

a Unless otherwise noted, data were obtained from the U.S. FDA Summary Basis of Approval Documents (http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm) or the EMA European Public Assessment Report (EPAR) (http://www.ema.europa.eu/htms/human/epar/eparintro.htm).
b Unless otherwise noted, labeling statements were obtained from nonclinical section(s) of U.S. Package Inserts (USPI) or Summary of Product Characteristics (SmPC); most recent version available up through December 2008 accessed.
Results
Review of Marketed Biotechnology-Derived Pharmaceuticals
General Overview
Tables 1 through 6 provide a listing of products (or groups of products with different names for the same molecule) that were reviewed. An initial review was conducted of the EMA and FDA Web sites with a cutoff approval date of December 2007. In addition, recent review articles that provided listings of biologic drugs were consulted (Cavangaro 2008; Leader, Baca, and Golam 2008; Snodin and Ryle 2006; Walsh 2006). Products were assigned to one of several classes, including peptide hormones and growth factors (Table 1; twenty-five listings), monoclonal antibodies (Table 2; twenty-one listings), fusion proteins and other immunomodulatory proteins (Table 3; five listings), hematopoietic factors (Table 4; seven listings), coagulation factors (Table 5; ten listings), or interferons and other factors (Table 6; twelve listings).
For the majority of biotherapeutics reviewed (fifty-one of eighty), no assessments related to carcinogenicity or tumor growth promotion were identified. Consistent with principles outlined in ICH S1A, common factors among molecules for which no assessments related to carcinogenicity were conducted include (1) limited duration of use in patients, (2) use as a replacement for an endogenous factor or hormone, or (3) use for serious, life-threatening conditions with significant unmet medical need, such as oncology indications. In addition to the factors listed above, a variety of technical issues may have precluded the conduct of two-year bioassays in these cases, including (1) immunogenicity (i.e., anti-drug antibodies) resulting in considerable reduction of exposure over time, and (2) the drug not having biological activity in rodents because of receptor–ligand species specificity or lack of mechanisms relevant to human risk assessment. In most cases the rationale for not conducting a carcinogenicity assessment was not available in the publicly available information.
For the twenty-nine biotherapeutics in which assessments related to carcinogenicity or growth promotion were identified, a variety of experimental approaches have been conducted. Only nine (11%) of the biotherapeutics in this review, consisting predominantly of peptide hormones and growth factors, were evaluated in an assay designed as a two-year rodent bioassay. In four of these cases, standard two-year bioassays were conducted in both rats and mice, and in five cases, two-year bioassays were conducted in a single species (three in rats, two in mice). This review did not identify any formal carcinogenicity bioassays in a transgenic mouse model. The only two-year study conducted with a rodent homologue noted in approval documents was for epoetin beta, in which the murine homologue was used in mice. As noted in the case review section, two-year bioassays were conducted with human growth hormone; however, these studies did not appear to support the regulatory approval but were reported in the peer-reviewed literature. It should be noted that six-month toxicity studies were conducted for two biotherapeutics, efalizumab and infliximab, with rodent-specific homologues; however, it is likely that these studies were conducted primarily to evaluate chronic toxicity end points. One-year rat studies, which appeared to be relevant to carcinogenic assessments, were conducted for four molecules. Three of these biotherapeutics were insulin analogs, as discussed later in this paper.
In addition to rodent models, a variety of alterative experimental approaches was used on a limited basis to aid assessment of carcinogenic potential or evaluation of enhanced tumor growth, including seven biotherapeutics with in vitro assessment of cell proliferation in normal cell lines, four biotherapeutics with in vivo assessment of cell proliferation in normal animals, four biotherapeutics with in vitro assessments of growth enhancement of tumor cell lines, five biotherapeutics with in vivo assessments of growth enhancement of tumor xenografts, two biotherapeutics with in vitro assessments in transformation assays, and one biotherapeutic assessed in a modified rasH2 model.
Product labeling related to carcinogenicity or tumor growth promotion was reviewed. Labeling statements typically included descriptions of the nonclinical findings and potential clinical relevance (or lack thereof), statements of theoretical concern, or presentation of numbers and rates of clinical cancer cases. In cases in which two-year rodent bioassays were conducted, a description of these studies was included in product labeling; however, descriptions of alternative approaches were not always included in product labeling. Selected examples regarding the presentation of these data in product labeling are provided in the case examples described later in this paper.
Peptide Hormones and Growth Factors
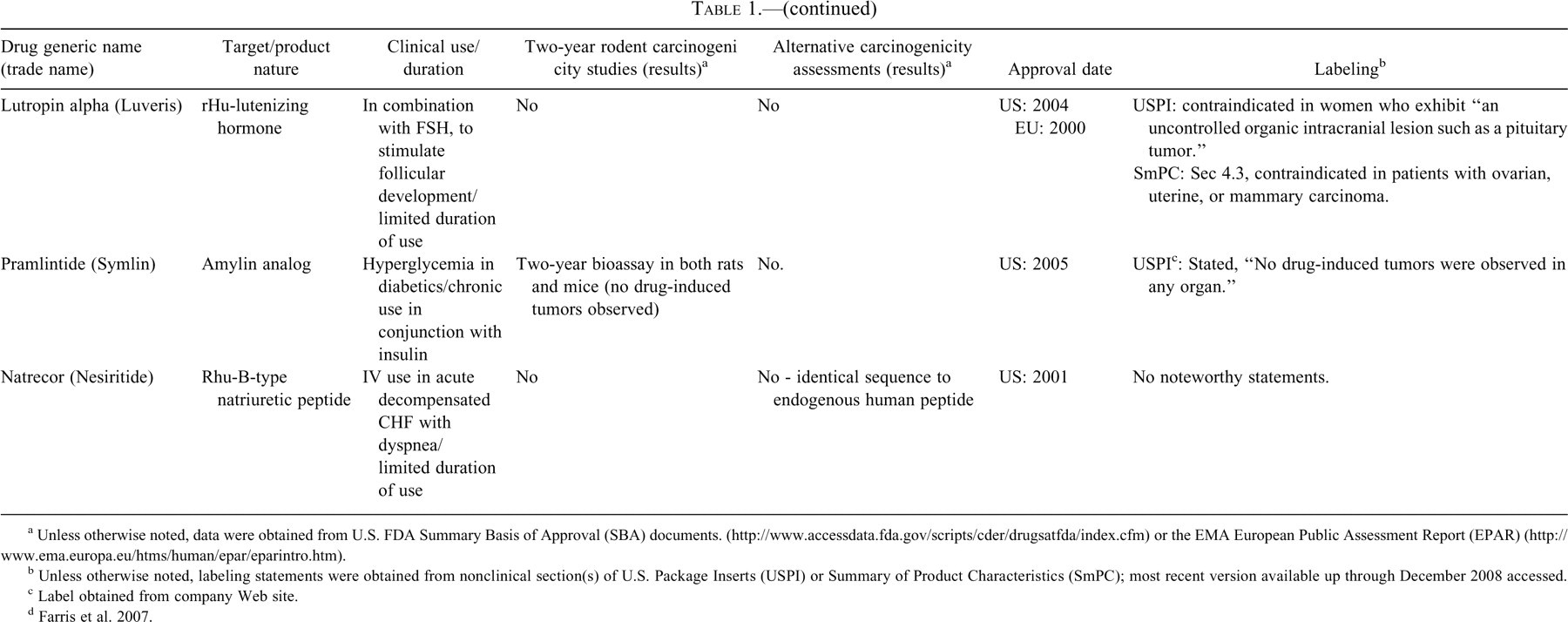
In Table 1, information on carcinogenicity assessment of approved hormones and growth factors are presented. 1 Consistent with ICH S1A, when native hormones or recombinant hormones having the same amino acid sequence as the endogenous hormone are given at essentially physiologic levels and where there is existing clinical experience, a nonclinical evaluation of carcinogenic potential was typically not conducted. Two-year bioassays in both rats and mice were conducted for four products (glargine, exenatide, octreotide, and pramlintide), and in rats only for three, products (teriparatide [Forteo], parathyroid hormone [Preotact], and mecasermin [Increlex]) (Table 1). A modified version of an alternative carcinogenicity mouse bioassay (rasH2) was conducted for one product, palifermin, to address a specific concern regarding potential impact on existing or secondary tumors. Several other products, most notably some insulin analogs but also calcitonin, included findings indicative of carcinogenic potential from one-year studies. In other cases, such as for insulin aspart, insulin detemir, and inhaled insulin, cell proliferation indices were quantified in six-month studies as indicators of carcinogenic potential. The use of novel approaches in the assessment of carcinogenic potential of insulin analogs is discussed in more detail later in this paper. A number of products, depending on targets, have included in vitro binding characteristics and/or effects on xenograft models. In some cases, such as for pegvisomant, the potential for stimulation of tumor growth has been evaluated in several xenograft models. Labeling information regarding tumor growth promotion or carcinogenicity for this class varied from bolded, “black box” warnings regarding findings in animal studies to brief descriptions in the carcinogenicity section.
Monoclonal Antibody and Fusion Proteins
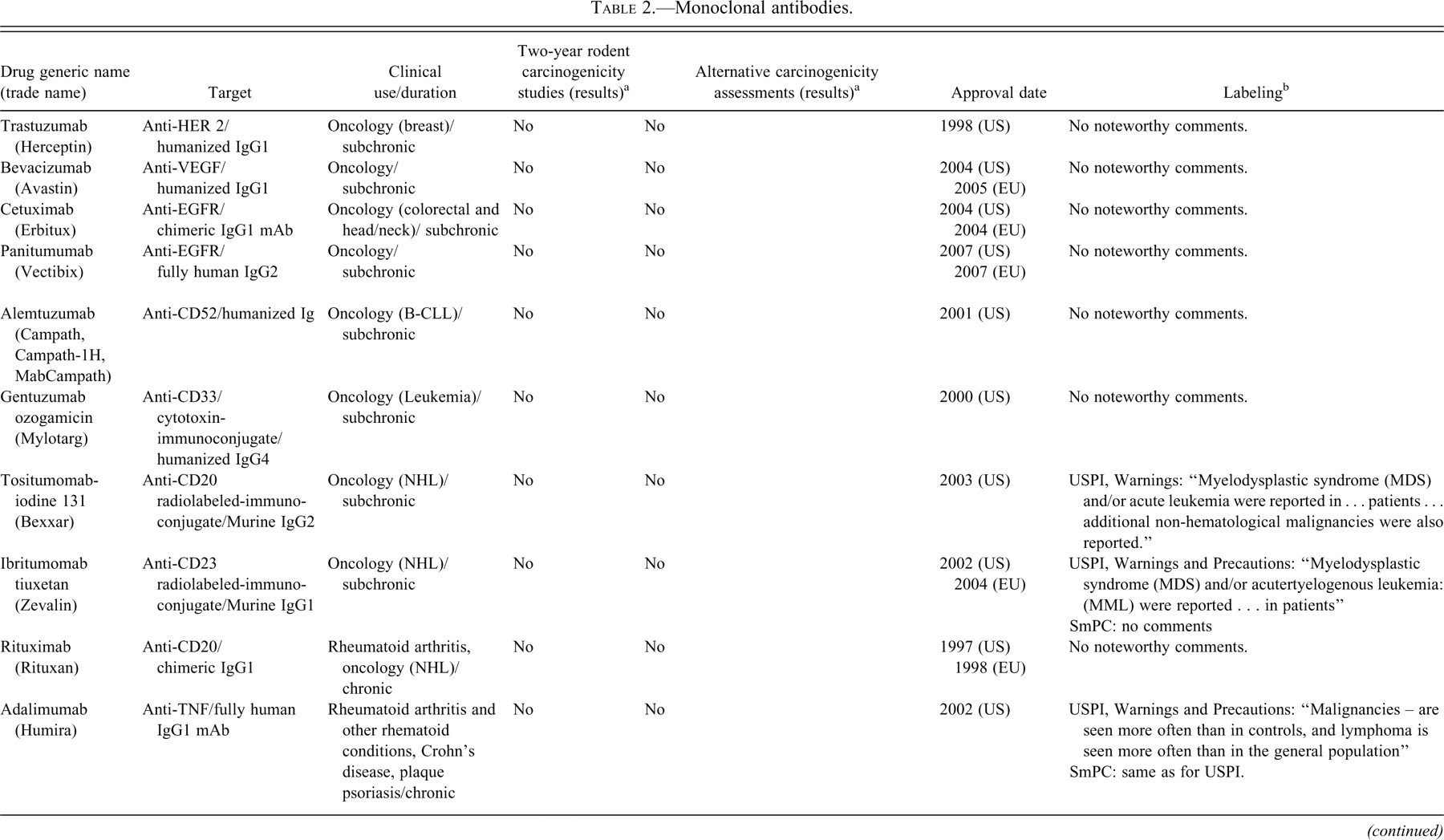
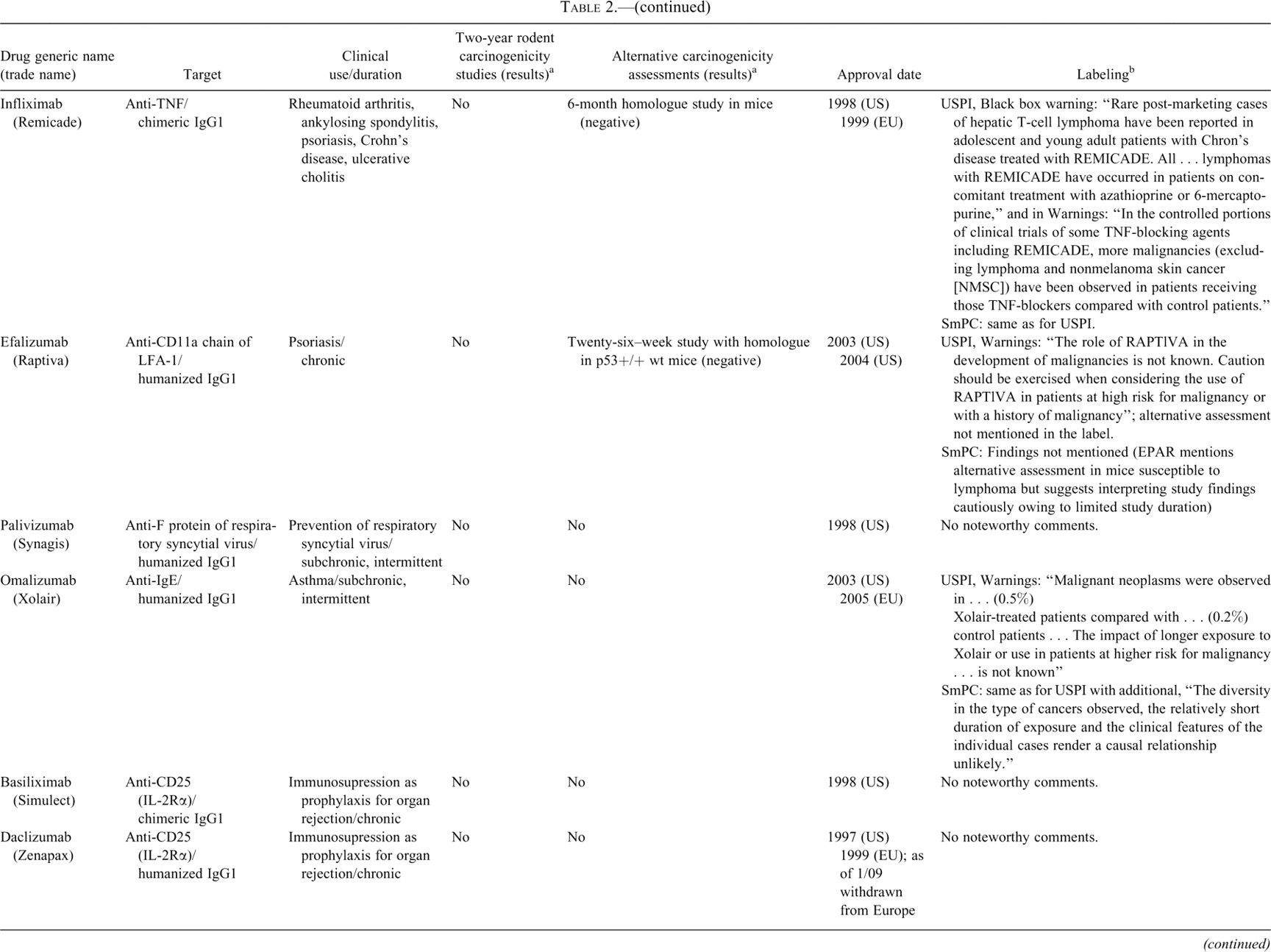
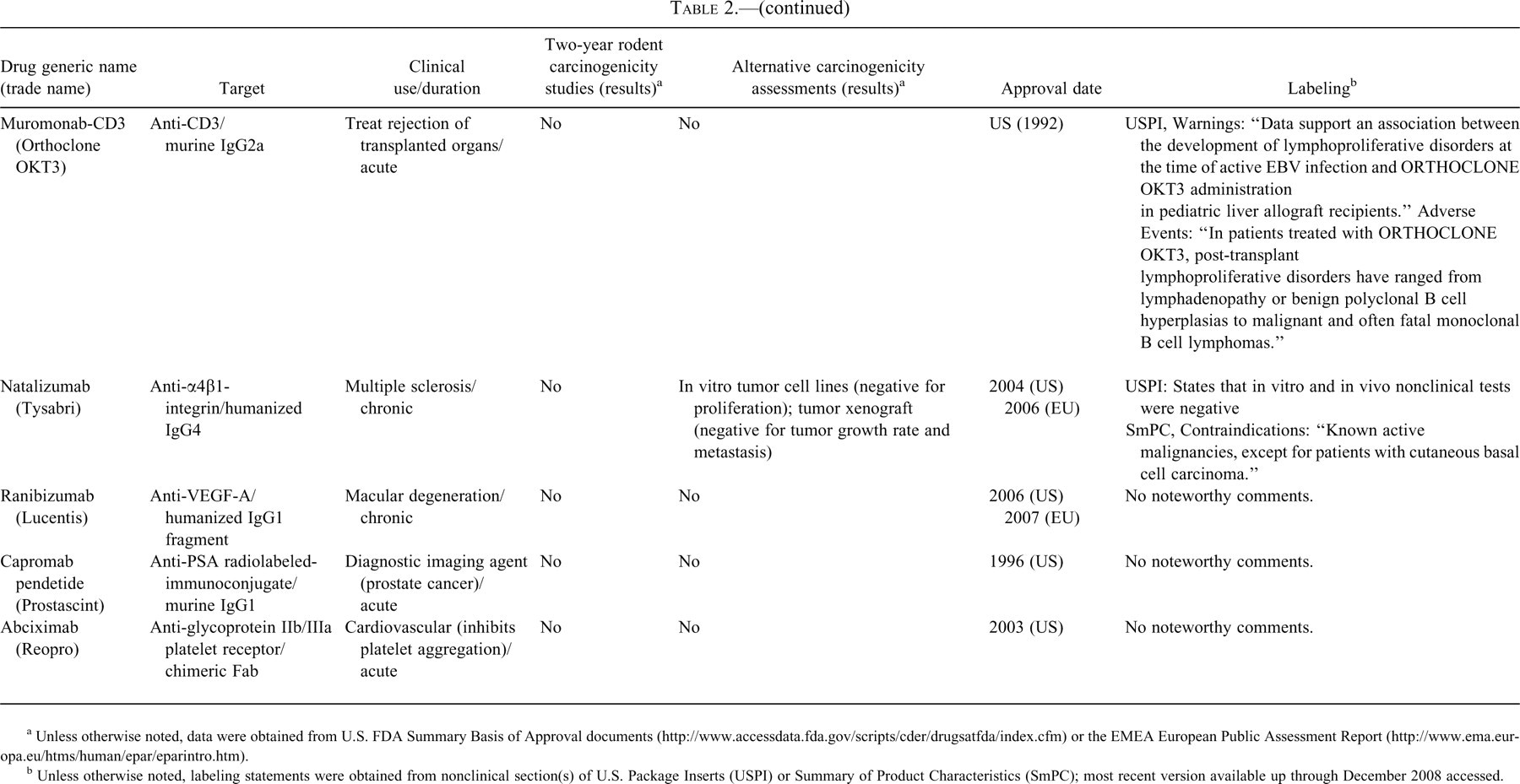
In Tables 2 and 3, information on carcinogenicity assessment of approved monoclonal antibodies and fusion proteins are presented. There is little precedent for carcinogenicity testing of therapeutic antibodies and fusion proteins. With the exception of abatacept (Orencia), which is pharmacologically active in rodents (discussed in more detail later in the paper), rodent carcinogenicity studies have not been conducted for any of the approved monoclonal antibodies (mAbs) or Fc-fusion proteins. Although the reasons for not conducting such studies are not always available, it is likely that rodent studies were not deemed appropriate owing to lack of relevant pharmacology and/or generation of antidrug antibody responses in rodent models. The nature of clinical use was also likely an important consideration as many of the mAbs and Fc-fusion proteins were approved for oncology indications or are not indicated for chronic administration in patients. Experiments other than two-year studies have been used to provide information regarding carcinogenicity or tumor growth promotion including the use of in vitro and in vivo tumor promotion studies for natalizumab. Various forms of label warnings and precautions were noted for immunomodulatory molecules and are described in additional detail in the case study section of this paper.
Hematopoietic Growth Factors
In Table 4, information on carcinogenicity assessment of approved hormones and growth factors is presented. Several hematopoietic growth factors and their analogs or derivatives have been developed as biotherapeutics. All of these therapeutics were developed to replace or augment endogenous levels of hematopoietic growth factors and encompass proteins that enhance the growth and/or maturations of both red and white blood cells. Only in the case of Recormon (epoetin beta) was a two-year rodent bioassay conducted; murine epoetin beta was used as a surrogate molecule in mice, and no carcinogenic effect was identified. Cellular proliferation and/or rodent xenograft studies were conducted with Recormon as well as Dynepo (epoetin delta) because of their use as supportive care products for oncology patients as additional means to assess the potential for tumor growth promotion. It is notable that many of the hematopoietic growth factors have precautions or warnings in their labels with regard to potential effects on tumor progression in the absence of any nonclinical testing supporting a carcinogenic effect. These labeling statements are associated with products that were tested for carcinogenic effects (i.e., Recormon and Dynepo), as well as several products in which additional carcinogenicity assays were not conducted. Thus, it is unclear that negative results in carcinogenicity or tumor growth promotion assays abrogate the theoretical concerns with hematopoietic growth factors.
Other Classes
Information on carcinogenicity assessment of marketed blood coagulation factors is presented in Table 5 and for other classes in Table 6. Table 5 lists the currently marketed blood coagulation factors. These proteins are used as replacement therapy proteins that treat patients’ endogenous protein factor deficiency and in accordance with ICH S1A, standard carcinogenicity assessments were not conducted.
The interferons listed in Table 6 were among the earliest biotherapeutics marketed and approved between 1993 and 2002. The duration of their clinical use varies, but in many cases, it exceeds six months. Furthermore, since they are not replacement therapies and because some of these products are not identical to the endogenous human protein, they would meet S1A criteria for required consideration of carcinogenic potential. Despite this fact, this review identified only two products (Betaseron and Neumega) out of twelve products in this group that explored alternative approaches to provide information related to a carcinogenicity assessment. Documents from the EMA provide some additional insight; for example, the EPAR for Avonex includes the statement, “Carcinogenicity studies were not performed because rh IFN beta-1a has no mutagenic or mitogenic potential. Its close similarity to the endogenous human protein that is not thought to be carcinogenic is also taken into account.” The EPAR for betaseron states that no chronic studies were conducted owing to formation of antidrug antibodies. A similar statement appears for Pegasys, a pegylated interferon.
The other products in Table 6 are not used for chronic treatment or are primarily used in oncology settings, and thus would not have met S1A criteria. Of note, the USPI for Neumega, a variant of IL-11 used to enhance platelet production, includes results from an in vitro study in which the molecule did not stimulate the growth of tumor colony-forming cells. These negative results were referred to in the carcinogenicity section (Table 6).
Selected Case Studies
Insulin Analogs
Insulins have been marketed for many years as replacement therapy for the treatment of diabetes, and consequently an extensive clinical safety database exists for native insulins from various sources (e.g., human, bovine, porcine). When originally developed for clinical use, native insulins were considered endogenous substances used for replacement therapy and thus were not tested in standard two-year bioassays to assess carcinogenic potential. Insulin analogs were considered to be novel molecules because they have structural changes in which the amino acid sequence is different from that of native human insulin. These changes result in different receptor potencies and an altered time course of action for regulation of glucose metabolism. Thus, there was concern that new safety issues might arise with their chronic use. In particular, evidence for a weak mitogenic effect of insulin (some of which was thought to be mediated through the closely related insulin-like growth factor-1 [IGF-1] receptor pathway) raised concern regarding chronic use of newly engineered insulin analogs. In some cases, the analogs were found to have different mitogenic effects compared to native insulin. Early data that heightened the concern related to increased mitogenicity and carcinogenic potential were obtained with an AspB-10 substituted insulin analog. Compared to native insulin, this analog was found to possess stronger affinity to the IGF-1 receptor, had greater in vitro mitogenic activity, and was associated with a significant increase in mammary tumors in rats treated for one year (Dideriksen, Jorgensen, and Drejer 1992).
The primary safety concern with chronic use of the new analogs was whether they produced a significantly greater risk relative to the well-characterized standard of care, native human insulin. Consistent with the draft of the ICH S6 Guidance for Biologics available at the time, it was not considered appropriate to perform standard two-year bioassays to assess carcinogenic potential of biologics. However, the availability of a large body of data from insulin use in human supported a unique approach of carcinogenicity assessment by comparison of the effects of the new analog with the known entity, human insulin. This approach is summarized in the EMA “Points to Consider” (http://www.emea.europa.eu/pdfs/human/swp/037201en.pdf). A common approach includes an in vitro assessment of mitogenic potential relative to native human insulin, Asp-B10 insulin analog and IGF-1 to provide a relative assessment of mitogenic potential. In vivo, an assessment of mitogenicity (e.g., BrdU or post-staining of tissues for nuclear cell cycle antigens) in the chronic toxicity studies of six-month duration may also be evaluated. If these studies indicate cause for concern, follow-up studies to explore this issue are performed.
Standard chronic toxicity testing of most insulin analogs included six-month studies in dogs and rats (insulin-aspart, -glargine, -glulisine, -lispro) (Table 1). In most cases, these studies were followed up with one-year studies in rats, which included an insulin comparator to assess carcinogenic potential relative to the standard of care. In only one case (Lantus, an insulin analog with a long duration of action), two-year standard carcinogenicity tests, including insulin comparator arms, were performed in both rats and mice (Table 1). In the case of glulisine, cell proliferation in mammary gland tissue was evaluated in the six-month and 1-year studies, and no differences in proliferation were observed between glulisine-treated groups, native insulin-treated groups, and untreated controls (Table 1).
The one-year assays in rats with the analogs Novolog and Apidra compared to native insulin detected an increase in mammary tumors in female rats for both the analog and native insulin relative to controls (Table 1). However, the overall risk of the analogs relative to insulin, with the exception of Insulin AspB10, was similar. Interestingly, the two-year bioassays performed with Lantus demonstrated no increase in mammary tumors relative to controls for both Lantus and native insulin. Thus, these two-year studies demonstrated that relative risk of Lantus and native insulin was similar, to allow a decision to be made about relative risk, but the overall results are different than those observed in the one-year studies. Compared to the results of the one-year rat studies in which the analogs and insulin caused an increase in mammary tumors relative to controls, these findings with Lantus in the two-year studies suggest that standard chronic studies or two-year carcinogenicity studies without appropriate comparators or clearly defined end points may not provide clear information for understanding the relative risk of insulin analogs. The results of the one-year rat studies are listed in the precautions sections of FDA product labels under the heading “Carcinogenesis, Mutagenesis, Impairment of Fertility.” In general, the multiples of human exposure are discussed and, in the case of approved analogs, it is noted that the incidence of tumors with the analog is similar to the incidence observed in the insulin comparator arm.
There are several key considerations that allowed a useful approach to assessing carcinogenic risk for these molecules, including an extensive safety database of a long-standing replacement therapy with a native molecule; the ability to include a native insulin control arm for direct comparison of the new insulin analogs to the well-characterized standard of care; and that it was possible to clearly define the question that needed to be answered in the assessment of carcinogenicity potential. Instead of focusing on the general question, “Does the new entity cause cancer?” the assessment endeavored to answer the question, “Does the new entity have increased risk relative to a well-characterized standard of care?”
Because of the extensive clinical information available for insulin prior to the advent of insulin analogs and the availability of a well-characterized comparator, this is a unique instance for demonstrating the feasibility of designing alternative approaches for assessing relative carcinogenic potential.
Parathyroid Hormone
Parathyroid hormone (PTH) is an endogenous eighty-four–amino acid hormone that plays a key role in calcium homeostasis. Parathyroid hormone and various analogs have been shown to have potent anabolic effects on the skeleton when given by intermittent injection and represent a class of bone anabolic drugs for the treatment of osteoporosis (Hodsman et al. 2005). The first approved agent was teriparatide (recombinant human parathyroid hormone [1-34]), a polypeptide composed of the first thirty-four amino acids of human PTH (Forteo, USPI). Full-length human recombinant PTH (1-84) has received regulatory approval in Europe as Preotact (Moen and Scott 2007).
The intended therapeutic use of PTH and PTH analogs is chronic and intended to elicit a supra-pharmacologic response rather than to serve as replacement therapy. Therefore, some consideration of carcinogenic potential was warranted. Because these peptides were biologically active and not highly immunogenic in rodents, two-year bioassays were possible. The first PTH analogue evaluated in a two-year rat carcinogenicity study, teriparatide, demonstrated an excessive magnitude of increases in bone mass and focal hyperplastic and neoplastic bone lesions (Vahle et al. 2002). This finding was followed by the issuance of draft guidance stating that the carcinogenic potential of PTH and related peptides should be evaluated in nonclinical studies (FDA 2000).
For teriparatide, studies related to assessing carcinogenic potential included two 2-year rat studies and a unique long-term monkey study. The initial carcinogenicity study was of more traditional design and included special procedures to quantify the pharmacodynamic effects, including histopathology of multiple bone sites and quantitative computed tomography (QCT) to determine bone mineral density and content (Vahle et al. 2002). In this study, bone mass was markedly increased and bone proliferative lesions, including osteosarcoma, were observed in all teriparatide-treated groups. This study demonstrated the value of specialized, quantitative procedures to evaluate the pharmacodynamic effects and their relationship to the carcinogenic process.
Although the rat was considered more sensitive than humans to the effects of PTH on bone growth because of fundamental differences in bone physiology between rodents and primates and the fact that these changes in rats occur over a lifetime of treatment compared to a relatively short (2–3%) portion of the normal span of treatment in humans (twenty-four months), an additional long term study of teriparatide was conducted in female F344 rats to determine the relative importance of dose, treatment duration, and age at initiation of treatment on the incidence of bone proliferative lesions (Vahle et al. 2004). This study used varying treatment groups consisting of various combinations of dose, treatment duration, and age at initiation of treatment. The study identified that treatment duration and administered dose were the most important factors in the teriparatide-induced bone tumors in rats. Age at initiation of treatment had a minor role, if any. The study also identified a treatment regimen that did not induce bone tumors but nevertheless resulted in a substantial increase in bone mass. This finding demonstrated that the threshold for the development of bone neoplasms in the rat that is greater than the threshold required to produce the desired pharmacological effect on bone. This dose, duration, and age at initiation study is a classic example of additional, well-designed long term studies occasionally needed to clarify human risk assessment.
Finally, teriparatide was given at 0 or 5 ug/kg/day subcutaneously for eighteen months to ovarectomized, skeletally mature female cynomolgus monkeys (thirty/group) with a three-year observation period following cessation of treatment (twenty-four/group) (Vahle et al. 2008). Radiographs and histology did not identify any bone proliferative lesions or microscopic lesions of osteosarcoma at the end of the treatment or observation period; however, increases in bone mass were observed. The lack of bone tumors in this study provides some reassurance regarding the safety of teriparatide for the primate skeleton, although the limitations of this study, including small group size, limit the ability to draw definitive conclusions regarding the risk of bone tumor developments in patients.
For PTH (1-84), a single rat two-year study has been reported (Jolette et al. 2006). Unique parameters in addition to bone densitometry of femur and lumbar vertebrae were radiographs to enhance the detection of hidden or “occult” tumors that might otherwise have gone undetected at necropsy and determine whether additional bones needed to be collected besides those listed in the protocol. All PTH doses caused widespread increased bone mass and increased bone mineral content and density. In the mid- and high-dose groups, proliferative changes in bone increased with increasing dose. Similar to teriparatide, osteosarcoma was the most common bone neoplasm. Small tumors such as skeletal fibrosarcomas were detected by radiography, and these tumors were not observed with teriparatide, presumably because radiography was not conducted. There were no drug-related increases in incidence of bone neoplasms at the lowest dose tested. This example demonstrated the general class effect of the continuum of receptor-based proliferative, benign and malignant neoplastic bone lesions with PTH compounds and the value of radiography in detecting small skeletal tumors.
In the case of PTH and related peptides, two-year rodent studies identified a carcinogenic response in rats and additional studies demonstrated dose and temporal relationships in this species. For the United States, these findings were given a black box warning in the label, citing the “uncertain relevance of the rat osteosarcoma finding to humans” and the recommendation that “teriparatide should be prescribed only to patients for whom the potential benefits are considered to outweigh the potential risk” (Table 1). Also of interest is that in this case, the animal findings led to the development of a long-term, post-marketing clinical surveillance program (Tashjian and Gagel 2006).
Human Keratinocyte Growth Factor
Keratinocyte growth factor (KGF) is a member of the fibroblast growth factor (FGF) family that is produced by mesenchymal cells and acts specifically on epithelial cells through the KGF receptor (KGFR) (Finch et al. 1989; Rubin et al. 1989). Keratinocyte growth factor has cytoprotective and regenerative effects on epithelial tissues and is believed to play a role in an organism’s response to epithelial tissue injury (Farrell et al. 1998; Rubin et al. 1995). Accordingly, a recombinant version of KGF, known as rHuKGF (recombinant human keratinocyte growth factor) or palifermin was developed to evaluate its potential to treat oral mucositis (Finch and Rubin 2006). Palifermin was approved in 2004 in the United States and in Europe in 2005, and subsequently in other regions of the world, and is currently marketed as Kepivance to reduce the incidence, duration, and severity of oral mucositis occurring as a side effect of chemo-/radiotherapy in patients with hematologic malignancies undergoing hematopoietic stem cell transplant (Table 1). Palifermin is a 140-amino acid protein identical to endogenous human KGF except that the first twenty-three N-terminal amino acids have been deleted. In the approved setting, palifermin is administered as an i.v. dose for three consecutive days before the conditioning regimen and for three days following transplant, for a total of six doses.
Because palifermin is an epithelial growth factor, there is a theoretical concern that treatment with the drug could adversely influence the growth of existing tumors expressing the KGFR (Finch and Rubin 2006). Since solid tumors of epithelial origin can express the KGFR, whereas hematologic malignancies have not been shown to express KGFR (Oelmann et al. 2004), the theoretical risk of an adverse impact of palifermin on tumor growth was initially restricted to the solid tumor setting.
To address this theoretical risk, additional nonclinical studies were conducted during clinical development to evaluate the effect of palifermin on human tumor cell lines representative of the tumor types occurring in the intended patient population (Hovland 2007). The studies first characterized the expression of the KGF receptor in a series of human tumor cell lines, then evaluated the effect of palifermin on the in vitro growth rate of the cell lines by 3H-thymidine, BrDu, and/or MTT incorporation (Ning et al. 1998). Thirty-eight of the forty-one cell lines were from epithelial tumors (e.g., lung, breast, colon, prostate, and tongue), whereas three were from hematological malignancies (e.g., leukemias). Of the thirty-eight epithelial tumor cell lines, only eleven showed any appreciable increases in growth rate, whereas palifermin did not influence the growth rate of the three leukemia cell lines. Subsequently, seven epithelial tumor cell lines showing increases in in vitro growth rate were evaluated in xenograft models in immune-deficient nude mice. In these studies, the cell lines were implanted subcutaneously, and palifermin was administered for three consecutive days for four to six weeks (duration varied according to the specific growth rate of the implanted tumor cell lines). This treatment regimen was selected to model one of the clinical treatment paradigms being considered in the solid tumor setting. No statistically significant increases in growth rate were observed in six of the seven tumor cell line xenografts. However, a modest dose-dependent increase in growth rate was observed in one of the seven tumor cell line xenografts.
In addition, palifermin was evaluated in a modified transgenic rasH2 (Tg.rasH2) mouse model to assess the drug’s potential to influence the rate of spontaneous tumor development. The rasH2 model is a validated short-term carcinogenicity bioassay for genotoxic and nongenotoxic carcinogens (ICH S1B 1998; Morton et al. 2002; Tamaoki 2001; Usui et al. 2001; Yamamoto, Urano, and Nomura 1998). The study with palifermin was intended to model the potential impact of the drug on existing (primary) tumors, as well as secondary tumors, which can be produced by cytotoxic chemo- and/or radiotherapy.
Notably, because secondary tumors developing in patients treated for hematological malignancies can be solid (e.g., epithelial) as well as hematological (Varady et al. 2001), the study was believed to provide meaningful information for the hematological transplant setting as well as for the solid tumor setting. The study’s intended goal, as stated in the FDA’s original approval letter, was “to evaluate the potential of palifermin to enhance the incidence of spontaneous tumors in the Tg.rasH2 transgenic mouse model.” The unique goal of the study is manifest in its atypical design. In the study, a nine-week treatment period involving single IV injections (to mimic a potential clinical regimen in the solid tumor setting) was followed by a seventeen-week untreated period before scheduled necropsy (Hovland 2007). The study was negative; no treatment-related increases in the incidence of neoplastic lesions were observed.
Although the mouse models mentioned above (xenograft and Tg.rasH2) demonstrated the limited preclinical potential of palifermin to influence the growth of tumor cells/tumors, concerns over the theoretical clinical risk were not entirely mitigated. At the time of approval in the hematological transplant setting, tumor promotion was one of the major potential risks noted by the FDA, which recommended that the risk be addressed with directed pharmacovigilance and appropriate labeling. The pharmacovigilance portion translated to a requirement for post-approval surveillance, with special attention to malignancies, that was manifest as two postmarketing commitments requiring that all subjects previously enrolled in any study conducted in the hematological malignancy and head and neck cancer settings be monitored for ten years. From the labeling perspective, the theoretical potential for Kepivance’s adverse impact on tumors was mentioned in the Precautions section of the U.S. package insert, as well as in the Special Warnings and Precautions section of the European SmPC.
Growth Hormone
The nonclinical studies supporting market authorization of human growth hormone (hGH) were completed prior to ICH S6 (IHC 1997b) implementation. Chemical and biological equivalence of recombinant and pituitary-derived growth hormone was established for Somatropin and Somatrem, which appeared to limit the extent of the nonclinical testing required. Repeat-dose toxicity studies in the rhesus monkey were completed for up to three months with Somatropin and thirteen weeks with Somatrem. Likely because hGH is replacement therapy, carcinogenicity assessments were not conducted. Product labels indicated that assessments were not conducted with a contraindication for use in patients with active tumors (Table 1).
Traditional two-year carcinogenicity bioassays in mice and rats were recently reported in the literature for GH (Farris et al. 2007). These studies used recombinant mouse GH and recombinant rat GH, homologues of human GH (hGH). Use of species-specific GH likely increases activity in that species and theoretically reduces the immunogenic potential. Activity of the homologues was demonstrated by increased weight gain in separate five-week hypophysectomized rodent studies. The top doses were “up to” approximately tenfold of basal GH levels. These doses exceeded the human dose by at least ten-fold on a mg/kg basis, but a quantitative comparison of activity or binding affinity across species was not provided. Although immunogenicity was measured and not increased in the five-week studies, it was not measured in the carcinogenicity studies. Exposure to GH was measured in the five-week studies and not in the carcinogenicity studies. Anticipated pharmacology of increased body weights were observed at the middle and high doses in the rat study, but not at any dose in the mouse study. Thus, exposure to GH and pharmacology in the murine study was not confirmed. Tumors were not increased with GH treatment in either rats or mice.
The carcinogenicity risk in patients treated with hGH therapy has been extensively evaluated. In 1988, five new cases of leukemia were reported in Japanese patients previously treated with cadaver hGH. Subsequently, the same group analyzed data from more than 32,000 Japanese patients receiving hGH between 1975 and 1997 and concluded that the risk for leukemia in otherwise healthy patients was no different from that in the general population (Nishi et al. 1999). The NIH followed 6,272 patients who received cadaver hGH from 1963 to 1985 to determine whether they had an increased risk of Creutzfeldt-Jacob disease. An evaluation of all deaths in these patients to 1996 was published in 2004 (Mills et al. 2004). Thus, the analysis includes at least ten years after the last patient received cadaver hGH. The overall death rate was almost four times higher in hGH recipients than expected. No increase in tumor risk was identified in patients who did not have a previous tumor. However, increased mortality was observed in patients with existing neoplasms or a history of tumors. Long-term observational or postmarketing studies by major manufacturers of hGH have failed to identify a signal in increased risk of tumor occurrence or recurrence. These observations include at least ten years of experience from 24,417 patients followed in the Genentech database (NCGS) (Allen et al. 1997), 25,977 patients from the Pharmacia (Pfizer) KIGS database (Wilton 1999), and 1,727 patients in the Lilly HypoCCS database (Attanasio et al. 2002). Taken together, a substantial amount of clinical data indicates that hGH treatment is not a significant risk factor for spontaneous tumors.
In addition to the clinical use of GH, acromegaly patients, who frequently have GH levels up to ten times physiological levels for many years, provide insight into the potential carcinogenic risk. A comprehensive review of multiple epidemiological studies indicates that the association of GH with an increased risk of malignancy remains controversial. The review included a retrospective analysis of epidemiological studies for cancer prevalence in 4,886 patients collected in publications that spanned a forty-five-year period (1957–2002) (Colao et al. 2004). An increase in colon polyps was most commonly reported, especially in patients with uncontrolled GH levels following treatment. However, an association of increased malignancy in other tissues was not clear. One of the largest multicenter retrospective studies included in the review (Orme et al. 1998) concluded that acromegaly may hasten the progression of preexisting neoplasms, but there was no evidence for increased occurrence of new neoplasms. Although there may be an increased risk of colon polyps, the risk of increased malignancy in acromegaly patients is uncertain and a large increase in risk seems unlikely. Because acromegaly patients frequently have GH levels that are more than tenfold of normal physiological levels, these data suggest that an increased risk of malignancy with hGH replacement therapy is negligible.
The epidemiological data described above are an important consideration in assessing whether a two-year rodent carcinogenicity study is warranted. The intent of the rodent studies is to provide a risk assessment for patients. If an extensive amount of epidemiological data is available, then the risk to patients can be inferred from this data set. Moreover, any result from the rodent study that is contrary to the epidemiological data would be difficult to interpret. Data from patients, from both previously approved similar biopharmaceuticals and disease conditions that overexpress the native protein, should always be taken into consideration when determining the need for, and designing, the carcinogenicity assessment.
Immunomodulatory Cases: Abatacept and Alefacept
Although there are a number of immunomodulatory biotherapeutics, it is interesting to compare the studies conducted for alefacept and abatacept, as they highlight some of the challenges of assessing immunomodulatory agents for carcinogenic potential (Table 3).
Alefacept
Alefacept (Amevive) is an immunoglobulin fusion protein approved in 2003 for treatment of psoriasis that nonspecifically blocks T-cell activation and produces marked T-cell depletion. Alefacept consists of the CD2-binding domain of the human leukocyte function antigen-3 (LFA-3) fused to the Fc portion of IgG1. Alafacept has biological activity only in humans and nonhuman primates and as such, the nonclinical safety studies were restricted to studies in nonhuman primates. In a twelve-month toxicity study in cynomolgus monkeys, alefacept treatment was associated with lymphoma in one female animal. The lymphoma was identified after twenty-eight weeks of dosing, and B-cell hyperplasia was identified in other monkeys. Lymphoproliferative changes, including plasmacytoid hyperplasia, polymorphic B-cell hyperplasia and lymphoma, were attributed to recrudescence of latent γ–herpes virus infections (lymphocryptovirus) and were characterized as posttransplant lymphoproliferative disease (PTLD). All animals in the study were positive for the endemic primate γ–herpes virus known as lymphocryptovirus (LCV). Latent LCV infection is generally asymptomatic but can lead to B-cell lymphomas when monkeys are immune suppressed. The label for alefacept (2006) indicates that it may increase the risk of malignancies and refers to both clinical data as well as the nonclinical data from the chronic cynomolgus toxicology study. In placebo-controlled studies, the incidence of malignancies was increased (1.3%) for alefacept-treated patients compared to the placebo (0.5%). The Carcinogenesis, Mutagenesis, and Fertility section of the label indicates that no formal carcinogenicity studies were conducted, but that lymphoma and hyperplasia were observed in the chronic cynomolgus toxicity study. The following disclaimer is also included: “The role of Amevive in the development of the lymphoid malignancy and the hyperplasia observed in nonhuman primates and the relevance to humans is not known. Immunodeficiency-associated lymphocyte disorders (plasmacytic hyperplasia, polymorphic proliferation, and B-cell lymphomas) occur in patients who have congenital or acquired immunodeficiences including those resulting from immunosuppressive therapy.”
Abatacept
Abatacept (Orencia), a soluble fusion protein that consists of the extracellular domain of human cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) linked to the modified Fc (hinge, CH2, and CH3 domains) portion of human immunoglobulin G1 (IgG1), was approved for use in rheumatoid arthritis in 2005. As a selective costimulation modulator, this molecule inhibits T-cell (T-lymphocyte) activation by binding to CD80 and CD86, thereby blocking interaction with CD28. Interaction with CD28 provides a costimulatory signal necessary for full activation of T lymphocytes. Abatacept is a unique example of a biopharmaceutical that is pharmacologically active in mice and rats and that long-term dosing is not limited by immunogenicity. This molecule is the only example of a monoclonal antibody or fusion protein that appears to have had a relatively traditional rodent bioassay conducted in the form of an eighty-eight-week mouse study. In the study, abatacept resulted in increases in the incidence of malignant lymphomas in all dose groups and mammary gland tumors in females in the mid- and high-dose groups. Further pathology evaluation of mice from this study revealed they were infected with murine leukemia and mouse mammary tumor viruses, which increase incidence of lymphomas and mammary gland tumors, respectively, in immunosuppressed mice. In a one-year toxicity study in cynomolgus monkeys, slight decreases in serum IgG and severe depletion of germinal centers in lymph nodes and spleen were observed; however, no lymphomas or preneoplastic morphologic changes were observed despite the presence of lymphocryptovirus, a virus known to induce these lesions in immunocompromised monkeys within the one-year time frame of this study.
The observed rate of lymphoma in the cumulative abatacept clinical trials was approximately 3.5-fold higher than expected in an age- and sex-matched general population. However, the rheumatoid arthritis patient population in general is known to have a higher incidence of lymphoma than the general population and the risk of lymphoma has been reported to increase with conventional, cytotoxic, and biological therapy (Zintzaras, Voulgarelis, and Moutsopoulos 2005). Thus, the relevance of the carcinogenicity findings in the mouse to the clinical observation of increased lymphoma in patients receiving abatacept is uncertain. The label for abatacept contains cautionary language regarding immunosuppression and increased risk of infection: “The possibility exists for drugs inhibiting T cell activation, including ORENCIA, to affect host defenses against infections and malignancies since T cells mediate cellular immune responses. The impact of treatment with ORENCIA on the development and course of malignancies is not fully understood. In clinical trials, a higher rate of infections was seen in ORENCIA treated patients compared to placebo.” The Carcinogenesis, Mutagenesis, and Fertility section of the label describes the eighty-eight-week mouse carcinogenicity study and the one-year toxicity study in cynomolgus monkeys but makes no statement correlating the results of these studies with patient risk of cancer.
It is interesting to note that for cyclosporine, a drug of choice for transplantation that is also used in the treatment of psoriasis, malignancy risk was also identified in monkey studies. Following pharmacologic cyclosporine immunosuppression and cardiac or renal transplantation in cynomolgus monkeys, lymphomas have been reported in as little as thirty-six days in association with Epstein-Barr Virus (EBV) seropositivity (Gaschen and Schuurman 2001). The contrasting results obtained in monkeys infected with viruses associated with lymphoma following exposure to cyclosporine, or immunosuppressive biotherapeutics (alefacept and abatacept) illustrates that although viral transformation is a known mechanism by which immunosuppressive agents can underlie carcinogenicity, demonstration of the associated risk in monkeys infected with viruses linked to cancer is difficult. Thus, even negative data from monkey toxicology studies do not eliminate the concern of malignancy in patients treated with immunomodulatory drugs.
In contrast to the nonclinical observation of lymphoma in monkeys, there have not been excessive numbers of malignancies noted in patients treated with alefacept (Callen 2007) and an alefacept clinical trial sponsored by the National Cancer Institute in relapsed or refractory cutaneous T-cell lymphoma or peripheral T-cell non-Hodgkin’s lymphoma is ongoing (http://www.clintrials.gov). Thus, positive malignancy data in animal studies should not preclude further development but should be considered in the context of clinical experience as part of an analysis of risk benefit.
Discussion and Recommendations
General Considerations
The flexibility provided by a science-driven, case-by-case approach presents challenges in determining the amount and type of data that constitute an appropriate assessment of carcinogenicity. The review of marketed molecules indicates that even for molecules that appear to have similar theoretical risks, the amount of animal testing done and the approach to labeling can be quite different. The current experience indicates that it is not appropriate to consider standard two-year rodent carcinogenicity studies as the only valid approach to assessing carcinogenicity. In contrast, the most appropriate approach is one in which the biology of the proposed therapy is carefully considered, and a weight of evidence approach is used to characterize carcinogenic or tumor growth potential. In some cases, a variety of experimental methods may be available and needed, whereas in others it may be acknowledged that in vitro and animal models are not useful in defining the risk to patients. As a weight of evidence approach is developed, the following areas should be carefully considered.
Identify Potential Concerns
Early in the discovery and development process, sponsors should carefully assess the proposed target and determine what, if any, theoretical risks there are for tumor induction or progression. The target assessment should include a thorough review of the literature and, if available, prior clinical experience with the therapeutic class. Important information may be available from genetically modified mice in which the target has been knocked out or overexpressed or from inherited conditions in humans. Data from the literature must be scrutinized carefully for the validity of the model, adequacy of the evaluations, potential confounding factors or bias, and other experimental conditions that might affect the relevance of the data to the safety of the target. In some cases, key experiments may need to be replicated or modified to better inform risk assessment. The intended clinical use is a primary consideration and should factor in duration of use and patient population. The nature of the theoretical concern should drive what, if any, additional assessments are warranted. As discussed in more detail below, approaches to address concerns related to growth factor biology will differ from those in which the concern is immunomodulation and decreased tumor surveillance.
Continuously Assess Emerging Data for Signals of Concern
As the discovery and development process continues, a large amount of preclinical pharmacology and toxicology data is generated, and additional data may emerge in the literature from related molecules in the class. These data should continually be assessed to determine whether there are signals of concern, such as identification of proliferative or preneoplastic changes in subchronic and chronic toxicity studies, prominent effects on innate or acquired immune function, or sustained changes in circulating hormones.
Develop a Carcinogenicity Assessment Strategy
Using the information described above, the sponsor should then develop a plan to assess carcinogenic potential, tailoring it to the molecule and therapeutic class under consideration. Depending on the nature of the target, mechanism of action, existing data, and clinical use of the biotherapeutic, there may already be an adequate understanding of carcinogenic potential and additional experimentation is not warranted. Alternatively, it may be determined that although there are theoretical concerns, there are no appropriate animal models or other experiments that will provide clinically meaningful information. In either case, the sponsor should provide a balanced assessment in the carcinogenicity section of the submission dossier. The approval documents for enfluviritide provide an interesting example where such an approach was employed (reviewed in Cavagnaro 2008). A similar approach has been outlined in the case of recombinant human interleukin-10 (Rosenblum and Dayan, 2002). In other cases, the sponsor may determine that additional experimentation is warranted to assess carcinogenic potential as discussed in the following section. The following sections present important factors to consider in designing and implementing a carcinogenicity strategy.
Approaches for the Assessment of Carcinogenicity
The review of marketed biotherapeutics indicated that a variety of experimental methods, including, but not limited to, two-year rodent bioassays have been used to assist in assessing carcinogenic or tumor growth potential. The purpose of the following section is to briefly review some of these alternative approaches and comment on their utility in predicting clinical risk. It is important to note that these approaches are part of a weight of evidence approach. No single approach, including two-year bioassays, provides a definitive prediction of human risk.
Additional Characterization in Standard Repeat-dose Toxicity Studies
Standard repeat-dose toxicity studies are useful for detecting many signals for increased carcinogenic risk. Traditional parameters evaluated in these studies, as well as non-routine parameters incorporated based on target concerns (cell proliferation, hormone levels, pharmacodynamic measures) have value in assessing carcinogenic risk in many situations. Evidence of tissue hyperplasia or dysplasia, increased organ weight, cell proliferation, immunosuppression, or endocrine changes may indicate a greater concern regarding carcinogenic risk. Likewise, lack of such evidence in a chronic toxicity study at a significant multiple of the maximal recommended human dose would lessen perception of risk.
The duration of the chronic toxicity studies for biotherapeutics is frequently debated. In some of these cases, carcinogenicity or tumor growth enhancement concerns have been included in the rationale for extending the duration of these studies. As indicated in ICH S6 (IHC 1997b), a six-month duration of treatment is generally appropriate, although there may be case-by-case circumstances for considering extending the treatment period to nine months or one year to see if progression of a specific toxicity occurs (Clarke et al. 2008). In our review of marketed biotherapeutics, it was difficult to determine if extended duration chronic toxicity studies were conducted to aid in assessing carcinogenic risk. One-year studies of insulin analogs in rats were conducted to compare relative carcinogenic risk to native insulin, which were surprisingly more sensitive than two-year carcinogenicity studies (Section 3.2). As previously discussed, lymphoma and lymphoproliferative lesions were observed by twenty-eight weeks in monkeys given alefacept; however, no carcinogenic effects were observed following one year of treatment with abatacept. Despite the fact that these were both immunomodulatory agents, it is not clear that the duration of the monkey study influenced risk assessment. Consistent with the conclusions of Clarke et al. (2008), we believe that in most cases, a six-month duration of treatment is adequate. Extending nonhuman primate studies from six months to nine or twelve months (which translates into <5% of additional treatment duration relative to lifespan) are not likely to be helpful in assessing carcinogenic potential or ability to enhance tumor growth. In addition, an evaluation of the carcinogenic potential of human carcinogens in monkeys over a thirty-six-year period showed that in most instances treatment periods of 5 to 10 years or more were required before a low incidence of tumors was detected (Schoeffner and Thorgeirsson 2000).
Assessments of Cell Proliferation
In some cases, it may be desirable to provide a quantitative assessment of cell proliferation, either in vitro or in vivo (Alison 1995). Cell proliferation assays are based on the assumption that an increase in carcinogenic risk occurs if there is an increase in the number of DNA replications, not necessarily the rate of DNA replication (Cohen 2004). Cell proliferation assays have been used with therapeutic growth factors (Section 4.5). Increased cell division with therapeutic growth factors or hormones is related to mitogenesis rather than toxicity with ensuing regeneration (Cohen 2004).
In vitro mitogenicity assays have been used to address theoretical concern with some biologics, as discussed for insulin analogs, keratinocyte growth factor, and natalizumab. The advantage of in vitro studies in this context is the relatively easy access to animal and human tissue, including cancer cell lines, and the ability to address hypotheses concerning potential mechanisms. A key disadvantage is that without a comparator with known clinical relevance (i.e., native human insulin), it is extremely challenging to determine the clinical relevance of the in vitro findings. Because of these limitations, positive results often warrant the conduct of in vivo studies to further understand the importance of the in vitro data. As an example, human KGF (described above) resulted in the growth of human epithelial tumor cell lines in vitro. This positive in vitro finding led to additional experiments in xenograft models in immune deficient mice and in transgenic mice to evaluate tumor growth enhancement risk.
The correlation between in vitro and in vivo cell proliferation is variable among biotechnology-derived pharmaceuticals. With insulin and insulin analogs (insulin-aspart, -detemir, -glusine, and -lispro), positive in vitro mitogenicity assays usually correlated with modest proliferative in in vivo effects in mammary tissue at six months or one year, with no differences between native insulin and its analogs. With human KGF, an increase in in vivo growth rate was observed in only one of seven tumor cell line xenografts that were positive in in vitro studies. With natalizumab, the few cell lines that were positive in vitro were negative for tumor-promoting potential in the similar tumor cell line xengrafts. Although in vitro cell proliferation data may be warranted in certain cases, it is important that these studies be rigorously conducted using appropriate controls and well-characterized cell lines that are known to possess the relevant receptors and downstream cell-signaling pathways.
The question of whether to measure cell proliferation in vivo depends on several factors. If there are findings from standard toxicity studies suggestive of increased cell proliferation (hyperplasia, preneoplastic lesions, or in some cases increased organ weight), then measuring labeling indices will provide a quantitative estimate of the labeling index. If there are no findings in the standard toxicity study suggestive of cell proliferation, the probability of detecting increased replicative rats is low; however, the sponsor or regulatory reviewer may determine these data are warranted based upon a specific theoretical concern. These in vivo measures of cell proliferation should be conducted only in a targeted manner, based on either theoretical concerns or in vitro or in vivo data that suggests a specific tissues or group of tissues may be affected.
Assessment of Apoptosis
Since decreased apoptosis can also increase carcinogenic risk, there may be a theoretical concern to measure this parameter in target organs similar to that conducted with carcinogenic chemicals or small-molecule pharmaceuticals (Chang, Chapkin, and Lupton 1997; Liao et al. 2002; Skov et al. 2007; Smith, Nyska, and Portier 2004). Measuring apoptosis is much less common than assessing cell proliferation in nonclinical safety studies and is usually of limited value, since (1) many organs have a low apoptotic rate (<1%) and therefore attempting to demonstrate a relative decrease compared to proliferation is difficult; (2) apoptotic rate increases with increasing proliferative rate, which confounds attempt to demonstrate a decreased apoptotic rate; and (3) mathematical modeling to understand the altered balance between apoptosis and cell proliferation and resulting predictive carcinogenic risk is difficult. For these reasons, it is not recommended to measure apoptosis in safety studies of biotechnology-derived pharmaceuticals.
In Vivo Assessments Using Rodent-Specific Homologue
As discussed previously, for many biotherapeutics, rodents are either not a pharmacologically relevant species or are of limited use in longer-term studies because of immunogenicity consequences. One potential approach to circumvent the species specificity could be to conduct studies using a homologous protein with pharmacological activity in rodents. A full discussion of the utility of rodent-specific homologous proteins in nonclinical safety evaluation is beyond the scope of this paper and the reader is referred to a recent review (Bussiere et al. 2009). Important considerations in determining the utility of a homologue include (1) recognition that the homologue is a unique protein from the clinical molecule with a different amino acid sequence and may have different binding affinity and/or bind a distinct epitope and (2) the distribution of the target, downstream mechanisms, and off-target and secondary effects may be different between rodent and human. In our review, the only biotherapeutic that appeared to use a homolgous protein in an assessment of carcinogenicity was epoetin beta, in which a murine version was tested in a two-year bioassay. Rodent-specific homologous proteins were used in two-year rodent bioassays for growth hormone; however, these studies were not conducted as part of the safety evaluation supporting marketing authorizations. As stated in the case review section, the two-year studies of growth hormone did not appear to substantially inform the safe clinical use of these molecules. The six-month toxicity studies with homologous proteins have been conducted in rodents for infliximab and efalizumab; however, it is likely that these studies were done primarily to characterize chronic toxicity, rather than inform regarding carcinogenic potential. Because of the inherent limitations of rodent-specific homologues (unique protein from clinical candidate, difference in affinity and binding epitope), we do not recommend the use of these proteins in two-year rodent studies to screen for carcinogenic potential. These molecules may be useful experimental tools to probe specific mechanistic questions and therefore appropriate on a case-by-case basis.
Transgenic Mice
In discussing the use of transgenic mice, it is important to distinguish between the use of one of the alternative mouse carcinogenicity bioassays (p53+/− knockout, rasH2 transgenic, TgAC transgenic, homozygous XPA knockout) and the use of target-specific knock-in or knockout mice. To date, use of transgenic mice in formal carcinogenicity studies with biotechnology-derived pharmaceuticals is nonexistent in published literature (Jacobson-Kram, Sistare, and Jacobs 2004). The only known example identified in our review of marketed biotherapeutics was the use of rasH2 mice to evaluate rHuKGF (described above). However, as noted previously, this study was not a formal carcinogenicity bioassay (six months of drug treatment), but rather an assay to evaluate the potential of the drug to influence spontaneous tumor incidence. Based on the limited experience with and lack of validation of biotherapeutics in the alternative mouse carcinogenicity assays, their use is not recommended to screen for carcinogenic effects.
In contrast to alternative mouse carcinogenicity bioassays, target-specific transgenic mice are modified to specifically knock out or knock in a specific gene thought to be important in the mechanisms of action. Our review did not identify the use of any of these models in support of registration of a biotherapeutic. The value of these mice in informing safety assessment is highly dependent on how well characterized the model is in terms of both phenotype and biologic similarity to the intended pharmacology. Although the literature may provide useful information regarding the transgenic animals, it may be necessary for sponsors to conduct an in-depth phenotypic characterization prior to determining that the transgenic model is useful to answer a specific question regarding carcinogenic potential. Although we do not recommend the routine use of these models, they may have utility for hazard identification if there is a specific, mechanism-based cause for concern and the model is rigorously characterized.
Special Considerations for Immunomodulators
Several lines of scientific investigation have demonstrated a notable relationship between overt immunosuppression and cancer, particularly in the surveillance of tumor antigens on transformed cells and oncogenic viruses. Epidemiological studies of cancer rates among organ transplant recipients, who are routinely treated over prolonged periods with immunosuppressive drugs to counteract host rejection, demonstrate an increased risk of melanoma, lymphoma, non-Kaposi’s sarcoma, and various other solid tumors. These studies generally report long-term relative risks ranging from two-fold to twenty-five-fold over the general population (Agraharkar et al 2004; Jensen et al. 1999; Kehinde et al. 1994; Marcen et al 2003; Penn 1988, 1991, 1996; Pham et al. 1995; Sheil 1986). These data are supported by epidemiological studies of other human populations, including individuals with underlying congenital or acquired immune system abnormalities (e.g., AIDS), and mechanistic studies in animals, including experiments using animal models with specific immunodeficiencies and tumor challenge models. The association with cancer is less clear for those agents that produce selective modulation of specific pathways of the immune system in the absence of overt immunosuppression (Wolfe and Michaud 2007). Cytotoxic T cells, NK cells, dendritic cells, and macrophages are believed to be largely responsible for immunosurveillance of tumors and oncogenic viruses (Dunn, Old, and Schrieber 2004; Penn 1988, 1998; Rosenberg 2001; Whiteside 2006) and are exploited in the use of cytokine therapies designed to stimulate anti-tumor immunity (Burkett et al. 2004; Davis et al. 2007; Moroz et al. 2004). Among approved biotechnology-derived therapeutics, review of the drug label suggests some increased risk of hyperplasia, lymphoproliferation, or cancer associated with abatacept (Orencia), an Fc-fusion protein that contains the extracellular domain of CTLA-4, Amevive (an Fc-fusion protein that contains the CD2-binding portion of human leukocyte function antigen-3), OKT-3 (an anti-CD3 monoclonal antibody), and anti–TNF therapies (Leonardi et al. 2006; Nair, Raval, and Mehta 2007) based on data available in either animals or humans. However, increased cancer risk among various patient populations receiving these treatments is not well established, and baseline cancer rates may differ by disease population. For example, lymphoma rates are approximately two times higher in rheumatoid arthritis patients as compared to the general population, thus complicating epidemiological associations of treatment and cancer (Baecklund et al. 2004; Kaiser 2008).
Because of the recognized importance of a functional system for immunosurveillance of tumor antigens and oncogenic viruses, there is interest in evaluating the potential for carcinogenicity associated with chronic immunosuppressive therapy in a nonclinical setting under ICH S6 (ICH 1997b). Based on established mechanisms of action for immune-mediated detection and clearance of tumor antigens and oncogenic viruses, special characterization of therapeutic agents that suppress activity of NK cells, macrophages, and/or cytotoxic T cells is of particular importance. However, validated animal models suitable to assess carcinogenic risk associated with impaired immunosurveillance of tumor antigens or oncogenic viruses are lacking. In the absence of such data, regulatory agencies have reviewed spontaneous cancer incidence gathered in the course of chronic studies evaluating unchallenged animals with various background rates of latent viral infections. To date, no comprehensive evaluation has been conducted to ascertain the utility of these data for informing cancer risk assessment.
Neoplasia, presumably associated with viral recrudescence, has been observed in cynomolgus monkeys following administration of immunosuppressive agents such as cyclosporine (Gaschen and Schuurman 2001) and alefacept. However, the ability to detect neoplasia will depend heavily on the background of the specific primate colony, the nature of the immunosuppression, and specifics of the study design, such as study duration and number of animals, as illustrated by the lack of findings with known oncogenic immunosuppressive agents (Haustein et al., 2008; Lerche and Osborn 2003; Mahalingam et al. 2007; Sasseville and Diters 2008; Wachtman and Mansfield 2008). Whereas viral and bacterial host resistance models have been developed in rodents, these have not been well characterized for their ability to predict clinical neoplasia risk with immunosuppression. Moreover, rodents may present an additional inherent challenge for testing biologically derived immunosuppressive agents when the agent is not pharmacologically active in the species.
Despite these challenges, the sponsor must consider means of establishing the potential carcinogenic risk of administering the therapeutic agent to human patients. This should minimally consist of a review of the pertinent immunology as it pertains to clearance of oncogenic viruses and tumor antigens, and review of mechanism of action and pharmacology data. Where possible, evaluations of the immunological effects of the therapeutic agent in normal animals or in host resistance studies (i.e., viral challenge, tumor xenografts) provide evidence directly applicable to an assessment of immunological surveillance and cancer risk. In these cases, the intent and design of such specialized studies should be driven by the specific informational needs based on identified risks and knowledge of mechanism of action and pharmacological class effects.
In the end, one should consider whether animal studies would provide meaningful additional information to guide clinical or regulatory decision making and whether other risk-management strategies, including informed consent, package labeling, and patient monitoring, should be considered.
Special Considerations for Therapeutic Growth Factors
Growth factors are a diverse set of molecules that are capable of stimulating cellular proliferation and differentiation. Growth factors typically act as signaling molecules and include both cytokines and hormones that bind to specific receptors on the surface of their target cells. Their ability to promote cell differentiation and maturation varies between the different growth factors. For example, bone morphogenic proteins stimulate bone cell differentiation, whereas fibroblast growth factor and vascular endothelial growth factor stimulate blood vessel differentiation. Growth factors are used clinically to modulate the growth and maturation of diverse cells types including fibroblasts, hematopoietic cells (both red and white blood cell lineages), and other specific cell types, for example, chondrocytes or osteocytes.
There is a theoretical concern that therapeutic growth factors may also enhance the growth and/or progression of tumor cells that express their receptors. Receptors for various growth factors may, in some circumstances, be constitutively expressed or up-regulated on tumor cells of similar origin to their normal counterparts or, in some cases, on divergent tumor cell types. The implication is that if tumor cells express the growth factor receptor that they will respond to exogenously administered therapeutic growth factors. Adding to this concern is the fact that some pathways important to tumor cell growth can be blocked by exogenously administered antigrowth factors, for example, anti-VEGF has been show to be effective in blocking tumor angiogenesis. The effectiveness of using antigrowth factors to treat various cancers has further heightened concern that the systemic or local use of growth factors in other disease processes (i.e., VEGF in cardiovascular disease) may increase the risk of progression of undetected cancer. However, it is uncertain whether increased levels of growth factors from exogenous administration will physiologically influence tumor cells that may already be operating under autocrine or paracrine mechanisms of cell growth. It is also unclear which growth factor receptors and signaling pathways may actually be driving tumor progression for a given neoplasm.
Because of the theoretical clinical risks of exogenous growth factor administration, sponsors have taken a proactive approach to address concerns. This approach has involved the utilization of nonclinical (in vitro and in vivo) models because the actual clinical consequences of altered tumor cell life cycles are difficult to quantify. This difficulty is stems from the typically small number of likely events in clinical trials (owing to limited sample sizes) and the extended period required (e.g., latency) for such effects to manifest in humans.
Some of the nonclinical strategies employed by biopharmaceutical companies are outlined in global regulatory guidelines describing nonclinical safety evaluation practices for biotechnology-derived pharmaceuticals (ICH S6). ICH S6 (1997b) specifically mentions growth factors as products that may require assessment of carcinogenic potential by some means. The guideline says that for drugs having the potential to support proliferation of cells, expression of the product’s receptor in relevant tumor and normal human cells should be characterized (e.g., presence or absence). Furthermore, it states that the ability of the product to stimulate the in vitro growth of relevant tumor and normal human cells should be evaluated. It goes on to say that if in vitro data show activity, further studies may be needed, and that these studies might include a two-year rodent bioassay if the molecule is biologically active and nonimmunogenic in that species.
These regulatory expectations have been addressed in a number of ways by sponsors and several of the approaches have been discussed previously in this paper. Assessment of growth factor receptor expression has been accomplished by measuring mRNA or protein levels by various techniques including immunohistochemistry, Western blotting, reverse transcriptase polymerase chain reaction, or RNAse protection assays. Although informative, these studies do not address the functionality of growth factor receptors on tumor cells. To assess biological activity, human tumor cell lines are grown in vitro, and exogenous growth factor is added to look for increased growth as an indicator of functional activity. Both of these approaches are consistent with expectations set forth in ICH S6 (summarized above), although extrapolation of these results to potential in vivo outcomes and human risk is difficult at best and in many cases may be irrelevant.
Accordingly, some companies have used in vivo models of tumor cell growth to obtain more physiologically relevant information. Some recombinant growth factors have been assessed in rodent tumor xenograft models. For example, it has been hypothesized that erythropoiesis-stimulating agents (ESAs) may enhance tumor progression directly by binding to the erythropoietin receptor on tumor cells or indirectly by increasing oxygenation to tumors (Osterborg et al. 2007). These concerns have led investigators to evaluate ESAs in both syngeneic and xenograft tumor models in rodents. Increased tumor and tumor vessel growth have been evaluated in these models with ESAs alone and in combination with increased oxygen and/or a variety of other chemo/radiotherapies (LaMontagne et al. 2006; Osterborg et al. 2007). A review of over twenty independent in vivo studies concluded that ESAs do not promote the growth of tumors and a careful analysis of detection of erythropoietin receptor on tumors may, in some cases, reflect a methodological artifact (Elliott et al. 2006). Xenograft models have also been applied to other therapeutic growth factors, such as palifermin (rHuKGF). Use of these xenograft models is facilitated by their immunodeficient nature (e.g., nude mice), in many cases eliminating concerns of a confounding immunogenic response directed against the therapeutic molecule. It is unclear how to best interpret these results, since these models were originally developed, and have been optimized, to measure inhibition of tumor cell growth rather than an increase in cell growth. Furthermore, the relevance and extrapolation of the results of these models, even for their designed purposes, is under debate.
As discussed previously, other approaches including studies with surrogate proteins (growth factor) and modified transgenic models (rHuKGF) have been described. All of these strategies can be used in risk assessment for a therapeutic growth factor. However, in many cases, completion of the above approaches has not altered the need for precautionary text in product labels regarding the theoretical risk of enhanced tumor growth with administration of the growth factor. Consequently, the value of these approaches is questionable.
Conclusion
The assessment of a carcinogenic potential or the ability to promote tumor growth are among the most challenging areas in the nonclinical assessment of biotherapeutics. In the initial development of these therapies, there appeared to be a perception that biotherapeutics were exempt from carcinogenicity concerns. This perception was largely based on the fact that two-year rodent studies were often not possible and genotoxicity concerns typically do not exist for biologics. With the rapid expansion of new biotherapeutics and targets, increased attention has been directed toward carcinogenicity assessments. Since classical two-year bioassay approaches are not applicable for most biotherapeutics, industry and regulatory authorities have relied on an ever-expanding array of experimental approaches to better characterize risk.
It is interesting to note that despite the array of theoretical concerns, the only molecules in our review in which there was at least some clinical evidence of an increased risk for malignancies in product labeling was for immunosuppressive drugs. Although frequently debated and often supported by in vitro data suggesting a potential risk, current clinical data with a variety of molecules which have effects on growth factors do not clearly demonstrate an increased risk. These observations must be viewed with caution, as detecting increased rates of cancer in postmarketing patient populations is often problematic because of the relatively low incidence rate, difficulty in case ascertainment, and long latency periods.
As in any scientific inquiry, scientists engaged in safety assessment should consider all of the available approaches and balance the need for animal data, the technical feasibility of the proposed experimentation, the design of the proposed study to answer specific questions, the ability to interpret the data generated, and most importantly, the ability to apply the results to human health risk assessment. Keeping in mind a science-driven weight of evidence approach, we offer the following recommendations: The mechanism of action and pharmacology should be carefully considered and relevant information from the scientific literature should be evaluated to identify what, if any, theoretical risks the molecule may have for inducing cancer or promoting the growth or spread of an existing tumor. Repeat-dose toxicology studies should be carefully evaluated for any signs that may give insight into concern regarding proliferative potential or immunosupression. Incorporation of mechanistic endpoints (cell proliferation, apoptosis, hormonal analysis) into repeat-dose studies is often valuable, but should be determined based on the biology of the molecule, rather than as a default for all therapeutics. When additional experimentation is warranted, two-year rodent bioassays should not be the default mechanism to assess carcinogenic potential. Experimentation should be hypothesis-driven and may include a variety of experimental methods to generate a weight-of-evidence assessment. In some cases, two-year bioassays may be appropriate given the mechanism of action or prior experience with the class. Alternative approaches should be justified. For immunomodulatory agents, the nature of immunomodulation should be characterized in animal studies, consistent with principles in ICH S8. For classes of agents for which there is already a known clinical risk, additional animal studies are not warranted as they are not likely to inform clinical risk. For novel agents with highly specific effects on the immune system, a consideration of the pathway affected and implications of affecting the pathway must be considered in determining what, if any, additional animal experimentation is warranted. For growth factors, a careful analysis of the literature and mechanistic understanding is important in determining if additional in vitro or in vivo studies are warranted. Although there is often a perceived need to characterize the potential of an agent to enhance tumor growth in animal models, we strongly believe that these studies should be interpreted with a great deal of caution, as their ability to predict human responses remains to be determined. Although data from animal studies may be useful in exploring the potential for risk in humans, current labeling practices indicate that these studies are not used to quantify risk relative to use in humans. As such, when a concern for human risk remains, we believe that
product labeling is a critical risk communication tool and should provide useful and relevant information to the prescribing physician. Descriptions of a conflicting array of in vitro and animal findings followed by statements that the relevance of the animal findings is not known is not useful to health care providers, and Further evaluation of human risk is best addressed in the postmarketing setting, through the use of data collection and/or prospective human studies conducted under a product risk management program.
Clearly it is no longer appropriate to indicate that carcinogenicity assessments for biotherapeutics are “not applicable.” Predicting carcinogenic and related risks will remain challenging for biotherapeutics. However, we suggest that an approach that incorporates thoughtful and rigorous consideration of the theoretical risks of a particular mechanism of action followed by hypothesis-driven experimentation is more rigorous than defaulting to the standard approach for small molecules of conducting 2-year bioassays.