
Abstract
Bococizumab is an anti-PCSK9 monoclonal antibody that was intended for the treatment of hypercholesterolemia. After reviewing the 6-month rat toxicity study data, in which there was a low spontaneous tumor incidence, unrelated to bococizumab administration, the U.S. FDA granted a carcinogenicity waiver request based on a weight-of-evidence assessment of low carcinogenic risk. Subsequently, after reviewing 6-month rat toxicity study data from another anti-PCSK9 antibody, RN317, with a similar low tumor incidence (unrelated to RN317), the U.S. FDA rescinded the bococizumab carcinogenicity study waiver and requested a full 2-year rat carcinogenicity study be conducted. The resulting 2-year carcinogenicity study demonstrated no bococizumab-related increase in tumors, confirming the weight-of-evidence evaluation and alleviating concerns regarding the carcinogenic potential. Here we report the scientific and regulatory background that led to the request for a rat carcinogenicity study, the feedback on the design of the carcinogenicity study, and the results from this study which affirmed the original weight-of-evidence assessment of low carcinogenic risk.
Introduction
Bococizumab, also previously known as RN316 or PF-04950615, is a humanized IgG2 monoclonal antibody targeted to the Proprotein Convertase Subtilisin Kexin type 9 (PCSK9) protein. This molecule, and a similar molecule (RN317) anticipated to have enhanced pharmacokinetic characteristics relative to bococizumab, 1 were being developed for the treatment of familial hypercholesterolemia. Both bococizumab and RN317 bind to the LDL-R binding region of PCSK9 and inhibit its activity. PCSK9 reduces the expression of low-density lipoprotein receptor (LDL-R) on hepatocytes, so inhibiting its activity results in increased LDL-R expression and an associated greater clearance of low-density lipoprotein cholesterol (LDL-C) from the blood. The enhanced clearance of LDL and lower blood LDL are associated with slowing or stopping the progression of atherosclerosis potentially leading to improved cardiovascular health.
Bococizumab and RN317 were under development for use as add-on therapies to standard-of-care treatment in patients with hypercholesterolemia who are unable to reach target LDL reduction goals and for patients where standard-of-care is not well tolerated. Bococizumab was evaluated in 6 lipid-lowering clinical trials as well as 2 cardiovascular outcomes trials (SPIRE-1 and SPIRE-2) and was found to be generally safe and effective in certain high-risk populations.2-5 In 2016 Pfizer discontinued the development of bococizumab 6 due to disappointing clinical results which saw an unanticipated attenuation of its cholesterol lowering efficacy, likely related to increased immunogenicity (antidrug antibodies 7 ), and injection site reactions compared to competitors. 6
Bococizumab was evaluated in nonclinical studies in the Sprague-Dawley rat and cynomolgus monkey (NHP). Bococizumab lowered LDL cholesterol in the rat and NHP, and, in rats, resulted in increased adrenal weights, adrenal hypertrophy, and adrenal cystic angiectasis/degeneration. Other bococizumab-related findings were considered immunogenicity-related (the result of a species-specific immune response to the administration of a humanized monoclonal antibody in these nonhuman species) and immunogenicity in nonclinical species is not considered predictive of immunogenicity in humans.8,9
The toxicology data was used in support of a carcinogenicity waiver request that was granted. However, following the submission of an additional 6-month chronic toxicity study with RN317, the carcinogenicity waiver for bococizumab was rescinded, and the U.S. FDA requested a 2-year carcinogenicity study be conducted prior to approval of bococizumab for marketing. This study was subsequently conducted by Pfizer, resulting in a definitive lack of carcinogenicity risk for bococizumab. Nevertheless, because 2-year carcinogenicity studies with monoclonal antibodies are rare and/or not often detailed in the literature, this manuscript describes the regulatory background leading up to the initiation of the carcinogenicity study and the carcinogenicity study results. Specifically, we summarize the 6-month toxicity data in rats that led both to the U.S. FDA’s grant of a carcinogenicity waiver for bococizumab and then the subsequent rescinding of the waiver. We then discuss the 2-year carcinogenicity findings and implication for future development strategy and carcinogenicity risk assessment of monoclonal antibodies.
Regulatory Background
The nonclinical toxicology program for bococizumab was developed based on the principles of ICHS6(R1).
10
The rat and NHP were justified as appropriate toxicity species based on conserved homology of PCSK9, comparable binding affinities, and functional pharmacological effect of bococizumab in both of these species. The initial bococizumab clinical studies were supported with short term rat and NHP 1-month toxicity studies with twice weekly dosing. The 1-month toxicological and pharmacodynamic profiles in the rat and NHP were considered comparable. Since similar toxicological findings were seen in both species, ICHS6(R1) suggests that longer term studies in a single species are generally sufficient to characterize sub-chronic and chronic toxicity leading to the conduct of a 6-month study of bococizumab in NHP. However, upon regulatory request, a 6-month study in the rat with a 3-month interim necropsy was also conducted. This 6-month rat report was submitted to the U.S. FDA in September 2011 and is shown as the starting point for regulatory interactions that led to the start of the 2-year rat carcinogenicity study (Figure 1). Timeline of regulatory interactions.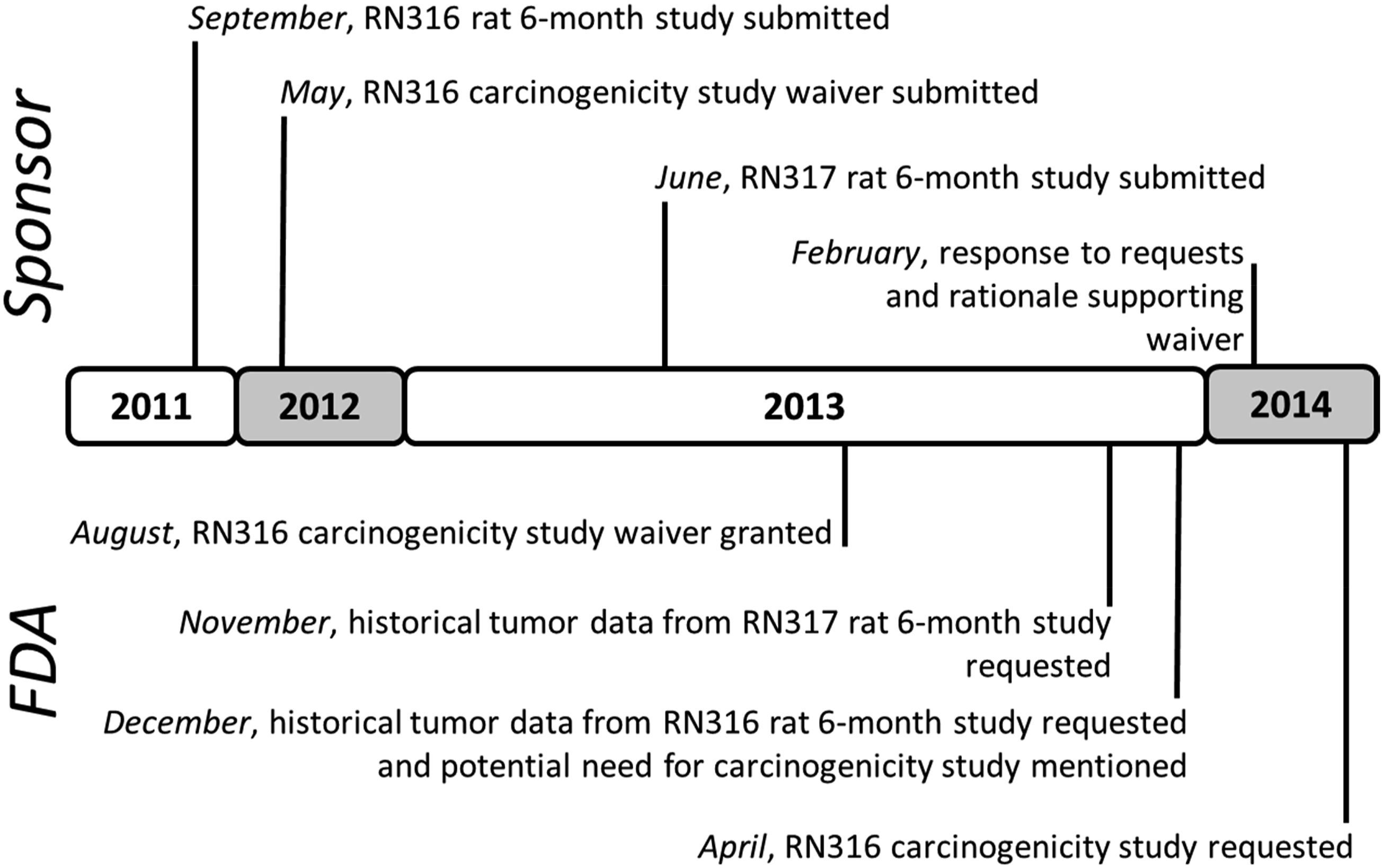
Tumor incidence in 6-month (26-week) studies with bococizumab and RN317.
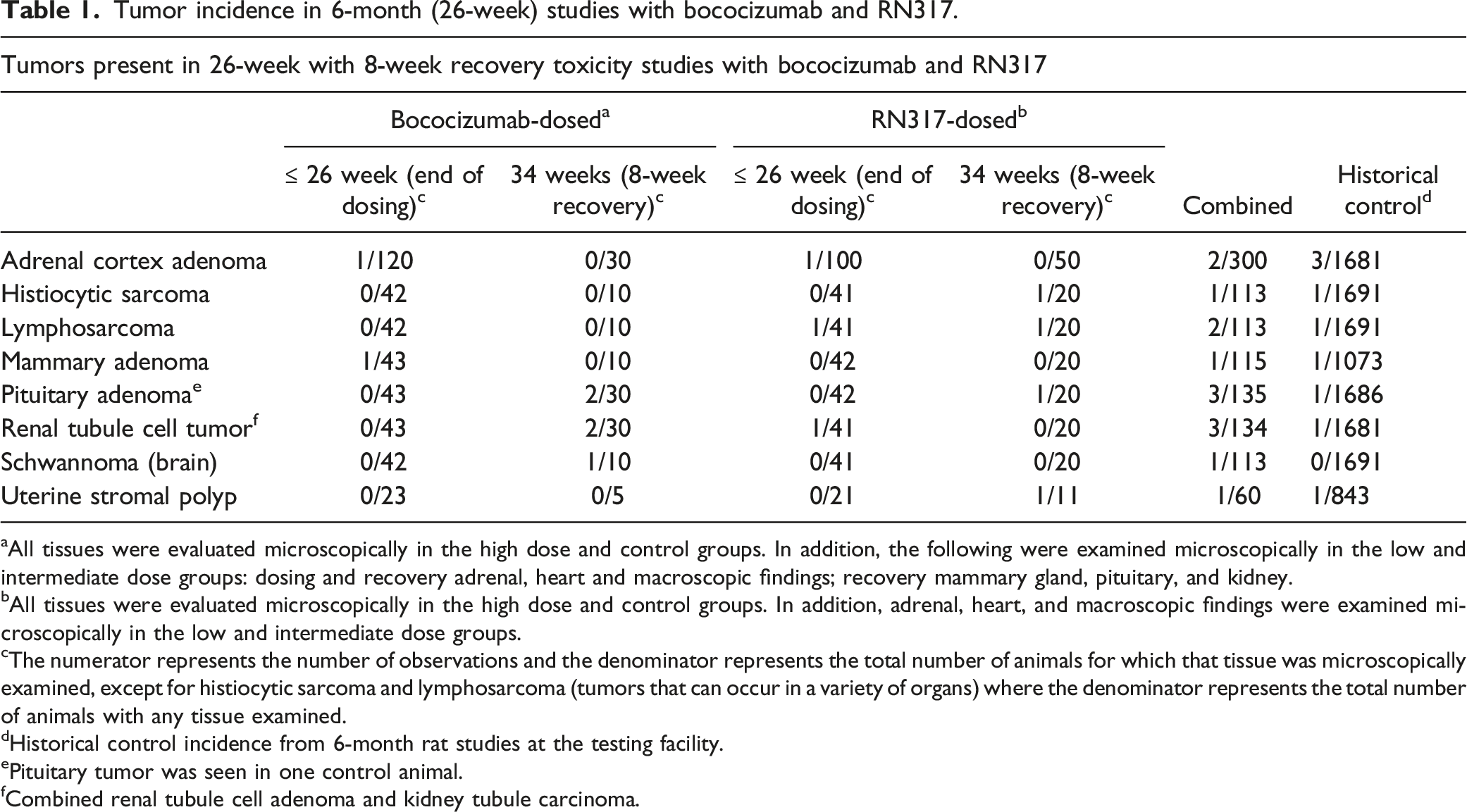
aAll tissues were evaluated microscopically in the high dose and control groups. In addition, the following were examined microscopically in the low and intermediate dose groups: dosing and recovery adrenal, heart and macroscopic findings; recovery mammary gland, pituitary, and kidney.
bAll tissues were evaluated microscopically in the high dose and control groups. In addition, adrenal, heart, and macroscopic findings were examined microscopically in the low and intermediate dose groups.
cThe numerator represents the number of observations and the denominator represents the total number of animals for which that tissue was microscopically examined, except for histiocytic sarcoma and lymphosarcoma (tumors that can occur in a variety of organs) where the denominator represents the total number of animals with any tissue examined.
dHistorical control incidence from 6-month rat studies at the testing facility.
ePituitary tumor was seen in one control animal.
fCombined renal tubule cell adenoma and kidney tubule carcinoma.
Key points included in the request for a carcinogenicity waiver submitted to the U.S. FDA in May 2012.

The RN317 chronic 6-month studies in rat and NHP were submitted to the U.S. FDA in June 2013. Similar to bococizumab, RN317 was well-tolerated and RN317-related findings included lowered LDL cholesterol and HDL cholesterol; in contrast to bococizumab, there were no adrenal gland findings while there were subcutaneous injection site findings (minimal to slight infiltrates, edema/necrosis, fibrosis, and/or hemorrhage). There were no signs of proliferation in the NHP study. Although there were several tumors in the chronic RN317 rat study, these were not considered related to RN317 administration. Through the end of the dosing phase, tumors present in rats administered RN317 included adrenal adenoma, lymphosarcoma, and renal tubule cell tumor; through the end of the 8-week recovery period additional tumors present were histiocytic sarcoma, pituitary adenoma and a uterine stromal polyp (Table 1). Similar to bococizumab, the tumors in rats administered RN317 were considered background findings and not RN317-related due to similar findings in concurrent controls (pituitary adenoma), low incidences of findings, the lack of a dose response, and/or different tissue types being affected.
In November of 2013 Pfizer received a request for information for the RN317 program in which the U.S. FDA provided several recommendations based on the “potential risk for immune modulation by PCSK9 and cholesterol depletion and the unusual incidence of 6 tumors in the 6-month rat study, all at high-dose.” The recommendations included similar information and data that had been requested for bococizumab including an immunotoxicology assessment evaluating the TDAR, Natural Killer cell activity and cytotoxic T cell activity. The lack of effect on these immunotoxicity endpoints had already been provided to the U.S. FDA for bococizumab and were in planning stages for RN317. In addition to this request, the U.S. FDA also requested historical control tumor and leukemia virus incidence data from the CRO and to “explain why these neoplasms are not considered drug related.”
In December 2013, Pfizer received a written request for information/advice from the U.S. FDA stating “we acknowledge prior concurrence on a waiver for carcinogenicity assessments. This concurrence was based on a preliminary weight of evidence assessment. Recently, a concerning imbalance in tumorigenicty was observed in your related compound RN317, in the chronic rat toxicity study. We note a similar imbalance in tumors at the recovery necropsy in your 6-month rat toxicity study for bococizumab as well. Further explanation is needed for why these observed neoplasms (malignant schwannoma, renal tubule cell adenomas, and pituitary adenomas) are not considered drug-related as they occurred in the mid- and high-dose groups. Some of these tumors are rare in 2-year carcinogenicity studies and potential PCSK9/cholesterol mechanisms could be implicated in the lymphocyte dysregulation. Historical background incidence of these tumors from the CRO that performed the 6-month rat toxicology study for bococizumab is requested. This information should include tumor incidence covering the total duration of the study period (treatment and recovery). Based on this new information and after consultation with CDER’s Executive Carcinogenicity Assessment Committee a carcinogenicity study will likely be needed with your Biological License Application (BLA) submission.”
In our written response to the U.S. FDA in Feb 2014, we maintained that despite the apparent tumor imbalance with a total of 14 tumors (Table 1) in rats administered bococizumab or RN317 relative to controls [1 control animal across both 6-month studies], that based on a weight of evidence rationale, that the tumors observed were incidental to treatment. Although there was an apparent overall imbalance, other factors should be considered when assessing carcinogenic potential including location/cell type (particularly for tumor combinations), 22 incidence of each tumor type, presence or absence of other proliferative (potentially preneoplastic) lesions, and context compared with historical control data. To fulfill the U.S. FDA’s request for further explanation as to why the tumors, particularly schwannoma, renal tubule cell tumor and pituitary adenoma, were not being considered bococizumab-related, additional rationale as discussed below was provided for considering specific tumor types incidental.
Summary of the Tumors Present in the 6-Month Bococizumab and RN317 Studies
Renal tubule cell adenomas were present at the end of the recovery period in 2 female rats administered bococizumab and renal tubule carcinoma was present at the end of the dosing period in 1 female rat administered RN317; thus, across the 2 studies, 3 female rats had renal tubular cell tumors. The historical incidence of renal cell carcinomas in controls at the test facility was 1/838 male rats, with none in females and none for tubule cell adenomas. Although the combined incidence of 3 renal tubule cell tumors across both 6-month studies is high compared with the test facility historical control data, the variant that was present (amphophilic-vacuolar) is a known familial condition in SD rats23-26 and has been seen in studies as short as 2 weeks. Other proliferative kidney findings, while typically seen with kidney tumor-causing test articles, were not observed in either 6-month study. Therefore, the kidney tubule cell tumors present were considered incidental and consistent with the described familial kidney tumors of rats.
A schwannoma was present in 1 recovery male (8-week recovery period, week 34 of study) administered bococizumab and in no animals administered RN317. There were no incidences of schwannoma in similarly aged historical control rats at the test facility. However, based on reports of schwannoma in younger rats27,28 and the single occurrence in the 6-month study, this finding was considered incidental.
Pituitary adenomas were present at the end of the recovery period in 1 male and 1 female administered bococizumab and 1 male administered RN317. Pituitary adenoma was also present in 1 control rat from the RN317 study. Although the test facility historical control incidence in similar aged rats was 1 male out of 845 rats (and no females), pituitary adenoma is a commonly occurring tumor in SD rats and has been reported in control rats of similar age and younger than in the 6-month toxicity studies.29-31 Furthermore, pituitary hyperplasia, a non-neoplastic proliferative lesion that is part of a morphologic continuum in the development of pituitary adenoma, was present in 2 control rats across the 2 studies and 1 rat administered RN317. Thus, the overall incidence of proliferative pituitary findings (hyperplasia and adenoma) across the 2 studies was 2/80 control rats and 4/150 bococizumab or RN317 dosed rats. Therefore, because pituitary adenomas have been reported in similar aged control rats, and the overall incidence of proliferative pituitary findings in concurrent control vs bococizumab and RN317 administered animals was similar, the pituitary adenomas were not considered test article-related.
U.S. FDA Request for Carcinogenicity Study
In April 2014, we received the feedback from the U.S. FDA that “Based on the tumor imbalance observed with bococizumab, which included tumors rarely observed or absent in 2-year lifetime rat studies in the laboratory’s historical control database, as well as the absence of demonstration of a rodent specific mechanism of neoplasia, a carcinogenicity study in rats administered bococizumab is anticipated with the BLA to address the carcinogenicity concern.”
At the time of initiation of the carcinogenicity study there were no published or publicly available information on the appropriate design of a carcinogenicity study with a monoclonal antibody. As such, we proposed a 2-year carcinogenicity study in Sprague-Dawley rats with once weekly intravenous dosing of bococizumab at 0, 10, 30 and 100 mg/kg in 20 mM histidine buffer containing 84 mg/mL Trehalose, 0.05 mg/mL EDTA and 0.2 mg/mL polysorbate 80 (pH 5.5). The high dose was estimated to result in drug exposures based on modeled projections that represented 49X over a human efficacious exposure. A key concern was understanding the impact of ADA development on exposure as well as potential for tumor formation or lack thereof. As such we included main study animal blood collection predose and every few months to characterize exposure, PD (cholesterol levels), and ADA.
The Executive Carcinogenicity Assessment Committee (CAC) agreed with the proposed study design and had minimal concerns. They agreed with the proposed dose levels based on the projected rat to human AUC ratios. In addition, they suggested using both a vehicle- and a saline-control group due to the use of a somewhat unique vehicle. Interestingly, despite the idiosyncratic nature of an immune response to a humanized antibody in another species, the CAC advised that bleeding of the main study animals was undesirable and instead suggested that a satellite group of 10 animals be used for exposure, PD, and ADA endpoints to reflect what is happening in the main animals. We incorporated these suggestions and conducted the bococizumab 2-year study.
Materials and Methods, 2-Year Rat Carcinogenicity Study
Animal Housing, Husbandry and Dose Rationale
Six-to 7-week old Sprague Dawley rats (CRL:CD; Charles River Laboratories, Raleigh, North Carolina) were acclimated for up to 10 days before study initiation, were group housed at 20o-26oC and 30%-70% humidity under a 12-hour light/12-hour dark light cycle, and provided food (Certified CR 14% Rodent Diet #5CR4 [PMI Nutrition International Certified LabDiet®]) and water ad libitum. Animals (60-100/sex/dose level) were administered 0 (saline), 0 (vehicle), 10, 30 or 100 mg/kg/week intravenously for up to 98 weeks. Dose levels were selected because they were well tolerated in the previous 6-month study with a no observed adverse effect level of 100 mg/kg. All procedures in the study were in compliance with applicable animal welfare acts and were approved by the local Institutional Animal Care and Use Committee (IACUC).
Test Article and Control Formulations
Bococizumab was prepared at concentrations of 153.5 or 148.6 mg/mL and was stored frozen (-60o to -80oC, protected from light). Bococizumab was thawed, sub-aliquoted into smaller volume containers, and refrozen until removed for dose formulation. Bococizumab containers were limited to 3 freeze/thaw cycles and used until empty/inaccessible. The vehicle (diluent) was a solution of 20 mm histidine, 84 mg/mL Trehalose, 0.2 mg/mL polysorbate 80, 0.05 mg/mL EDTA, pH 5.5. The saline-control was 0.9% sodium chloride for injection, USP (sterile saline). Bococizumab dose formulations were prepared and dispensed using aseptic techniques. Bococizumab aliquots were thawed in a refrigerator (2° to 8°C, up to 75 hours) and diluted with vehicle to a final concentration of 15 mg/mL (based on the actual concentrations as supplied). The 15 mg/mL dose formulations were sterile-filtered (0.2 or 0.22 micron polyethersulfone membrane) and apportioned for once weekly dosing for each cohort of animals. Dose formulations were stored in a refrigerator (2° to 8°C, protected from light) until removed for dosing and were used within 8 hours of removal from the refrigerator. Dose formulations prepared on the day of use were stored under ambient conditions and used within 8 hours of preparation completion.
Study Endpoints
Rats were monitored via daily health checks and weekly clinical examinations. Body weight and food consumption (group-housed) were recorded weekly through week 25 and once every 4 weeks thereafter. Blood was collected from animals assigned to the toxicokinetic (satellite) subgroups for evaluation of high-density lipoprotein cholesterol, low-density lipoprotein cholesterol and total cholesterol at intervals from weeks 12 through 91. At necropsy, terminal body weights and macroscopic findings were recorded, and a standard comprehensive tissue list was collected, formalin fixed, paraffin embedded and processed to slides. Slides stained with hematoxylin and eosin were microscopically examined by a board-certified veterinary pathologist.
Bioanalytical, Toxicokinetics and Anti-Drug Antibody Sampling
Scheduled blood samples for assessing bococizumab plasma exposure and anti-drug antibody (ADA) were collected from animals via a jugular vein on Day 1 and during Weeks 12, 26, 39, 52, 65, 78, and 91, and 96 (males only). Bioanalytical collections from satellite animals (22/sex/dose group) were as follows: predose, 0.5-, 24-, 72-, 120-, and 168-hours postdose for Day 1, Week 52, and Week 96 (males only), using subsets of animals to spread out blood volumes; and once predose during Week 12, 26, 39, 65, 78 and 91. Blood samples for ADA from satellite animals were collected once predose. Bioanalytical and ADA samples were also obtained on days of early terminal euthanasia for satellite and main study female animals in Weeks 86 (mid-dose), 95 (low-dose), and 96 (saline-control, vehicle-control, and high-dose), and from early terminal euthanasia satellite and main study male animals during Week 98 (all dose groups). Blood was collected and processed to obtain plasma (potassium (K2) EDTA anticoagulant). Harvested plasma was stored frozen (-60 to -80°C), maintained on dry ice for shipping, until analyzed for bococizumab or ADA.
Statistical Analysis
All statistical analyses were performed separately for each sex of the main study animals. Survival data were analyzed using two-sided log-rank and Wilcoxon tests. Tests were performed for dose response (vehicle-control and dosed groups only) along with pairwise comparisons for the saline-control group and each dosed group against the vehicle-control group. Additionally, Kaplan-Meier 32 product-limit estimate survival curves were produced for graphical evaluation of survival.
Occult tumors were analyzed by the IARC asymptotic fixed interval-based prevalence test. 33 One-sided tests were performed for dose response (vehicle-control and dosed groups only) along with pairwise comparisons for each group against the vehicle-control group. In the case of sparse tables, the exact form of the test was used.
For males, the terminal necropsy was Week 98 for all groups. Due to having groups terminated earlier because of poor survival, the cut-off points for the interval-based test were weeks 0 to 52, 53 to 78, 79 to 88, 89 to 97, and the terminal necropsy. For females, the terminal necropsy was Week 96 for Group 1 (saline-control), Group 2 (vehicle-control) and Group 5 (high-dose). For Group 3 (low-dose) the terminal necropsy was Week 95 and for Group 4 (mid-dose) the terminal necropsy was Week 87. Due to having groups terminated earlier because of poor survival, the cut-off points for the interval-based test were weeks 0 to 52, 53 to 78, 79 to 86, 87 to Day 659, and terminal necropsy. The terminal necropsies at weeks 95 and 96 for Groups 1, 2, 3 and 5 were combined into a single terminal necropsy starting on Day 660.
Observable or palpable tumors were analyzed using the methods previously described for analyzing survival, using the time to death or time of detection of the tumor as a surrogate for the tumor onset time. One-sided tests were performed. Benign and malignant neoplastic incidences were evaluated separately and combined, where appropriate. The study pathologists determined which tumor combinations were statistically analyzed, using the criteria for combination in Guidelines for Combining Neoplasms for Evaluation of Rodent Carcinogenicity Studies 34 as a guide. Indication of possible treatment effects were assessed on the basis of rare or common tumor type, in line with the current U.S. FDA guidelines. 35
Results
Bococizumab Exposure in Plasma
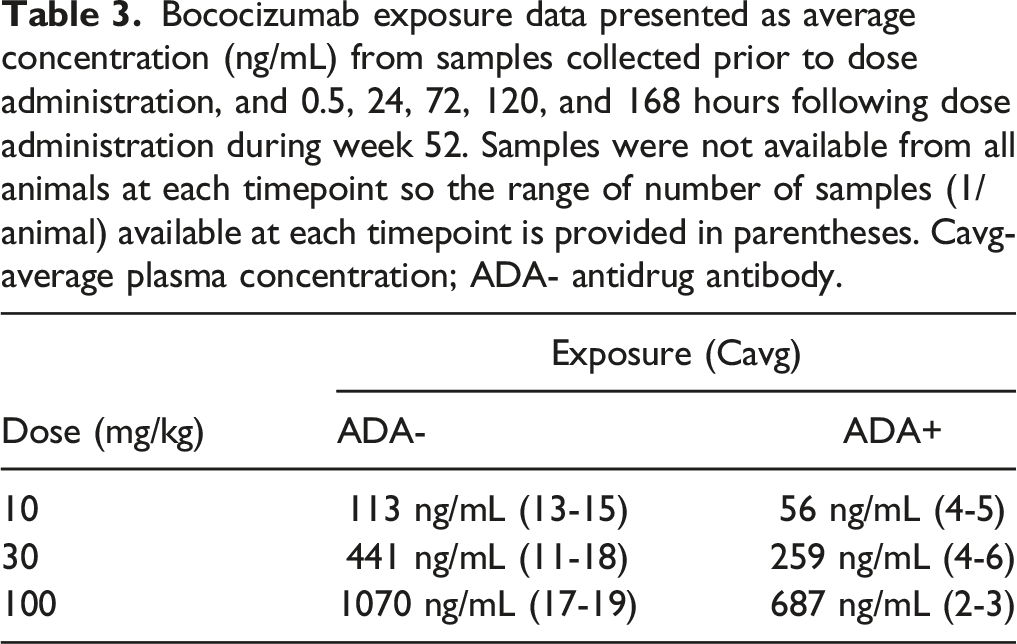
Bococizumab exposure data presented as average concentration (ng/mL) from samples collected prior to dose administration, and 0.5, 24, 72, 120, and 168 hours following dose administration during week 52. Samples were not available from all animals at each timepoint so the range of number of samples (1/animal) available at each timepoint is provided in parentheses. Cavg- average plasma concentration; ADA- antidrug antibody.
Pharmacodynamic Assessment
Pharmacologic activity was measured by the assessment of lipid level, total cholesterol, low-density lipoprotein (LDL) and high-density lipoprotein (HDL) and results were similar to those in earlier, shorter term studies. There were no differences in lipid test results between saline-controls and vehicle-controls. Subsequent comparisons were between animals administered bococizumab and saline-controls. Lipid parameters were lower throughout the dosing phase at ≥10 mg/kg compared with saline-control, but a dose response was generally not present. Total cholesterol concentration, compared with saline-control, was mildly to moderately lower in males and females (0.26-0.64x and 0.44-0.85x of the saline-control value, respectively). The HDL cholesterol concentration was moderately lower in males and females (0.12-0.48x and 0.11-0.52x, of the saline-control value, respectively). The LDL cholesterol concentration was also moderately lower in males and females (0.11-0.46x and 0.14-0.64x, of the saline-control value, respectively). Thus, although there was a concern that immunogenicity would preclude achieving sufficient exposure over a 2-year period, a clear pharmacodynamic effect was evident.
Survival and Clinical Observation During the 98-Week Study Duration
No bococizumab-related difference in survival was detected among any of the groups administered bococizumab during the 98-weeks of the study when compared to the vehicle-control group, although statistical differences among the groups were found. Survival for males and females is presented in Figure 2. Female rats administered 30 mg/kg/dose had statistically significant lower survival rate than female rats receiving vehicle-control (15 of 60 vs 35 of 100 in vehicle-control) with P = 0.0075 and P = 0.0173 for the Log-Rank and Wilcoxon test, respectively. Because no dose relationship was noted, the significant lower survival rate in females administered 30 mg/kg/dose was not considered related to bococizumab administration. Kaplan-Meier survival curves for males (top) and females (bottom) over the 98-week study.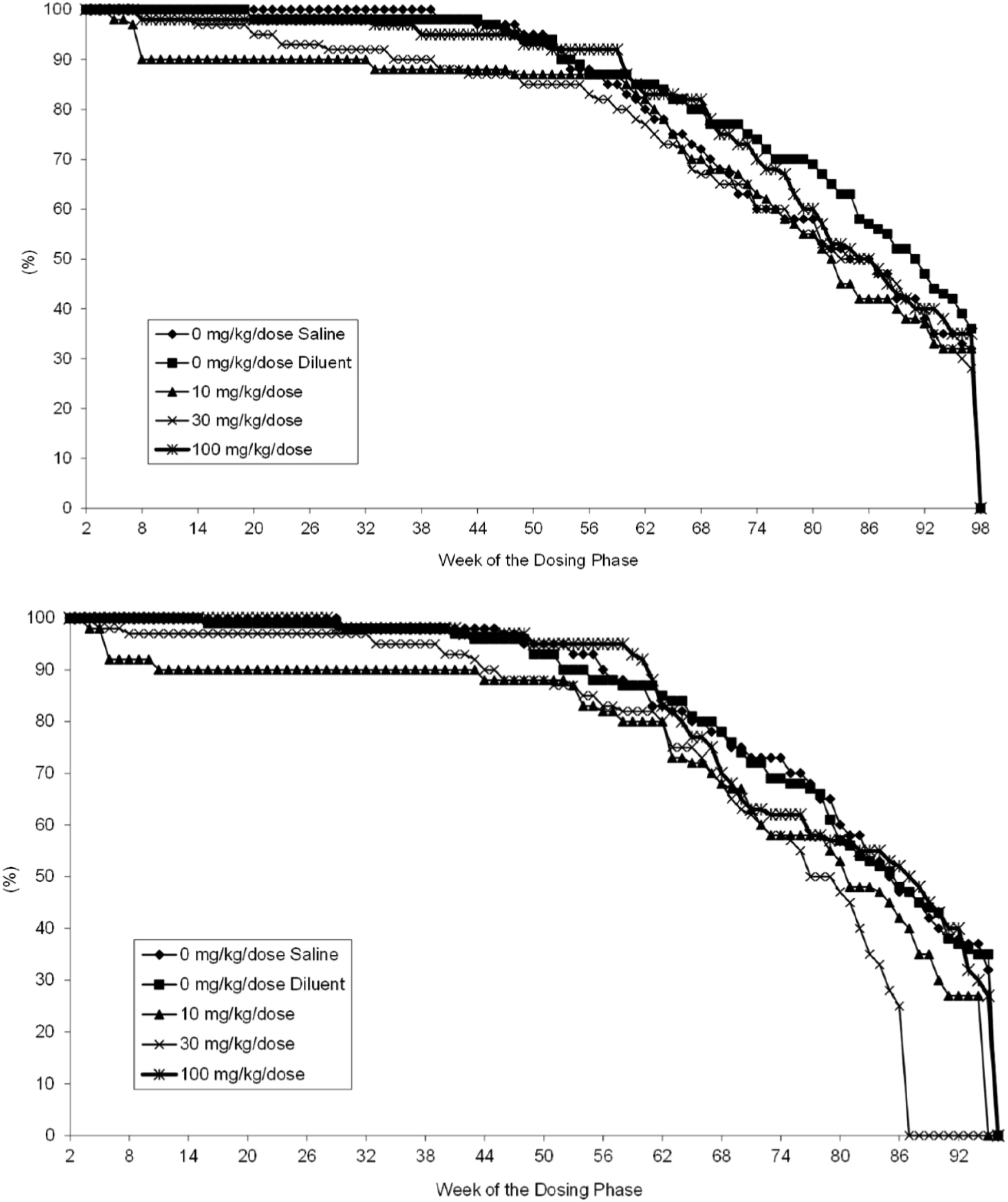
No clinical observations were noted that were attributed to bococizumab administration at any dose level. All clinical observations over the course of the 98-week study were consistent with anticipated findings for group-housed aging rats and generally occurred at comparable incidences as control animals; therefore, they were considered not bococizumab-related.
Macroscopic and Microscopic Observations
No macroscopic findings were observed that were considered bococizumab-related.
Incidence of subset of tumors from 2-year bococizumab carcinogenicity study. Tumors included are those that were present in the 6-month bococizumab and RN317 rat studies, similar tumors, and tumor combinations. For incidence of all tumors see Supplemental Results Tumor Incidence Table.
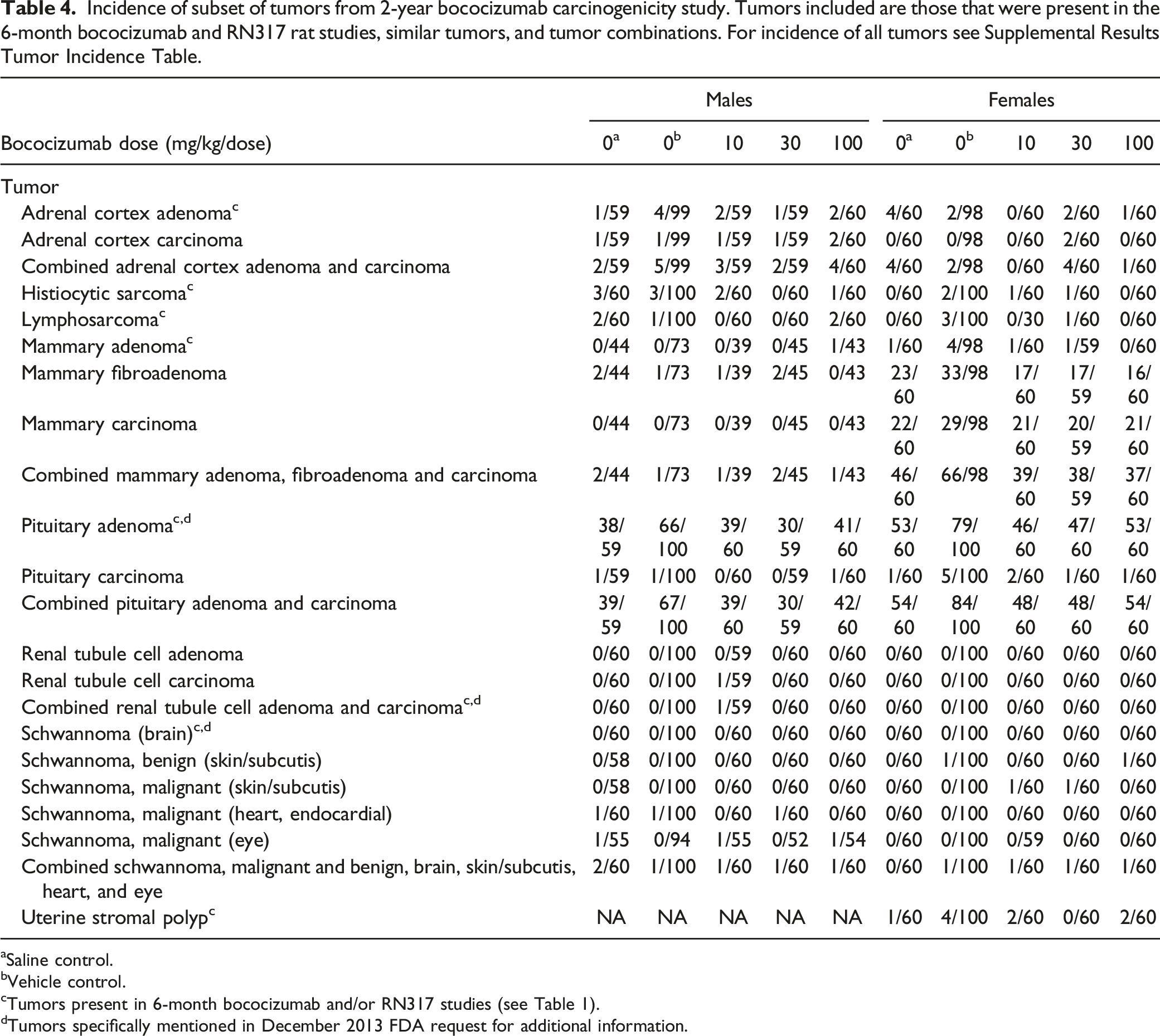
aSaline control.
bVehicle control.
cTumors present in 6-month bococizumab and/or RN317 studies (see Table 1).
dTumors specifically mentioned in December 2013 FDA request for additional information.
A statistically significantly increased incidence in fibrosarcomas of skin/subcutis was noted for females administered 100 mg/kg/dose (2/60 = 3.3%) relative to vehicle-controls (0/100 = 0%). No statistically significant increased incidence was present in females for fibroma of the skin/subcutis alone, or for a combined analysis of tumors of the skin/subcutis that included fibrosarcoma, sarcoma not otherwise specified (NOS), and fibroma. There was no overall dose relationship for skin/subcutis fibromas in either sex or the combination of fibrosarcoma, sarcoma (NOS), and fibromas for females in the current study; a similar incidence of fibrosarcomas was noted in control females in comparable studies conducted at this test facility; several skin/subcutis fibrosarcomas were present in both male control groups for the current study (ie 8/100 = 8% fibrosarcomas in male vehicle control group); the number of skin/subcutis fibrosarcomas in females administered 100 mg/kg/dose was low; and the overall incidence of fibrosarcoma was similar to the reported incidence in the literature. 36 Therefore, this finding was not bococizumab-related and consistent with background variation in incidence for this strain. The low absolute numbers of benign mammary adenoma in males, the lack of a dose relationship for incidence of benign mammary adenoma in both sexes, and the lack of a dose relationship for incidence of the combination of benign mammary adenoma and benign mammary fibroadenoma in both sexes indicates the differences in incidence of these findings were not bococizumab-related. Single incidences of neoplasms not present in controls included malignant schwannoma in the skin/subcutis of 1 female administered 10 mg/kg/dose and 1 female administered 30 mg/kg/dose. These small differences were attributed to variability of background findings and were not considered bococizumab-related because of the low incidence in affected groups, similarity in incidence to that reported in the literature, 36 and the lack of a dose response.
Bococizumab non-neoplastic findings were noted in the adrenal cortex (diffuse hypertrophy) and liver (bridging fibrosis) of both sexes. In the adrenal cortex of males administered ≥10 mg/kg/dose and females administered 30 mg/kg/dose, diffuse hypertrophy was noted at a higher incidence compared with vehicle controls. Although the incidence was not clearly dose-related, these findings were considered bococizumab-related because they had been seen in shorter term bococizumab studies and are likely due to reduced serum lipoproteins. In the liver of animals administered 10 or 30 mg/kg/dose, bridging fibrosis was noted at a higher incidence, compared with saline- or vehicle-controls. Although the incidence was not dose-related and the finding was also present in some control animals, similar findings occur in rats administered foreign proteins and are thought to be secondary to the anti-foreign protein antibody.37-41 Therefore, this finding was considered bococizumab-related and secondary to anti-drug antibody. All other microscopic findings were considered spontaneous and/or incidental because they occurred at a low incidence, lacked a dose relationship, and/or their severity was as expected for this animal age and strain; therefore, they were considered not bococizumab-related.
Discussion
Consistent with the weight-of-evidence assessment for low risk of carcinogenicity, there were no bococizumab-related effects on survival or tumor incidence in the 2-year carcinogenicity study in rats. This outcome supported the initial weight-of-evidence recommendation that a 2-year carcinogenicity study was unnecessary. Findings that were identified as a potential concern in the 6-month studies either were not noted in the high-dose bococizumab group (renal tube cell tumor, schwannoma) or occurred at incidence lower or similar to controls (adrenal, mammary, and pituitary adenomas) indicating that the findings in the 6-month rat study were incidental to bococizumab administration.
Comparison of carcinogenicity assessments of PCSK9 targeting antibodies. Data for Repatha and Praluent obtained from publicly available FDA approval documents.

Carcinogenicity studies for monoclonal antibodies are uncommon. Carcinogenicity studies conducted for mAbs that the authors are aware of include a lifetime bioassay in rat with romosozumab which promotes bone growth by targeting sclerostin, 44 a lifetime bioassay in hamsters with evolocumab (PCSK9 inhibitor) mentioned above, and a 6-month study in Tg. rasH2 mice with reslizumab (interleukin-5 antagonist). 45 No drug-related tumors were noted in any of these studies. In the case of romosozumab and reslizumab the carcinogenicity concern was based on the mechanism of action. Romosozumab is a bone-building agent, although there were no neoplastic risk factors observed in monkey and rat toxicity studies 44 and reslizumab has immunomodulating activity 46 ; the reason for carcinogenicity concern for evolocumab is unknown. A carcinogenicity study in mice has also been conducted with the Fc-CTLA-4 fusion protein abatacept where an increased incidence of malignant lymphomas and mammary gland tumors were observed but were attributed to viral infections of leukemia virus and mouse mammary tumor virus, respectively, due to the immunosuppression in the mice. 47
The bococizumab carcinogenicity waiver request was based on the approach outlined in ICH S6 guidance for Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals; “Standard carcinogenicity bioassays are generally inappropriate for biotechnology-derived pharmaceuticals. However, product-specific assessment of carcinogenic potential may still be needed depending upon duration of clinical dosing, patient population and/or biological activity of the product (e.g., growth factors, immunosuppressive agents, etc.)”. Bococizumab is a monoclonal antibody with no expected genotoxicity, growth promotion, or increased cell proliferation potential. Based on available genetic and epidemiological data, no causal relationship between the pharmacological effect of bococizumab on lowering of LDL-C and cancer risk has been established in humans. Immunotoxicity assessments conducted in rats dosed with a combination of bococizumab and atorvastatin for 1- or 3-months indicated that there was no impact on immune function. Furthermore, the concerns with potential increase in intestinal bile acid loading were also alleviated by lack of bococizumab-related effect in rats dosed for 3 months in combination with a high dose of atorvastatin.
The U.S. FDA requested a carcinogenicity study based on a numerical imbalance in neoplasm incidence in 6-month toxicity studies with bococizumab and the closely related compound RN317. There were a total of 7 tumors observed in 150 bococizumab-dosed animals within the 6-month bococizumab study and 7 tumors observed in 150 RN-317-dosed animals within the RN317 study. Additionally, 1 control animal in the RN317 6-month study had a pituitary adenoma. The majority of these neoplasms were observed in high-dose groups and at the end of the recovery phase (34 weeks) while a few were seen at or before the dosing phase necropsy. Each of the neoplasms that appeared in bococizumab and RN317 6-month studies were considered incidental and not test article-related as they are reported to have occurred spontaneously in studies of similar duration in the test facility historical control data and/or reported in the literature. Furthermore, there was no evidence of direct degenerative/regenerative or cytotoxic changes in any of the tissues evaluated in the chronic bococizumab and RN317 studies that would typically precede proliferative lesions. In total, there were 8 different tumor types observed in the 2 rat studies, each with a different cell type and tissue of origin. There is no apparent plausible mechanism common to the 8 tumor types observed. Nonetheless, the occurrence of these tumors after 6 months of exposure to 2 monoclonal antibodies against the same target raised regulatory concern.
The evidence presented here for bococizumab indicates that using a weight-of-evidence assessment to determine carcinogenicity risk was appropriate. The lack of carcinogenicity in the 2-year evolucumab study further supports the weight-of-evidence assessment as sufficiently predictive of risk for PCSK9 inhibitors. A weight-of-evidence assessment may have supported the alirocumab approval without the conduct of a carcinogenicity study. More recently, inclisiran (liver-targeting double-stranded small interfering RNA that results in lower PCSK9 levels) also demonstrated no carcinogenic risk in a 2-year carcinogenicity study in rats and a 26-week study in RasH2Tg mice. 48
Technical feasibility is also an important consideration for the design and conduct of a carcinogenicity study with any protein therapeutic. One often cited rationale for not performing carcinogenicity studies with biologics in rodents, which was also cited in the bococizumab waiver request, is that the occurrence of immunogenicity makes traditional carcinogenicity studies unfeasible or of limited value for assessing risk to humans. 49 A high incidence of immunogenicity in the bococizumab 6-month rat study suggested exposure would not be sustained in a sufficient number of animals in the 2-year carcinogenicity study. However, in the bococizumab 2-year study, exposure was maintained throughout the study although a subset of animals demonstrated ADA (incidences of 24%, 24% and 14% in animals administered 10, 30, and 100 mg/kg/dose, respectively). Furthermore, there was clear evidence of pharmacodynamic activity based on the lowering of HDL and LDL cholesterol, and the presence of adrenal cortical hypertrophy consistent with earlier short-term studies. The occurrence of ADA had no impact on study interpretation other than an increased incidence of bridging fibrosis in the liver that was considered secondary to the ADA response.37-41
Summary/Conclusions
The data from the bococizumab 2-year carcinogenicity study presented here indicates that the weight-of-evidence assessment that determined low carcinogenicity risk was appropriate.
Supplemental Material
Supplemental Material - Regulatory Experience Assessing the Carcinogenic Potential of a Monoclonal Antibody Inhibiting PCSK9, Bococizumab, Including a 2-Year Carcinogenicity Study in Rats
Supplemental Material for Regulatory Experience Assessing the Carcinogenic Potential of a Monoclonal Antibody Inhibiting PCSK9, Bococizumab, Including a 2-Year Carcinogenicity Study in Rats by Bernard S. Buetow, Gregg D Cappon, Laura M. Aschenbrenner, Lawrence Updyke, Vince R. Torti, Mark Evans6\, Shana R. Dalton, Steven Bailey, and Christopher J Bowman in International Journal of Toxicology
Footnotes
Acknowledgments
Thank you to Rick Perry, Frank Geoly and Jon Heyen for critical review of this manuscript, and Christine Stethem for data review.
Author Contributions
Bernard S. Buetow contributed to design, contributed to analysis, drafted manuscript, and critically revised manuscript; Gregg D. Cappon contributed to conception and design, contributed to interpretation, drafted manuscript, and critically revised manuscript; Laura M. Aschenbrenner contributed to conception and design, contributed to acquisition, analysis, and interpretation, drafted manuscript, and critically revised manuscript; Lawrence Updyke contributed to design, contributed to analysis, and critically revised manuscript; Vince R. Torti contributed to conception, contributed to interpretation, drafted manuscript, and critically revised manuscript; Mark Evans contributed to design, contributed to analysis, drafted manuscript, and critically revised manuscript; Shana R. Dalton contributed to acquisition, analysis, and interpretation, drafted manuscript, and critically revised manuscript; Steven Bailey contributed to analysis, drafted manuscript, and critically revised manuscript. All authors gave final approval and agree to be accountable for all aspects. of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Pfizer, Inc.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.