
Abstract
Determining the adverse nature of findings from nonclinical safety studies often poses a challenge for the key stakeholders responsible for interpreting the results of definitive toxicity studies in support of pharmaceutical product development. Although there are instances in which responses to treatment clearly indicate intolerability or tissue injury associated with dysfunction; in practice, more often there is uncertainty in characterizing an effect of drug treatment as adverse or not. This is due to the inherent variability in responses of biological test systems to toxicological insults, leaving the ultimate analyses of adversity to individual interpretation and subjectivity. This article is a follow-up to the workshop entitled, “Adverse or Not Adverse?: Thinking process behind adversity determination during nonclinical drug development,” conducted at the 58th Annual Meeting of the Society of Toxicology, March 2019 in Baltimore, MD. In this paper, we further discuss and incorporate the perspectives of authors representing different roles, such as Study Director, Study Pathologist, Pharmacology/Toxicology Reviewer (U.S. Food and Drug Administration), and Sponsor in the determination and use of adversity. We also present a practical stepwise approach as an aid in this assessment, and further apply these principles to discuss 10 case studies with different therapeutic modalities and unique challenges.
Introduction
The main objectives of conducting nonclinical safety studies for investigative therapeutic agents are the identification of the target organs of toxicity, characterization of toxic effects, and determination of a dose in nonclinical studies that potentially could be used, in case of IND-enabling studies, to establish a safe first-in-human (FIH) dose for initiation of clinical trials, or to otherwise assist clinical development. To find such a dose, it is pertinent to determine whether the findings from nonclinical studies are adverse or non-adverse in nature. Characterization of the adverse nature of nonclinical findings can often be challenging as well as subjective. The characterization of a particular effect or a group of effects as adverse or non-adverse at a particular dose level may potentially impact clinical dose selection and incorporation of relevant endpoints into clinical protocols to monitor potential adverse events in human trials. Adversity determination facilitates the identification of a dose at which there is no adverse effect (NOAEL; no-observed-adverse-effect-level). The NOAEL has been the most common approach used to calculate a maximum recommended starting dose (MRSD) for FIH clinical trials. 1
Alternative approaches such as the MABEL (minimal anticipated biological effect level, particularly for immune agonists); the HNSTD (highest non-severely toxic dose, for anticancer drugs in non-rodents); and the STD10 (severely toxic dose in 10% of the animals, for anticancer drugs in rodents); are also used to calculate the MRSD for certain therapeutics.2-8 Thus, determination and characterization of adversity is critical to the process of translating toxicology results to human safety risk assessment in view of subsequent clinical trials.
Other factors, such as relevance of the predictive nature of the species, local and systemic exposure considerations, and the conditions under which an adverse effect is observed in a toxicology study are also included in the risk assessment process; however, the NOAEL is often the starting point for such an assessment. Despite its importance in this process, the characterization of adversity of an observed effect in a toxicology study can be subject to interpretation, and opinions differ among even the most seasoned nonclinical safety evaluation scientists. As such, this topic has been debated and discussed over past decades, and progress has been made in attempting to define, characterize, and standardize what constitutes an adverse finding in a nonclinical toxicology study.9-23
As experience with new drug modalities and molecular targets, as well as understanding of the pathophysiology of various types of target organ toxicities have increased over the years, challenges in adversity classification consequently should be declining; however, in practice this appears not to be the case. The reasons for this trend are likely multi-factorial and may include, for example, the historical lack of clear and consistent definitions of adversity; more complex study designs with additional endpoints, such as functional observational batteries, biomarkers, specialty pathology; and in some cases, inadequate training and understanding of the finding’s impact on an animal’s well-being. In addition, with novel therapeutics, the line between exaggerated pharmacology and toxicity in normal, healthy test animals can become blurred.
A recent “best practice” guidance from the Scientific and Regulatory Policy Committee of the Society of Toxicologic Pathology (STP) has helped in determining, communicating, and using adverse effect data from nonclinical studies.
15
Similarly, specific guidance for, and opinions on, determining adversity of clinical and anatomic pathological effects have also been published,12,13,23-26 and various definitions of adversity and the NOAEL have been proposed.10,14,15 For this publication, we highlight two definitions of adversity. The first is a more formal definition from a working group of the European Society of Toxicologic Pathology,
12
which states: “…an adverse effect is a test item-related change in the morphology, physiology, growth, development, reproduction or life span of the animal model that likely results in an impairment of functional capacity to maintain homeostasis and/or an impairment of the capacity to respond to an additional challenge.”
The second is from The Illustrated Dictionary of Toxicologic Pathology and Safety Science
27
and may be considered a simpler definition as a default way of thinking for the Study Director, who is ultimately responsible for determining the NOAEL: “…adverse effects (in toxicology studies) are any changes related to treatment with a test substance that are considered as potentially harmful to the well-being of the test species, resulting in dysfunction, or negatively impacting the ability of an animal to thrive or develop normally.”
This manuscript is a follow-up of a workshop conducted during the 58th Annual Meeting of the Society of Toxicology in Baltimore, MD, in March 2019. In this workshop, the authors presented general background on and principles of adversity determination and the NOAEL, followed by case studies that illustrated practical considerations for the roles of a Study Director, a study pathologist, and a Regulatory Authority pharmacology/toxicology reviewer. This manuscript will expand on the information provided in the workshop and further discuss not only the general process(es) involved, but the roles and perspectives of key stakeholders in adversity determination, and present case studies or sample scenarios that illustrate many of the challenges inherent in this process.
Starting at the Beginning: The Process
Step 1: Determine Whether the Effect is Test Article-Related
The evaluation of whether a finding in a toxicology study is adverse or not starts with the premise that the finding is truly related to the test article. Often, both the relationship of the finding to the test article and the adverse nature of the finding are obvious, but when they are not, the following questions may help guide the Study Director in evaluating the data or challenging what may seem obvious (Figure 1). 1. Is the effect within historical control data and/or statistically significant? Major steps involved and questions to be asked at each step in the determination of adversity and NOAEL in a toxicology study.
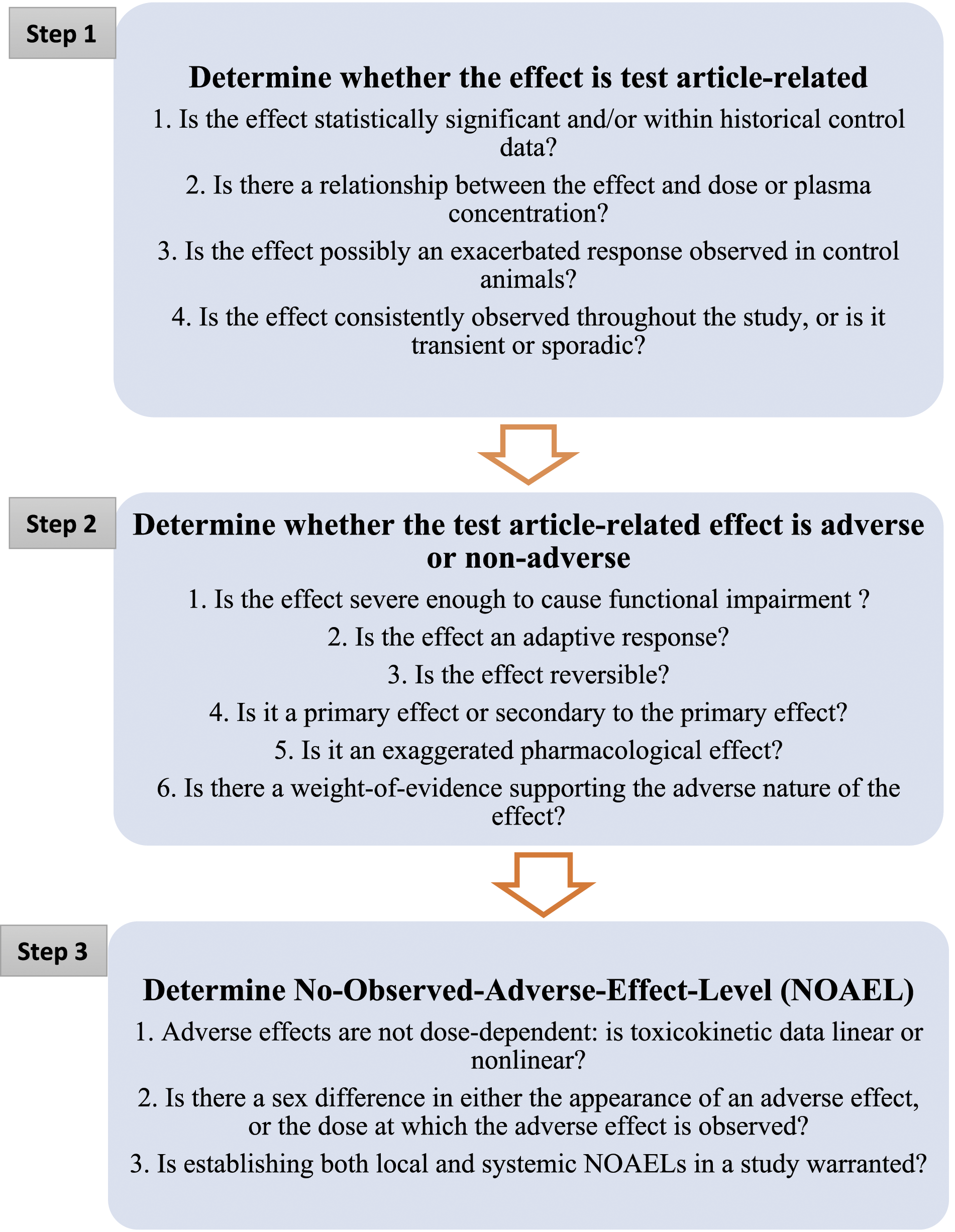
The first step after data acquisition is usually the statistical analysis and determination of statistical significance for numerical data, starting simply with looking at the high-dose group mean data versus that of controls and lower-dose groups. Although the differences in group mean between controls and test article-treated groups may not be statistically significant, the Study Director or reviewer must also consider whether the direction of a change reflects known class pharmacologic/toxicologic effects (either on- or off-target).
To simplify this process, particularly for rodent studies, it is important to evaluate group mean data first, and determine whether any statistical significance exists between the control and treated groups. The relatively greater number of animals per group in rodent studies (minimum of 5–10 per sex for shorter-term studies) and the genetic homogeneity within rodent strains somewhat diminish inter-animal variability. For non-rodent studies in which the number of animals per group may be as small as three or fewer per sex, it is important to look at the response of individual animals within each treatment group over time, as a significant effect can occur in only one animal per sex (representing one third of the group) with minimal effect on the group mean. Here, it is important to consider the totality of the data for an individual animal, including all evaluated parameters. In addition, exposure differences between animals, and potential pharmacokinetic (PK)-altering immune responses, that is, anti-drug antibodies (ADAs), affecting individual animals should be considered. Without this context, changes considered incidental or spurious in a small number of rodents, or isolated to a single nonrodent animal, might be overlooked, thereby causing potentially important findings in the study to be missed.
Last, while statistical analysis is generally the first step in analyzing most of the endpoints (e.g., i.e., organ weights), in a toxicity study, incidence or severity in the anatomic pathology tables are generally not statistically analyzed, with the exception of tumor incidence tables in carcinogenicity studies. Although there are statistical tests which can be applied to analyze the test-article related trends and the effects of pathology findings on severity, 28 their use is generally not recommended, as the severity criteria of pathology lesions (e.g., non-neoplastic findings) is often only semi-quantitative and nonlinear. In such cases, the study pathologist determines the significance of a test article-related effect based on the incidence and severity of lesions in the concurrent controls along with historical control data, rather than by statistical analysis.
Inter-animal biological variability is often well reflected in appropriately curated historical control data. The benefits and pitfalls of using historical control data (i.e., reference intervals) for interpreting clinical pathology data from toxicology studies have been described earlier.10,29,30 The consensus among toxicologic clinical pathologists is that comparisons with concurrent control groups are the most useful, and of primary importance in determining the potential effect of a test article on a clinical pathology parameter, particularly in rodents. However, appropriately qualified historical control data in a toxicologic clinical pathology setting can be helpful in determining if the suspected effect is within the biological (or analytical) variation of that parameter. For example, certain clinical chemistry analytes, such as serum creatine kinase levels, can have a broad reference interval. For such parameters, an effect may be clearly test article-related, yet still within the reference interval; it may not be considered adverse, especially in the absence of an associated non-adverse histopathological finding. Similarly, as mentioned earlier, properly established in-house histopathological historical control data for a given species are often helpful in determining if the incidence and severity of relatively rare findings or background lesions observed in the treatment groups are treatment-related, and whether an effect has crossed the threshold for consideration as adverse.
12
2. Is there a relationship between the effect and dose and/or plasma concentration?
The effect within historical control data and/or statistically significant findings observed in the absence of a dose- or exposure-related response are generally considered less likely to be test article-related, although there are exceptions. During the evaluation of toxicological data, there is often a good correlation between the incidence and/or severity of an effect with the dose level or with cumulative drug exposure over the course of the study. When systemic exposure data is available, it generally shows that plasma concentrations increase with dose, although the increases in exposure may or may not be dose-proportional. This is often the case where higher dose levels have saturable rates of absorption or plasma clearance resulting in either less than or greater than dose proportionality, respectively, or when receptor occupancy becomes saturated.
However, as discussed earlier, such caveats as immunogenicity, increased metabolic clearance, or other PK-altering factors may further confound dose-exposure relationships, especially with repeated dosing. To help confirm whether an effect is related to the test article, exposure-indicating toxicokinetic parameters such as area under the curve (AUC) and/or maximum plasma concentration (Cmax) should be considered. The AUC and Cmax should be evaluated with respect to the affected dose group(s), or preferably, individual animal(s), to assess whether there is a relationship between drug (or possibly metabolite) plasma concentrations and incidence or severity of the effect.
It is also important to schedule data collection at the projected time of maximum plasma concentration (Tmax) for functional endpoints such as functional observational batteries (FOB), electrocardiograms (ECGs), and blood pressure measurements. For these endpoints, Cmax is often more relevant in determining a correlation with the observed test article-related effect. An effect observed only at the high-dose, even in the presence of linear toxicokinetics (TK), may indicate that the toxicity-exposure threshold for that particular effect has been reached at the high-dose only. Hence, the effect observed only at the high-dose may in fact be related to test article administration. 3. Is the effect possibly an exacerbated response observed in control animals?
The decision to classify a potentially exacerbated background response in treated animals as test article-related depends on the type, incidence, and severity of the finding. It may be tempting to assume that a finding in control animals represents a true background incidence, but this may not always be the case. In an unpublished 6-month toxicity study in Sprague-Dawley rats, females showed dose-related increased incidence and severity of adrenal cortical cystic degeneration, with none of the control animals showing the lesion. Males had a similar increased incidence of multifocal adrenal cortical vacuolation, again with no control incidence. As both adrenal findings were well-described spontaneous age-related changes in rats, with a higher spontaneous incidence in Sprague-Dawley rats,
31
the relationship to treatment was uncertain but considered to be a possible exacerbation of a common background finding. Given that the adrenal histopathological findings were identical to spontaneous age-related changes in the adrenals, they were not considered adverse. In light of the uncertain etiology of these findings, they were discussed at length in the body of the report supported by literature, and likewise were explained in the concluding paragraph of the study summary. 4. Is the effect consistently observed throughout the study, or is it transient or sporadic?
This question is typically applicable to in-life data such as clinical signs, body weight, and food consumption, as well as clinical pathology or other endpoints measured repeatedly at multiple time points or intervals during a study. If some concerning clinical signs emergent upon treatment do not exhibit persistence with continued dosing and their appearance is sporadic in individual animals, those clinical signs could be just incidental and not related to test article exposure. However, a transient response in any repeated measure could just as well be test article-related and transient due to, for example, cellular tolerance, compensation, or reduced plasma exposure over time. An interim evaluation of a toxicological endpoint is essentially a “snapshot” assessment of that parameter at that specific time and may not be reflective of the treatment effect with continued dosing.
In addition, some parameters may reflect active injury, but not necessarily post-repair poorly functioning tissues. For example, alanine aminotransferase (ALT) and aspartate aminotransferase (AST) are considered “leakage enzymes,” reflective primarily of hepatocellular injury when elevated in serum or plasma, although one or both may be elevated with skeletal or cardiac muscle injury. In the case of a test article-related degenerative effect on the liver during early phases of a study, serum levels of ALT and AST may be increased, yet may return to baseline levels in later measurements as tissues with degenerative changes progress into necrotic tissue, which is replaced with compensatory hepatocellular proliferation or fibrosis (e.g., in longer term studies). The liver function may still be adversely affected, but that would not be apparent based on circulating ALT or AST values. Moreover, terminal liver histopathology may reflect prior injury and subsequent repair. Alternatively, other downstream serum markers of liver function (e.g., total protein and albumin) may aid in determining whether there was truly an effect on the liver during the study, or if the observed change was incidental. Since test article-related clinical pathology changes in toxicology studies have potential relevance in clinical safety monitoring, changes in these parameters occurring over the course of a study should be scrutinized carefully and not be casually dismissed as sporadic or transient.
Step 1 Take-Home Message: In determining a test article-related effect, the following should be considered: (1) historical control data and/or statistical significance in numerical data comparing treated versus control incidences of findings, including whether expected pharmacological- or class-related effects were observed, (2) trends or dose/exposure-related responses, (3) confirm or challenge any appearance of exacerbation of control or background incidence, and (4) whether the finding appears sporadically.
Step 2: Determine Whether the Test Article-Related Effect Is Adverse
In theory, the objective of a toxicology study is to determine adverse effects and to characterize those effects. The dose at which the test article produces an unwanted or adverse effect can only be defined within the confines of the study design.
Toxicology studies are often designed with three test article dose groups and a control group. This toxicology study design helps determine the highest “non-adverse dose” in that test species (NOAEL), and the lowest dose that produces adverse findings (lowest observed adverse effect level [LOAEL]). The NOAEL generally sets the limits for the clinical starting dose. However, the NOAEL and LOAEL can be used to communicate to the clinical team the nature and potential severity of the adverse findings, as well as the level of exposure associated with toxicity and steepness of the dose-response curve.
The toxicology study in a nonclinical drug development program is designed to determine tolerability and to evaluate the effects of test article via assessments of various endpoints such as mortality, clinical signs, food consumption, body weights, clinical pathology analytes, and histopathological examination identifying target organ toxicity and its reversibility. Changes in one endpoint, or in multiple endpoints combined, are important in determining whether a dose level is associated with an adverse event. The following considerations can aid in this assessment. 1. Is the effect severe enough to cause functional impairment?
Assessing the severity of an effect on body weight, food consumption, and/or clinical signs helps in determining the overall health status of an animal. When the severity of a change in these parameters is associated with or would be predicted to be associated with an impairment of the animal’s ability to thrive normally, or reflects distress to an animal, the change is considered adverse. For these types of changes, we defer to the definition of NOAEL that states, “adverse effects (in toxicology studies) are any changes related to treatment with a test substance that are considered as potentially harmful to the well-being of the test species, resulting in dysfunction, or negatively impacting the ability of an animal to thrive or develop normally.” 27 The Organisation for Economic Co-operation and Development (OECD) guidance document and other publications have described critical clinical signs and conditions as humane endpoints which might help the study team determine the threshold of concern for clinical signs in the test species.32-36 Further, OECD guidance ‘Document on the Recognition, Assessment, and Use of Clinical Signs as Humane Endpoints for Experimental Animals Use in Safety Evaluation’ states that a decrease in body weight of >10% by non-weight loss drugs raises the level of concern, while a decrease of >20% is considered adverse. 32 We urge caution in the exclusive use of such criteria to determine adversity; however, they are valuable in helping guide an overall assessment, and often can assist in justifying an adversity determination.
Assessing adversity associated with clinical pathology findings can be challenging. Changes in certain clinical pathology analytes, such as sodium, potassium, calcium, glucose, hemoglobin, and platelets can be adverse depending on the degree of the changes, due to the critical importance of their presence in blood for the normal functioning of organs and systems. On the other hand, changes in some parameters, such as ALT, AST, and lactate dehydrogenase, are not considered adverse but can serve as biomarkers of tissue injury, which often needs to be confirmed by other clinical pathology parameters and/or histopathology. Some clinical pathology analytes, such as total protein and albumin, may only serve as biomarkers of organ function (liver, in this case), but their changes below a certain critical level can indicate adversity. 23 Earlier published reviews and commentary can guide interpretation of clinical pathology in nonclinical toxicology studies.13,23,26,30,37
Severity grading of microscopic lesions helps in assessing the functional impact of the lesions and their adversity classification. In its “Points to Consider” article, the Scientific and Regulatory Policy Committee of the STP recommends consideration of such criteria as the extent of damage to the tissue/organ affected, complexity, or context of morphologic change and distribution (focal, multifocal, or diffuse), when assigning severity grades to the microscopic lesions.
38
In general, a lesion with a “minimal” severity grade may be non-adverse, while the same lesion with “marked” or “severe” grades may qualify as adverse. However, this may not be applicable to some lesions with minimal severity grades, such as neuronal necrosis which may be classified as adverse. While in some cases adversity may be defined by the severity of an isolated lesion, in many cases, it is defined by a combination of changes which individually might not be considered adverse. 2. Is the effect an adaptive response?
In response to a toxic insult, organ systems will often try to maintain homeostasis or mount a defense by adopting temporary compensatory changes that may be considered adaptive in nature. It may be difficult to distinguish between benign, quickly reversible changes due to increased tissue or metabolic burden, and adverse biological changes. Often the findings reflect the tissue’s response to a drug (or metabolite) and are not necessarily degenerative in nature.
A common example is induction of liver microsomal enzymes in response to prolonged presence of a drug in circulation. On a tissue level, this adaptation expresses in the form of increased liver weight, size, and microscopic hepatocellular hypertrophy (often but not exclusively centrilobular). Plasma pharmacokinetics are also often altered with a lowering of total exposure of the free drug. Examples of agents producing these types of metabolic adaptive changes are phenobarbital, methadone, and other microsomal enzyme inducers, or drug classes such as fibrates and peroxisome proliferators.
19
Such adaptive changes, if not coupled with degenerative changes, are considered non-adverse and often are widely variable between species. 3. Is the effect reversible?
Although an effect may be partially or completely reversible during the test article-free recovery period, its adversity classification should be evaluated in the context of all other available information and a weight-of-evidence approach. Reversibility of a lesion does not always indicate a non-adverse effect. Reversibility (complete, partial, or none) is only a factor when the effect is minor enough to be equivocally adverse, and when it helps in the assessment of the degree of harm the effect has on the animals’ overall well-being, or on tissue integrity. 4. Is it a primary effect of the test article, or secondary to the primary effect?
Whether the effect is considered primary or secondary in nature, all test article-related effects should be considered for severity; and thus, adversity determination. However, some secondary effects can be ameliorated by providing animals with additional enrichment based on humane reasons and standard veterinary care, as well as modification of feed time and duration based on post-dose clinical observations.
For example, in a hypothetical toxicology case study, clinical signs indicative of nausea, such as discomfort, salivation, retching, and emesis are observed in dogs immediately after dose administration. Moreover, due to the study design requirement, dogs have access to food only for 3–4 h post-dose, leading to a significant decrease in food consumption. Consequently, body weight is also decreased significantly in some animals (approximately 20%) during the first 2 weeks of the study. However, when the duration of food access is increased for up to 8–18 h post-dose without compromising study objectives, food consumption and body weight gain return to control animal levels. In this case, reduced food consumption and decreased group mean body weight during the first 2 weeks are indirect effects of a possible nauseating effect of the test article and is ameliorated by giving access to food over a longer time. In this scenario, decreased food consumption and decreased body weight gain during the first 2 weeks would not be considered adverse. 5. Is it an exaggerated pharmacological effect?
If a pharmacological effect of a test article is exaggerated in a toxicology study due to a higher dose or exposure, the exaggerated pharmacology, depending on the magnitude or severity of the effect, can become an adverse effect. For example, administration of immunomodulators (enhancers/agonists or suppressors) can either overstimulate or suppress the immune system, which can be detrimental to the animal’s ability to thrive normally and handle the test article’s effect. If there is an evidence of increased infections or cytokine activity after administration of immunomodulators, this may result in changes in clinical signs and body weight/food consumption, reflecting the animal’s inability to tolerate the immune modulation and indicating that the exaggerated pharmacology was adverse at a particular dose level. In those situations, it becomes critical to evaluate not only the etiology of the change (the pharmacology of the test article), but also to explain how that effect adversely affected the animal.
In a toxicology study, sometimes the desired pharmacologic effect can become problematic. For hypnotic agents (sleep aids, relaxants, and anesthetics), the dose at which the desired effect occurs may essentially incapacitate the animal, requiring additional supportive measures after the treatment. While this is not toxicity, the ability of the Study Director to characterize adverse changes under these conditions may completely depend on the rigor of the supportive measures provided to each individual animal at each dose level. On the other hand, a test article designed to be central nervous system (CNS)-active may at high dose levels cause recumbency or disturbances in balance and coordination. These changes in fact may be related to the pharmacology of the test article, but not desired at the therapeutic dose levels. Whether these findings would be considered adverse or not would depend on the severity, incidence, and frequency of the findings in the context of the animal’s overall clinical condition and its ability to function normally.
Often, the line between adverse and non-adverse levels of exaggerated pharmacology in a toxicology study can be challenging to identify. Just because an effect is reflective of exaggerated pharmacology does not mean it is non-adverse. Likewise, exaggerated pharmacology resulting in statistically significant changes in study endpoints may not necessarily have adverse sequelae. The threshold between adverse and non-adverse exaggerated pharmacology can vary by a wide margin across the classes of pharmacological effects and is beyond the scope of this paper. 6. Is the totality of the effects severe enough (weight-of-evidence) to result in dysfunction of the organs or organism?
Based on an evaluation of all applicable data and sub-reports from the study (as well as potentially relevant publications), the Study Director can make an initial assessment of whether test article effects should be considered adverse. This process includes not only consideration of the relative toxicologic significance of a finding, but also involves evaluating whether it is associated with various other changes that might represent further evidence of dysfunction or the animal’s impaired ability to thrive or respond to stress. At this stage, the Study Director may seek concurrence, counsel, or expert opinion from other subject matter experts (SMEs) as well as the Sponsor, who may be aware of additional information.
Step 2 Take-Home Message: Since the finding has been deemed a true test article-related effect, the following questions should be considered in determining whether that effect is adverse: (1) How far is the effect from the expected value, incidence, or severity of the change or historical reference value? Is it within or far outside of normal biological variation? (2) Is the effect reflective of the animal’s or a specific organ’s ability to alter its structure or adjust its physiology to adapt to the toxicological insult? (3) With cessation of dosing, would the effect be reversed such that there would be no overall negative impact on the tissue, organ, or the animal’s well-being? (4) Is the effect highly correlated with anticipated further degenerative injury or a downstream neoplastic change? (5) Is it an “acceptable” exaggerated pharmacology, or has it become adverse? (6) Could other indicators or responses in the study help substantiate or alternatively question whether the effect should be deemed adverse (weight-of-evidence)?
Step 3: Determine the NOAEL
Simply, the NOAEL is the highest dose at which there are no observed adverse effects. In practice, it is one dose level below the lowest dose at which adverse effects were observed (i.e., if adverse effects were observed at the mid-dose, but not at the low-dose, the low-dose would be the NOAEL). It is important in all cases to not only define the NOAEL clearly with a rational, well-crafted justification, but also to include exposure information (Cmax and/or AUC) and a statement about the data it was based on to help extrapolate relative risk across species. Factors to consider in establishing the NOAEL are discussed below. 1. Adverse effects are not dose-dependent: is toxicokinetic data linear or nonlinear?
Linear toxicokinetics occur when toxicokinetic parameters such as AUC and Cmax change dose-proportionately, and other parameters such as volume of distribution (Vd), half-life, and clearance remain constant across dose levels. In nonlinear kinetics, plasma concentrations do not increase dose-proportionately, while Vd and half-life values may change as the dose level increases. In these situations, the mid-dose might produce the highest plasma concentrations and adverse effects, while the high-dose might produce much lower plasma concentrations and no adverse findings. Under these conditions, the low-dose can be considered the NOAEL. 2. Is there a sex difference in either the appearance of an adverse effect, or the dose at which the adverse effect is observed?
It is not unusual to observe sex differences in pharmacokinetics and/or sensitivity in response to a pharmacologic agent. 39 If the data support a clear difference in response between the sexes, this may justify characterizing a separate NOAEL for each sex; in such a case, a rationale for the difference should be provided in the study report and in any associated summaries. This situation might be encountered more often when the test article has autonomic or endocrine-like effects, and the sex of the animal determines which target organs are affected and at which dose levels. This information can also help the Sponsor determine whether a higher risk may be associated with a particular sex in clinical trials, or if dose levels should be adjusted based on the sex of the subject or patient. As a conservative measure, the Sponsor and/or regulatory reviewer may still propose that the lower NOAEL dose be used to calculate the human equivalent dose (HED) required for interspecies scaling to calculate the MRSD.
In a hypothetical case of a non-oncology therapeutic intended to be used only in the male population, adverse effects are observed at mid and high doses in the ovaries of female dogs, and the low dose is determined to be the NOAEL in females. However, in males with no lesions, the high dose is determined to be the NOAEL. In this case, use of the male NOAEL dose for HED calculations could be logical and appropriate, given the intended human population; nonetheless, both NOAELs should be included in the toxicology study report. 3. Is establishing both local and systemic NOAELs in a study warranted?
There are cases in which it might be useful to differentiate a local (e.g., tissue or injection site) and a systemic NOAEL depending on the site of exposure (e.g., for ocular or topical dermal studies). In such cases, there may be an unintended systemic exposure that results in organ toxicity at a dose that shows no adverse findings in the eye or dermal site of application. To this end, identification of an ocular-specific NOAEL would be misleading without characterizing systemic exposure and effects.
Another example might be one in which injection site reactions are noted in a toxicology study at dose levels lower than those resulting in adverse systemic effects. It would be acceptable, depending on preference, to either caveat the systemic NOAEL with a mention of the injection site reactions, or to identify a lower local injection site NOAEL. In either case, unless the local reactions represented a severe or locally debilitating response, the injection site reaction may not necessarily drive the NOAEL, which would be used for calculation of the FIH dose. This is because either (1) the clinical indication is severe enough that the local reaction is anticipated to be tolerated, or (2) the local reaction observed in the clinical setting may be transient and could be managed or ameliorated with the use of local or systemic antihistamines or other agents that would allow the toleration of higher dose levels.
Although uncommon, there are instances in which it may be appropriate to report an organ-specific NOAEL. For example, specific investigational studies may be conducted to characterize the toxicity of a specific organ or organ system of interest, as demonstrated in the following case. A follow-up investigational toxicology study was designed to increase understanding of the cardiotoxic effects of a compound reported in a previous study. It was conducted in response to a regulatory authority’s concern about cardiotoxicity of the compound which was noted during the review of the Investigational New Drug (IND) application. Various parameters, such as clinical signs, body weight, heart weight, heart-specific serum biomarkers, gross pathology, and heart histopathology were studied in this follow-up Good Laboratory Practice (GLP) study. No other organs were evaluated microscopically. In this case, since no other data were available for overall assessment of animal health (other than body weight and clinical signs), a heart-specific NOAEL was determined.
Step 3 Take-Home Message: In determining the NOAEL, the following should be considered: (1) adverse findings, like any other change, typically follow a dose-response, but not always, (2) if there are sex-specific NOAELs, these should be explicitly stated and a rationale for the differences provided, (3) both local and systemic NOAELs may exist, and it may be important to caveat the NOAEL accordingly.
Roles of Key Stakeholders in the Determination and Use of Adversity and the NOAEL
Study Director
In a toxicology study, Good Laboratory Practice Regulations (GLPs) dictate that the Study Director “has overall responsibility for the technical conduct of the study, as well as for the interpretation, analysis, documentation and reporting of results, and represents the single point of study control.” 40 As stated in an OECD GLP consensus document, “the Study Director is responsible for drawing the final overall conclusions from the study . . .[and] must coordinate with other study scientists, and/or Principal Investigator(s) keeping informed of their findings during the study and receiving and evaluating their respective individual reports for inclusion in the final study report.” Thus, ultimately it is the responsibility of the Study Director to not only determine if a finding is related to treatment, but to interpret the results of the finding in an integrative report. The Study Director must rely on the expertise and diligence of numerous experts, including the Study Pathologist, clinical pathologist, attending veterinarian, research technicians, dose formulation technicians, analytical and bioanalytical chemists, DMPK (drug metabolism and pharmacokinetics) scientists, ophthalmologists, cardiologists, immunologists, neuroscientists, and others, as applicable.
Often, the Study Director reviews and evaluates clinical signs, body weight, and food consumption data for test article-related findings. Contributing scientists provide most of the other results and interpretive narrative in draft report format, typically include clinical pathology, anatomic pathology, bioanalysis and toxicokinetics, ophthalmology and safety pharmacology evaluations (ECG, respiratory, neurobehavioral). Since drug safety is greater than the sum of all the various individual parts of a toxicology report, to fully integrate the results, the Study Director must not only review the draft narratives from contributing scientists, but then reconcile the findings or interpretations with possible correlating findings from other reports or datasets in the same animal or dose groups. This is both an integrative and iterative process. Sometimes, the various datasets fit together nicely, like pieces of a flat jigsaw puzzle. However, more often it resembles a 3D puzzle, whereby the Study Director’s review may not only challenge some of the conclusions in the contributing scientists’ reports, but also may help expand or put them into perspective. Here, simple identification of a finding is not sufficient. A determination must be made as to whether the finding is truly related to the test article, and whether that finding is adverse in the test species.
As discussed earlier, in the final report appropriate justification should be given for classification of an effect(s) as adverse or non-adverse. This can be done within the main body of the report. However, it is critical to include a brief justification in the Summary section. This is important for several reasons: (1) it may seem obvious to the Study Director in drafting the final report based on the narrative included in the Results section, but bottom-line assessments such as the NOAEL are integrative in nature, and usually are reserved for the Summary and Conclusion sections of the main report, (2) often there are challenges or questions as to how the NOAEL was determined from stakeholders with access to only the Summary (in its use as a stand-alone document) and not the full report; the broader audience often seeks a rationale to support findings listed in the Summary, (3) reiterating the justification for the NOAEL in the Summary and Conclusion sections allows the Study Director to revisit the basis for the NOAEL designation in the integrative narrative, which can then easily be reviewed by senior management, the Study Pathologist, or other principal investigators that may not have had access to the rest of the data, (4) years later, a responsible party may question the Contract Research Organization (CRO) or Study Director to justify how the NOAEL was established, with perhaps only the Summary available. Given the massive amount of data being reviewed by Study Directors, especially in a busy CRO, the thinking behind the determination a NOAEL for an individual study after weeks, months, or years may no longer be as obvious to the Study Director as it was when drafting the final report.
It is important to state here that the report need not classify every effect as adverse or not. The report summary and conclusions need to clearly state the dose at which an adverse effect occurred, and the nature of that adverse change, and not attempt to classify every test article effect as adverse or not. In the body of the report, the section dealing with the results or findings for a given endpoint should highlight if an effect was deemed adverse and provide justification. If needed, this can be further distilled to a short justification in the Summary section, depending on whether this is needed. It is imperative, however, that the report’s Summary and Conclusions justify the NOAEL and add caveats where needed.
Although determination of a NOAEL in a GLP study is a common practice, there can be a difference of opinion on reporting the NOAEL in non-GLP dose range-finding studies. These are typically pilot and range-finding studies, which often lack the statistical power in animal numbers, and/or the full complement of endpoints to fully characterize whether an adverse effect is present at a given dose level. In many cases, they are designed to determine tolerability only, that is, maximum tolerated dose (MTD), or dose-range finding (DRF), with limited, if any, histopathology or necropsy data. In practice, the reports for these studies tend to list only a single or repeat dose MTD, with a short description of findings; however, in some cases the Sponsor may request reporting of a NOAEL. In those cases, if the Study Director agrees that the data allows for such a determination, the NOAEL should be clearly qualified with respect to limitations in either the number of animals, group, or sex, and/or parameters not evaluated that would typically be included in a definitive toxicology study. Thus, the decision to report a NOAEL in a dose range-finding study report should be made with consideration of a potential Sponsor request, but also be tempered by the adequacy of the data available to support it. In either case, it may be useful to include a discussion of the NOAEL from dose range-finding studies in the IND, in the context of the drug’s overall toxicological or pharmacological profile.
Contributing Scientists
In addition to the Study Director, other SMEs, sometimes called contributing scientists (CS), help evaluate various datasets of the study report within their areas of expertise. These individuals may include veterinary cardiologists, ophthalmologists, analytical chemists, immunologists, clinical and anatomic pathologists, as well as others, depending on the nature and scope of the toxicology study. It is often helpful for the Study Director to enlist the participation of one or more contributing scientists early in the study planning stage, and certainly by the time the protocol is drafted. As contributing scientists are drafting their own reports for integration into the full report by the Study Director, it is important that the Study Director be available to provide any additional information that can help them in their assessments, and to make sure they have all the pertinent information needed. All contributory information is important for a comprehensive evaluation of the study results.
However, it is the Study Pathologist’s assessment that often is most critical, as it enables the identification of target organs and helps characterize the nature of organ-specific effects as adverse or not. After reviewing the microscopic changes in the tissue, the Study Pathologist ascribes morphologic descriptive terms for the diagnoses and grades the severity of lesions, which is an inherently subjective process. The pathologist’s role in this case is not limited to generating tabulated morphological data, but to provide an interpretation in relation to test article exposure. In general, tissues from concurrent control group animals are used to set a background pathology range and help in distinguishing histopathologic changes from normal biological variations. This assessment of variations in background histopathology of control groups is critical in identifying thresholds for distinguishing treatment-related lesions or treatment-related exacerbation of a background finding.
Making an adversity call on histopathological findings in isolation, based on the nature and extent of severity, is challenging. 24 Suggestions have been made to classify the designation of adversity (clearly adverse, adverse only at higher severities, adverse only when seen in combination with deleterious findings, exacerbations of spontaneous findings) and non-adversity (adaptive changes, secondary changes, pharmacological effects, lower severity changes with no functional disturbances). These suggestions challenge the STP best practice guidelines 15 and have highlighted some of the practical guidance for the toxicologic pathologist in determining adversity. The pathologist should consider the pharmacological target, the clinical signs, hematology, clinical chemistry, urinalysis, organ weight, and gross pathology data in an integrated way, and take a weight-of-evidence approach to determine adversity. 41
Regulatory Toxicology Reviewer
Pharmacology/Toxicology (Pharm/Tox) reviewers at the US-FDA review and evaluate the pharmacological and toxicological data contained in INDs and New Drug Applications (NDAs). They assess the safety of drugs based on nonclinical toxicology data submitted by the Sponsor. This includes evaluation of data to ensure that nonclinical toxicology studies support the manufacturer’s claims for safety, and review of proposed dose levels to determine whether sufficient margins of safety exist for clinical use. The review process includes determination of a NOAEL in each pivotal study supporting an IND or NDA. Each study is individually reviewed and a NOAEL is determined based on the reviewer’s assessment of the data in the study report, the Sponsor’s conclusion, as well as discussion with the supervisor, and if necessary, other experts. A consensus on the NOAEL may be reached or the reviewer may disagree with the NOAEL in the study report and set a new NOAEL. This new NOAEL may be higher or lower than the original. Alternatively, the reviewer may determine that a NOAEL was not established in the study.
According to the FDA guidance on determining the starting dose in clinical trials, 1 “several definitions of NOAEL exist, but for selecting a starting dose (in human clinical trials), the following is used: the highest dose level that does not produce a significant increase in adverse effects in comparison to the control group. In this context, adverse effects that are biologically significant (even if they are not statistically significant) should be considered in the determination of the NOAEL.”
The FDA guidance 1 describes “three types of findings in nonclinical toxicology studies that can be used to determine the NOAEL: (1) overt toxicity (e.g., clinical signs, macro- and microscopic lesions); (2) surrogate markers of toxicity (e.g., serum liver enzyme levels); and (3) exaggerated pharmacodynamic effects.” Consideration of human relevance of the adverse effects defining a NOAEL in nonclinical studies is also addressed as follows, “… an adverse effect observed in nonclinical toxicology studies used to define a NOAEL for the purpose of dose-setting should be based on an effect that would be unacceptable if produced by the initial dose of a therapeutic in a phase 1 clinical trial conducted in adult healthy volunteers.” 1 As such, these concepts are all considered when selecting a NOAEL from nonclinical toxicology studies for FIH dose selection.
The NOAEL is a generally accepted benchmark for safety when derived from appropriate animal studies. Such NOAELs, from a nonclinical species, resulting in the lowest HED can serve as the starting point for determining a reasonably safe starting dose (the MRSD) of a new therapeutic in healthy human volunteers, or can also assist in determining the initial clinical dose in FIH studies involving patients. 1 In addition, starting doses and safety margins may be calculated based on clinical pharmacokinetic data, if available. For biologics, along with the NOAEL from the most relevant species, other values such as the MABEL and the pharmacologically active dose (PAD) are also considered in the calculation of the MRSD. It is important to note that in addition to adverse findings, selection of the FIH dose will be influenced by several other factors which will also influence the safety factors applied to the NOAEL. These include whether the effect is monitorable in the clinic, reversibility (or recovery of the effect), slope of the dose-response curve, and bioavailability variation in animal models, which may indicate uncertain human bioavailability. 1
Sponsor
The Sponsor communicates the results of the nonclinical toxicology studies to regulators in the context of published literature, previous nonclinical studies, and clinical data, if available. In this role, the Sponsor is usually the author of the IND and Investigator Brochure (IB) sections related to toxicology. Here, the Sponsor must evaluate the IND-enabling studies that have been conducted, along with the knowledge about the pharmacology of the molecule and any pilot studies, to formulate an assessment of the possible adverse effects of the molecule. In some situations, despite an adverse finding seen in one species, the Sponsor may develop an argument to regulators contending that based on information from more relevant species, the adverse finding may not be relevant.
In the nonclinical section of the IND, the Sponsor integrates all available data about the molecule and calculates a MRSD or clinical starting dose, based on NOAELs, the HNSTD, and/or STD10 from nonclinical studies. In the IND, the Sponsor discusses whether the adverse findings from nonclinical studies have any human relevance; and if so, can they be monitored during the clinical trials.
It is important that the Sponsor provide the study director with complete and current information on the molecule. A full understanding of the pharmacology of the test article will enable the Study Director to appropriately characterize findings observed in a toxicology study.
Case Studies
Case Studies.
IV, intravenous; SC, subcutaneous.
Case Study #1: Due Diligence Is Your Friend!
Study Type: 28-day study
Species: Rat
Route: Intravenous (IV)
Drug Class: Reformulated previously approved drug (small molecule) with new route of administration for non-oncology indication
Findings: Severe clinical signs such as tremors, altered gait, respiratory distress at high-dose; microscopic liver vacuolation, anterior pituitary degenerative effects at high-dose; and thyroid hypertrophy, with dose-related severity (minimal to moderate). Toxicokinetics was evaluated, but serum thyroid hormone analysis was not included in the study.
The Study Director issued a draft report to the Sponsor in which the low-dose was declared to be the NOAEL based on thyroid hypertrophy of greater than minimal severity at the mid-dose level. The Sponsor questioned the rationale for this and supplied a published reference in a different species showing the same drug, with a different route, higher doses, and the same thyroid histopathology findings, with no functional (thyroid hormone) changes. The Study Pathologist was unmoved by this and stated that only functional data in the rat would be relevant. The senior toxicology reviewer for the CRO, in reviewing the correspondence, looked up the original NDA (available online at FDA website) and found an oral 13-week study in rats with the same active ingredient, same dose levels, same histopathology, higher exposure levels than in the current study, and no effects on thyroid hormones. The Study Pathologist then agreed this was supportive data and that the mid-dose thyroid histopathology was likely not adverse. The Study Director revised the report and the mid-dose was determined to be the NOAEL.
Take-home message: The Study Director and Study Pathologist took a conservative approach to the NOAEL, which is not a bad thing, but did not do their due diligence in providing support or questioning their initial impressions. The Study Director could have asked the pathologist why the thyroid change was considered adverse, but instead took the opinion at face value and incorporated it into determining the NOAEL. This conclusion caused great consternation for the Sponsor, who rightly questioned the call in their review of the draft report. Help with the answers to this dilemma for both the Study Director and Study Pathologist would have been readily available had they asked questions about the drug they were testing, and then used the available resources in the public domain to guide their thinking. Given the importance of the NOAEL, it is useful to vet the initial assessments with other principal stakeholders in a study before sending a draft report to a Sponsor, especially if there is some uncertainty or potential challenges in justifying the call. The Sponsor to some extent was also somewhat accountable here, since they should be the SME on their own molecule; and had they been aware of the studies performed for the original NDA and furnished the information up front to the Study Director, the correct NOAEL might have been determined the first time.
Case Study #2. A Little ICH Guidance Goes a Long Way
Study Type: 3-week study
Species: Rat
Route: IV
Drug Class: Anticancer, Antibody-Drug-Conjugate
Findings: Histological degenerative changes in bone marrow, thymus, spleen, lung, and testes; minimal/mild at low-dose, severe/marked at mid- and high-dose levels; generally reversible after 6 weeks of recovery, except in the testes, at the mid- and high-dose levels.
Based on the histopathology findings at the mid- and high-dose levels, along with the absence of reversibility of the testicular findings in mid- and high-dose animals, the Study Director declared the low-dose as the NOAEL. Upon receiving the draft, the Sponsor was concerned that the NOAEL would translate to a lower than necessary starting dose in cancer patients; hence, they approached a toxicology consultant. The consultant reviewed the data and asked the Study Director if he had calculated the STD10. The Study Director stated that he was unaware of the rationale or process for determining the STD10. The consultant sent the Study Director the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) S9 guidance discussing optional use of the STD10 in lieu of the NOAEL for rodent studies with anticancer therapeutics and assisted the Study Director in plotting the data to estimate the STD10. Since the histological changes at the high-dose in females did not fit the definition of severely toxic (i.e., causing death/moribundity, life-threatening, or irreversible), and the severely toxic dose in males was greater than the mid-dose (due to the irreversible testicular findings), the STD10 in males was determined to be about halfway between the low- and mid-dose levels, and in females it was considered greater than the high-dose. The Study Director revised the next draft of his report accordingly. The STD10 in male rats turned out to be more in line with the NOAEL in monkeys, which allowed for a higher starting dose in the clinic compared to the original rat NOAEL determined by the Study Director.
Take-home message: It was unclear whether the Sponsor had neglected to provide adequate information regarding the indication for the test article, but the Study Director should have questioned the Sponsor. However, the critical piece missing for the Study Director (and possibly for the Sponsor), was knowledge of regulatory guidance on the subject of determining the starting dose for anticancer therapeutics, per ICH S9. Ideally, the Study Director and the Sponsor would have exchanged enough information prior to the study start so that at some level of protocol review the concept of the STD10 versus the NOAEL would likely have emerged. Both parties could then have discussed whether to report one or both up front; or alternatively, the Study Director could have just reported the NOAEL and the Sponsor could have made the adjusted calculation in the IND. In this case, it was early enough in the process for the Study Director to assist the Sponsor in reporting the STD10 versus a NOAEL, and the difference between males and females was large enough that it was useful to list both in the bottom-line assessment. Both the Sponsor and Study Director learned that the therapeutic indication is an important piece of information to be shared up front, and that in some cases, it can ultimately affect the bottom-line assessment and starting dose levels in the clinic.
Case Study #3. There Can Be Different NOAELs for Local and Systemic Toxicity
Study Type: 9-month study
Species: Dog
Route: Subcutaneous
Drug Class: Therapeutic peptide; non-oncology indication
Findings: At 2 months into a chronic study, moderate-to-severe erythema and swelling at the local injection sites in individual mid- and high-dose animals jeopardized the continued dosing in the study. The Sponsor had communicated this potential local adverse effect to the Study Director in advance of the protocol. While the absence of any such findings early on gave both parties a degree of assurance that the study would go to completion, they were now of major concern. Efforts were quickly made to find more animals to either re-initiate the study or add lower dose groups to the current study. Either approach would have been catastrophic for the Sponsor in terms of time and resources; but more importantly, the increase in animal use would have been very troubling. The initial approach was to immediately lower the mid- and high-dose levels, but to keep the low-dose level the same. The Study Director, senior management at the CRO, and the Sponsor Monitor and his team started daily calls to check on animals and investigate whether this approach, along with individual animal dosing holidays or finding alternative dose sites, could work. This plan worked in some cases; in others, it caused further consternation as to how to mitigate the issue. After some research, it was decided that these reactions might be histamine-related, and two different antihistamine regimens were tried in select affected animals. This approach, along with a short dosing holiday and alternate dose sites, ameliorated the local reactions enough that it was considered safe to continue dosing. Only affected animals were given the antihistamine regimen, as were a subset of controls. The study continued as planned for its complete 9-month schedule. Histopathology findings at injection sites included inflammation or hemorrhage, with near-complete recovery after 4 weeks treatment-free. The NOAEL was reported as the low dose (local) and high dose (systemic).
Take-home message: This brainstorming of a step-by-step approach to save the study is a good example of proper communication between the Study Director and the Sponsor Monitor, and proved to be in the best interest of the study and the animals. By sharing relevant information up front, the Sponsor was able to alert the Study Director about effects to watch for, prompting an immediate response when the effect first started to emerge. By thinking through the objectives of the study, the possible impact of therapeutic intervention in individual animals as they became affected, and how to deal with comparisons to control animals, a plan was enacted that enabled the study to continue. The local versus systemic NOAEL reporting was important, as the Sponsor needed to dose-escalate in the clinic and could feel comfortable doing so if no local reactions were to occur; or if any did, they could be controlled by local or systemic antihistamine treatment. This difficult situation could have been a disaster for both the CRO and the Sponsor; however, the clear thinking of both the Study Director and Sponsor enabled the study objectives to be met, and the drug to safely advance in the clinic.
Case Study #4. Characterization of the Clinical Signs Helps!
Study Type: 6-month GLP study
Species: Sprague-Dawley rat
Route: Oral gavage
Drug Class: Small molecule; non-oncology indication
The test article was administered at three dose levels.
Findings: High-dose group males and females started showing clinical signs of seizures after 30 days of dosing. After Day 100, the mid-dose animals, and after Day 130, the low-dose animals started showing seizures. Typically, the seizures were observed either within a few seconds after dosing or while handling the animals during routine procedures such as blood collection, body weight measurement, and cage change. Seizure episodes, on average, lasted for less than 1 minute.
Seizure Incidences in Rats (Numbers Are Hypothetical).
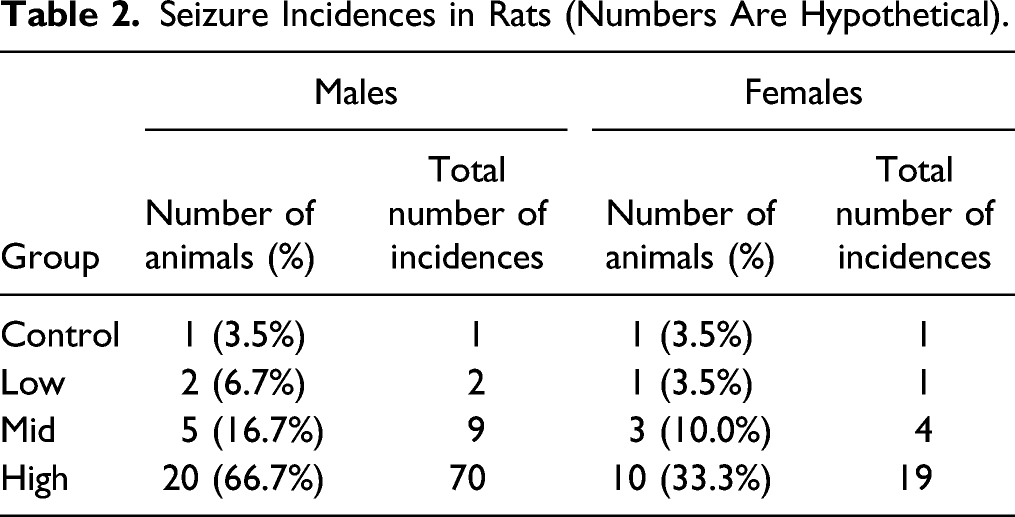
Although no moribundity was observed, due to increased incidences of seizures, the sponsor decided to stop the dosing in high-dose main group males after Day 90 to assess the probable histopathological changes in their brains. Dosing in the rest of the animals was continued as per schedule, and all animals survived until the end of the study. Vacuoles in the brain were observed in both males and females from the high-dose groups. In this study, brain lesions and higher seizure incidences at the high-dose were considered adverse. The incidences of seizures were clearly dose-dependent and test article-related. But the question remained, were these seizures adverse? If at all adverse, then at what dose level? Should a single incidence of seizure be considered adverse? The study director consulted the literature and discussed with SMEs; however, there was no clear consensus as to whether the seizure incidences at lower doses in this study should be considered adverse or not. Finally, seizures in the low-dose group were considered likely non-adverse for the following reasons, and the low-dose was considered the NOAEL: 1. Incidences in the low-dose group were fewer, and even comparable to those of controls. 2. All seizure incidences occurred during handling of the animals for routine procedures. The cageside clinical observation was performed at Tmax but seizures were not observed at the time when test article plasma concentration was highest (Cmax). Thus, there was no clear correlation of plasma concentration to seizure events. 3. The seizures were observed intermittently and with low frequency. The interval between two incidences of seizures in an animal, if any animal had repeat incidence, was many days to many weeks. 4. There was no post-ictal depression phase since animals quickly recovered from the seizure phase and started normal activities thereafter. 5. The animals (including high dose TK cohort males), which were treated for the entire treatment period, survived until the end of the study. 6. There were no other indications of negative effects, such as changes in body weight and food consumption, on the overall health of the animals.
Take-home message: 1. Detailed notes regarding the timing of the convulsion episodes were available, including exact timing of the convulsion relative to the dose time, relevant TK information, and length of each episode. This information was helpful in characterizing the seizures. 2. In the absence of electroencephalogram data, “seizure-like clinical behavior” should have been documented as convulsions rather than as seizures during daily clinical observations. 3. A “distraction test” with a clicker or pinch should have been performed when seizure incidences occurred to confirm whether the events were of psychogenic non-epileptic seizures origin. 4. Additional neurological evaluations were needed to understand why there was a dose-dependent increase in the incidences only while handling the animals for routine procedures such as dosing, and no incidences were observed cageside at Tmax for any dose level.
Case Study #5. A NOAEL Disagreement During the IND Review Process
Study Type: 12-week study
Species: Rat
Route: Oral gavage (daily)
Drug Class: Small molecule; non-oncology indication
Findings: Kidney toxicity (nephropathy, tubular basophilia, dilatation, vacuolation in collecting ducts), clinical signs (lethargy, intermittently observed in 50% of animals within 1 h of dosing), and decreased body weight (average of 20%) were observed in animals dosed daily at the mid- and high-doses; in addition, mortality was observed at the high dose. Increased liver weight and centrilobular hypertrophy of the liver were observed at all the dose levels in both sexes, with increased severity and in a dose-responsive manner. A dose-dependent increase in hepatocellular lipids were observed at all the doses. In the regulatory submission, the mid-dose was considered to be the NOAEL, and changes in lipid metabolism, clinical signs, or decreased weight were not considered adverse.
Regulatory perspective: The agency disagreed with the Sponsor’s NOAEL (mid-dose) and considered it to be the low-dose. This was based on kidney toxicity, changes in lipid metabolism, clinical signs, and decreased weight at the mid- and high-doses.
Liver hypertrophy was not considered a direct toxic effect. Low severity and absence of hepatocellular degeneration or cytotoxicity contributed to this decision. Microsomal enzyme induction was considered adaptive to promote hepatic clearance. Combined, the increased aminotransferases, increased hepatocellular lipids, and increased plasma cholesterol at the mid- and high-doses were considered adverse, and reflected a change in lipid metabolism. Clinical signs and weight loss were also considered adverse at the mid- and high-doses.
Take-home message: Changes that affect the well-being and development of an animal may be considered adverse based on their severity and duration. Here, the weight loss and lethargy were clearly related to the test article and negatively impacted the animal’s functioning; as such, they were considered adverse. In addition, changes in several clinical chemistry parameters can be considered adverse if they collectively impact a single integrated process, such as lipid metabolism.
Case Study #6. Adaptive Changes and Sex-Specific NOAEL
Study Type: 4-week study
Species: Rats
Route: Oral gavage (daily)
Drug Class: Small molecule; non-oncology indication
Findings: Death was observed at the high-dose in both males and females (cause not determined). A reversible reduction in body weight was observed in mid- and high-dose males. Increased cholesterol accompanied by increased triglycerides was observed in high-dose males and mid- and high-dose females. Non-reversible low epididymides and testis weights were noted in high-dose males. This was accompanied by degeneration of the seminiferous tubules and spermatozoa in testes and epididymides of mid- and high-dose males at the end of the study, and at the high-dose in recovery animals. In addition, reversible epithelial hypertrophy was observed in the kidneys of mid- and high-dose animals.
Regulatory perspective: In agreement with the Study Director’s evaluation, the epithelial hypertrophy in the kidneys was considered an adaptive response to an increase in active transcellular transport capacity, and the finding was not considered in the identification of the NOAEL. The mid-dose was considered the NOAEL based on mortality in both sexes and irreversible effects in the male reproductive system at the high dose.
Take-home message: An adaptive change may or may not be considered adverse, depending on the nature and consequence of the change. In this case, the hypertrophy did not lead to any cellular cytotoxicity or death, nor was it coupled to any degenerative effects. Death was considered a result of exposure to the test article; and as such, was considered crucial in determining the NOAEL. Last, irreversible and cytotoxic damage to tissues of the reproductive tract were also considered pivotal in setting the NOAEL. Had the male reproductive findings been the only adverse effect observed in the study, a male-specific NOAEL might have been considered.
Case Study #7. An Unusual Dose-Response Curve
Study Type: 2-week study
Species: Rat
Route: IV infusion (twice per week)
Drug Class: mAb to exogenous target; non-oncology indication
Findings: Mortality was observed in 4 out of 10 animals at the low-dose and 10 out of 10 animals at the mid dose, mostly within 2 hours after the fourth and final dose. There was no mortality at the high dose. A NOAEL was not identified. Results from a tissue cross reactivity study (TCR) showed membrane staining in the pancreas in both rodent and human tissues. In the regulatory submission, the deaths were attributed to anaphylaxis, but the Sponsor did not provide supportive data to exclude direct toxicity. The TCR findings were attributed to off-target binding.
Regulatory perspective: The cause of death was likely anaphylaxis with an inverse dose-response; no deaths at the high-dose, death after the fourth and final dose, and rapid onset of death (1–2 h post-dose). Direct pancreatic toxicity was a concern based on the TCR study results. A full clinical hold was imposed; the Sponsor was asked to conduct a toxicology study in a second species and include parameters to determine the mechanism of anaphylaxis in case it was observed. Also, the Sponsor was asked to investigate the cause of death in the rodent study. Upon further investigation, this was revealed to be immune complex formation and not direct organ toxicity. Immunohistochemical analysis revealed no membrane binding in the pancreas. A 4-week follow-up toxicology study was conducted in nonhuman primates with weekly IV infusions and a 6-week recovery period. No adverse findings were observed, and the NOAEL was the high dose. Clinical trials could proceed with this clarifying data, and no further follow-up studies were recommended.
Take-home message: Unexplained deaths should be investigated and not left unexplained, especially if the death is clearly test article-related and/or if there is an unusual dose-response curve. Follow-up studies may be needed in a second species. Membrane binding in a TCR study should be explained, and a rationale should be provided if follow-up studies are not planned. Immune complexes formed in nonclinical species may lead to non-dose-response effects. Moreover, ADAs observed in a nonclinical species may not be translatable to humans, and sometimes not even to other higher-order nonclinical species.
Case Study #8. Check Your Lab Before You Wreck Your Study
Study Type: 2-week study
Species: Rat and dog
Route: IV (daily)
Drug Class: Small molecule; non-oncology indication
Findings: In a 2-week rodent toxicology study, death was observed at the low- and mid-doses, and poor clinical condition was noted at all doses. Histopathology revealed vacuolation in nervous system tissues and lesions in lymphoid tissues, liver, kidney, lung, and spinal cord at all doses. Although dehydration was proposed as the cause of vacuolation, supporting data was not provided. As such, a NOAEL was not identified.
In a similar 2-week study in dogs, there was no mortality, however emesis and salivation were observed at all doses. Increased creatine phosphokinase (CPK) was noted at the mid- and high-doses and ST depression and increased R wave amplitude was observed during ECG evaluation at the mid-dose. There were no histopathology findings in the dogs. As such, the NOAEL in the dogs was the low-dose, based on CPK increases and changes in ECG parameters.
Regulatory perspective: A clinical hold was imposed because of the vacuolation in nervous system tissues in the rat, as well as a failure to rule out drug-related neurotoxicity. The sponsor was asked to repeat the 2-week study and include a peer-reviewed pathology evaluation by a board-certified pathologist. Also, addition of a recovery group was recommended to investigate reversibility of any drug-related effects, and the addition of lower doses was suggested in an attempt to establish a NOAEL. Additionally, evaluation of drug levels in nervous system tissues (or a distribution study) was recommended to determine whether the drug crossed the blood-brain barrier. Last, the Sponsor was asked to provide literature support for the conclusion that the vacuolation in the nervous system tissues was due to dehydration. It is noteworthy that the facility where the original rat study was conducted was newly established and had never been inspected.
The rat study was ultimately repeated in another laboratory, with a 2-week recovery period. This laboratory had recently been inspected by the FDA and was without issues. In contrast to the adverse effects reported in the original study, at the low- and mid-doses (similar clinical exposures across the dose range), there were no adverse effects in the second rat study. Safety margins were sufficient for all proposed clinical doses; as such, the clinical trial could proceed safely. Although the NOAEL was the low-dose in the dogs, there were sufficient safety margins at all proposed clinical doses; consequently, this was not an issue.
Take-home message: Strange or unexplained findings should be investigated and a rationale, including supporting data, should be provided to the agency upon study submission. The investigation should not only include analysis of the study results, but also consider potential issues with the study facility and common practices, which may impact study conduct and observed results.
Case Study #9. HNSTD Can Be Tricky!
Study Type: 1-month
Species: Dog
Route: Oral gavage
Drug class: Small molecule; oncology indication
Doses: 0, 2.5, 10 and 25 mg/kg/day
Findings: Male and female animals became moribund at 25 mg/kg/day around days 13-17. Significant decreased body weight was associated with a decrease in food consumption. All female animals at 25 mg/kg/day had to be sacrificed early. Two female animals at 25 mg/kg/day were given dosing holidays for 2 days at day 15, but neither their body weights nor food consumption were recovered, and they were euthanized at day 17. This is consistent with toxicokinetic data, as drug accumulation was observed. The male dogs, whose effects were similar to those of the females, recovered soon after decreases in the plasma drug concentrations. There were no primary test article-related microscopic findings, and any test article-related clinical pathology findings in all animals at 25 mg/kg/day were reversible and considered non-adverse. Microscopic findings were limited to lymphoid organs at 25 mg/kg/day and were considered secondary to stress from adverse clinical signs. There were no test article-related significant findings up to high-dose levels in rodent species.
Narrative: The NOAEL was considered to be 10 mg/kg/day due to the lack of adverse clinical signs seen at this dose. Considering the oncology indication, the team was asked to decide whether to take the NOAEL or the HNSTD approach. Per the ICH S9 guideline, “The HNSTD is defined as the highest dose level that does not produce evidence of lethality, life-threatening toxicities or irreversible findings.” Although 25 mg/kg/day was not tolerated, the HNSTD was considered to be this dose, as there was no underlying pathology or target organ toxicity to cause inappetence or body weight loss. It is worth noting that in this case, animals did not have sudden death of unexplained cause. The decision to euthanize these animals was based on animal welfare criteria, as animals were losing weight gradually. Clinical signs were reversible (weight loss continuation in females was likely due to drug accumulation). The HNSTD from dogs was used to determine the starting dose in the FIH clinical study and was acceptable in the IND submission.
Take-home message: A common approach for many small molecules is to set a clinical starting dose at one-tenth of the STD10 in rodents. If the non-rodent is the most sensitive species, then one-sixth of the HNSTD is considered an appropriate clinical starting dose. The HNSTD is defined as the highest dose level that does not produce evidence of lethality, life-threatening toxicities, or irreversible findings. It was debated and challenged whether the high-dose at which animals were euthanized due to adverse clinical signs (e.g., loss of body weight, hypoactivity) should be considered HNSTD or not. Despite evidence of animals not tolerating the high-dose, the Sponsor concluded that the high dose was the HNSTD. The rationale for this position was that the dogs did not have target organ toxicity, and test article-related clinical pathology findings were without any microscopic correlates and were reversible. Adverse clinical signs in dogs were nonspecific in terms of specific target organ toxicity and were monitorable in clinics. This case study shows that what is considered a “severe effect” for determination of the HNSTD may be subjective.
Case Study #10. Exaggerated Pharmacological Effects
Study Type: 28-day
Study Species: Cynomolgus monkey
Route: IV
Drug Class: Biologics; oncology indication
Findings: In this case study, cynomolgus macaques were administered an immunostimulatory biologic compound in a toxicology study under a fairly standard study design with three dose levels and recovery groups. The proposed clinical use of the molecule as an immuno-oncology product was to enhance immune responses in cancer patients, although this molecule could be developed for non-oncology indications as well. The animals tolerated the compound well; however, enhanced immune cell infiltrates were found in some tissues, most particularly in the liver and intestine, and at the injection site. The severity of these cellular infiltrates seemed to be dose-dependent, with minimal findings in the low-dose animals. At the high-dose level, there were only a few foci in the liver where necrotic cells were present. Liver enzyme levels remained within normal limits for all animals. The findings largely resolved during the recovery period.
The Study Director concluded that the findings observed in treated animals exceeded those observed in the control animals, as well as those recorded in the historical database. Thus, these findings were considered test article-related but non-adverse. Some other biomarkers assessed in this study indicated that the test article caused immunostimulation in the monkeys. These findings were consistent with the pharmacological mechanism of action of the test article and were classified as non-adverse in the toxicity study report. Immune cell infiltration could be expected at sites with some basal stimulus for immune stimulation. This conclusion was based on available knowledge of the molecule’s pharmacology. The particular test article had been shown to effectively act as an immunostimulant in in vitro assays and in research studies. In documents submitted to regulatory authorities, the Sponsor argued that the observation of infiltrating lymphocytes was consistent with the pharmacology of the molecule and could be viewed as an indication of biologic activity in the monkey.
In addition, other immuno-oncology products were associated with similar preclinical and clinical findings. In this study, an increase in infiltrating lymphocytes in tissues like the liver and intestine, and at the injection site, could be expected. Since the observation of foci of necrosis in liver was limited, with minimal severity and without clinical pathology correlates; and that histological observations tended to resolve in the recovery phase, it was reasonable to view these observations as expected non-adverse pharmacological effects. The high dose was considered the NOAEL.
The final conclusion was that these infiltrates were designated non-adverse, since they did not correlate with biomarkers of adversity or clinical observations of distress. Mild inflammation at the site of injection also was designated non-adverse since it did not have a significant impact on the welfare of the animals. It is a common observation, particularly with vaccines in humans, and is not associated with significant distress.
Take-home message: Adversity was considered in the context of several factors. First, the determination of adversity and the NOAEL were considered in relation to the molecule’s pharmacology. In this case, a finding that might be considered adverse was actually a desired effect of the drug. Exaggerated pharmacology does not mean the finding is not adverse, but that consideration of the expected pharmacology can be helpful in evaluating findings that are relatively minimal in severity. Second, in the regulatory submission, the particular finding was compared to similar findings in humans using licensed products that had similar mechanisms of action. Such a comparison can be helpful in determining the level of distress associated with a particular finding. Finally, the determination of adversity, particularly as it relates to clinical development, may take the clinical indication into consideration when discussing the adversity in the IND/CTA (Clinical Trial Application) submission. In this case, some injection site inflammation would likely be more acceptable for products with oncology indications, as opposed to, for example, injectable products targeted to a healthy pediatric population.
Discussion
While discussing various approaches to determine adversity in nonclinical toxicity studies, it is important to put the entire process in perspective, starting with restating the objectives of these types of studies. In the simplest terms, the primary objective of a nonclinical toxicity study is to characterize the toxicity, identify target organs if possible, determine the dose level at which the effects of the test article are truly harmful (adverse) to the test animal, and identify the dose at which no adverse findings are observed (the NOAEL). This process involves several steps, beginning with an assessment of whether a finding is truly related to test article treatment; and if so, then likely employing a weight-of-evidence approach 42 to determine the adverse nature of the effect and identify the dose at which the adverse effect is not present (see Figure 1). Here, we described a stepwise approach that should prove useful in differentiating adverse from non-adverse effects, although we fully acknowledge that in the end, determination of adversity is still subject to differences in interpretation among the key stakeholders for a given therapeutic. Ultimately, it is up to the Study Director to make the final call in the report based on their review of all the pertinent study data and the expert assessments of contributing scientists. Here, we should emphasize that contributing scientists’ reports should generally be devoid of any NOAEL declarations. However, if a finding is deemed adverse by an expert’s (e.g., study pathologist, veterinary cardiologist, or other contributing scientist) evaluation, the dose level at which the finding is no longer evident or no longer qualified as adverse should be stated in their reports.
It is also important to consider the two opposing perspectives on adversity determination in a toxicology study. One states that adversity should only be considered in the test species under the conditions of a specific study. 15 The opposing perspective states that extrapolation of adversity to humans should be considered to determine the adverse nature of the finding in the test species in a toxicology study. In this context, human relevance requires some discussion. By definition, nonclinical safety studies in support of human pharmaceutical development have an underlying objective of human safety, which may not be congruent with the study-specific NOAEL. At times, Study Directors may need to challenge or rethink their definitions of “adverse vs non-adverse” under the individual study conditions.
Many definitions of adversity cited across a broad range of published toxicology and pathology papers, some of which we have discussed here, equate adversity with harm to the test species. This is why, for example, many authors discount increased liver serum enzyme levels as non-adverse in the absence of correlating liver pathologies. Clearly increased serum liver enzymes in a human trial would be considered adverse as a marker for liver injury. 43 However, in a toxicology study there generally is a good rationale for considering them non-adverse in the absence of liver pathology.
The term “harm” becomes problematic and quite subjective in some instances. An example is recumbency, in which clinical signs are indicative of dysfunction but do not necessarily affect the animal’s overall ability to thrive. When an animal responds to treatment by being recumbent for short duration, temporarily losing its ability to maintain balance or its righting reflex, there may be no overall negative impact on food consumption or body weight, no microscopic changes in the tissues, and the animal may only be affected for a short time after each dose; thus, the animal is not really harmed. Conservatively, using this definition of harm, transient recumbency would not be an adverse event, yet like elevated liver enzymes in the clinic, it would likely be considered an adverse effect in a human trial unless it was an expected pharmacological effect. However, unlike in the liver enzyme example, which was non-adverse in the test species because of an absence of a quantifiable, correlating evaluation of harm (liver pathology), the episodes of recumbency have no such quantifiable correlate except for measures of the overall ability of the animal to thrive (e.g., food consumption, body weight gain). By the same token, if we used that reasoning for humans, recumbency would not be adverse as long as the human subject was able to get up and eat, gain weight, and did not have any apparent organ distress! But understandably that is not true for humans.
In these types of cases (typically clinical sign-related), the definition of harm should be challenged. In doing so, perhaps the better question to ask: is the animal’s behavior dysfunctional enough that if the same clinical sign occurred in a human, would that behavior or effect be considered adverse? Here, we refer to the FDA guidance 1 on estimating the maximum safe starting dose in initial clinical trials, which considers the difficulty in characterizing adverse effects in animal studies, and states: “Although the nature and extent of adverse effects can vary greatly with different types of therapeutics, and it is anticipated that in many instances, experts will disagree on the characterization of effects as being adverse or not, the use of NOAEL as a benchmark for dose-setting in healthy volunteers should be acceptable to all responsible investigators.” It concludes, “As a general rule, an adverse effect observed in nonclinical toxicology studies used to define a NOAEL for the purpose of dose-setting should be based on an effect that would be unacceptable if produced by the initial dose of a therapeutic in a phase 1 clinical trial conducted in adult healthy volunteers.”
On the other hand, and as discussed earlier, a good example in which human relevance should not be used as a benchmark for adversity determinations, would be in cases where the animal species has a known sensitivity or unique tissue responses, not reported in humans, to a class of test agents. This, however, does not mean the report cannot discuss a species’ sensitivity in the context of adversity. It may, in fact, be warranted to help put the adverse findings in perspective, but it clearly should not influence the determination of the NOAEL from the study.
As the final step in the safety evaluation process as it pertains to the NOAEL and adversity, the study Sponsor is responsible for putting the study findings into context with respect to assessing human safety in the IND/CTA documents. While writing the regulatory documents, Sponsors reevaluate the NOAELs in non-rodent and/or rodent species while considering the relevance of those adverse findings to humans. NOAELs from all the nonclinical species are converted to HEDs and MRSD for initial clinical trial. 1
The NOAEL standard for initial dose setting in human studies may not be the only scientifically justifiable approach covering all therapeutic classes. The most notable exceptions based on risk/benefit apply to anticancer therapeutics and certain classes of immunomodulators. For anticancer therapeutics, more risk is acceptable, allowing for alternative initial human dose-setting approaches based not on adversity, but on severity of toxicity (i.e., STD10 or HNSTD). On the contrary, for immunomodulators much less risk is allowable, because inherent difficulties in predicting hyper-exaggerated immune responses necessitate the Sponsor’s use of the MABEL or PAD to determine MRSD. Whichever benchmark is used, the Study Director must provide justification for determination of the NOAEL, STD, or HNSTD based on the study results; and when needed, the rationale for determination of adversity or severe toxicity.
Furthermore, risk tolerance for a given adverse effect (basis for the NOAEL), may be considered in the context of the clinical indication and the target population being treated. Generally, the sponsor, not the study director, assesses risk tolerance in the IND, typically in the Integrated Summary section. It is easy to imagine that a particular adverse event might be considered an acceptable or tolerable risk to one patient population but might be unacceptable to a different patient cohort or to healthy volunteers in a FIH clinical study. The actual clinical manifestations of the adversity might be similar in both cases, but the acceptance or tolerance of the risk of those manifestations might be different.
Despite the negative connotations of nonclinical adverse findings in drug development, it is important to remember that characterizing toxicity is the primary objective of our toxicology studies. Without characterization of nonclinical toxicity, clinicians have little or no basis for establishing safe dose escalation schemes in humans, nor would they know which adverse clinical signs or changes in biomarkers to monitor. If a Sponsor does not appreciate that a toxicology study seeks to induce toxicities at the high dose level, they may become concerned that the toxicology study is not “clean.”
It is not uncommon to receive regulatory approval for therapeutic agents with adverse findings in toxicology studies.16,17 The regulatory approval process of an investigational drug, despite adverse effects in nonclinical studies, depends on many factors including, but not limited to, risk-benefit given the therapeutic indication, currently available therapeutic options, the critical nature of the adverse effects, human relevance, depth of understanding about the adverse effects (including mechanisms of action), and ease of monitorability in human trials.
Conclusion
Despite challenges, practical stepwise considerations are available to the nonclinical scientists that can aid in the process of determining adversity and a NOAEL. These include determining (1) whether the effect is truly test article-related, (2) the overall severity of the effect, (3) whether the effect is adaptive in nature, (4) whether recovery would lessen any negative impact on the animal’s well-being, (5) whether further degenerative injury or a downstream neoplastic change could be anticipated, (6) how the primary or secondary nature of the effect might influence adversity, (7) if the effect is an acceptable exaggerated pharmacology or has become toxicity, and (8) if there are other related endpoints that weigh in determining adversity at a given dose level.
Case studies on issues concerning adversity determination and the NOAEL indicate that the best outcomes occur when the Study Director consults with the contributing scientists concerning their findings and understands the nature of the molecule and its indication. This requires good communication between the Study Director and other key players in the study, as well as with the Sponsor. Human relevance of adverse toxicological findings is important but should not influence the determination of the NOAEL in the study. Finally, translation of the adverse findings in a toxicology report directly impacts safe human starting dose levels and escalation in the clinic, underscoring the importance of diligence at all levels in evaluating nonclinical safety data.
Footnotes
Acknowledgments
The authors would like to thank Joanne Berger, FDA Library, for editing a draft of the manuscript.
Author Contributions
Kale, V, contributed to conception and design, contributed to acquisition, analysis, and interpretation, drafted manuscript, critically revised manuscript, gave final approval, agrees to be accountable for all aspects of work ensuring integrity and accuracy. Bebenek, I, contributed to conception and design, contributed to acquisition, analysis, and interpretation, drafted manuscript, critically revised manuscript, gave final approval, agrees to be accountable for all aspects of work ensuring integrity and accuracy. Ghantous, H, contributed to design, contributed to analysis and interpretation, drafted manuscript, critically revised manuscript, gave final approval, agrees to be accountable for all aspects of work ensuring integrity and accuracy. Kapeghian, J, contributed to conception and design, contributed to acquisition, analysis, and interpretation, drafted manuscript, critically revised manuscript, gave final approval, agrees to be accountable for all aspects of work ensuring integrity and accuracy. Singh, B, contributed to conception and design, contributed to acquisition, analysis, and interpretation, drafted manuscript, critically revised manuscript, gave final approval, agrees to be accountable for all aspects of work ensuring integrity and accuracy. Thomas, L, contributed to conception and design, contributed to acquisition, analysis, and interpretation, drafted manuscript, critically revised manuscript, gave final approval, agrees to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Disclaimer
The views expressed in this manuscript on the strategy of adversity determination are those of the authors, and do not necessarily represent their employers’ practices or agency’s recommendations.