
Abstract
The growth in drug development over the past years reflects significant advancements in basic sciences and a greater understanding of molecular pathways of disease. Benchmarking industry practices has been important to enable a critical reflection on the path to evolve pharmaceutical testing, and the outcome of past industry surveys has had some impact on best practices in testing. A survey was provided to members of SPS, ACT, and STP. The survey consisted of 37 questions and was provided to 2550 participants with a response rate of 24%. Most respondents (∼75%) came from the US and Europe. The survey encompassed multiple topics encountered in nonclinical testing of pharmaceuticals. The most frequent target indications were oncology (69%), inflammation (55%), neurology/psychiatry/pain (46%), cardiovascular (44%), and metabolic diseases (39%). The most frequent drug-induced toxicology issues confronted were hepatic, hematopoietic, and gastrointestinal. Toxicological effects that impacted the no observed adverse effect level (NOAEL) were most frequently based on histopathology findings. The survey comprised topics encountered in the use of biomarkers in nonclinical safety assessment, most commonly those used to assess inflammation, cardiac/vascular, renal, and hepatic toxicity as well as common practices related to the assessment of endocrine effects, carcinogenicity, genotoxicity, juvenile and male-mediated developmental and female reproductive toxicity. The survey explored the impact of regulatory meetings on program design, application of the 3 Rs, and reasons for program delays. Overall, the survey results provide a broad perspective of current practices based on the experience of the scientific community engaged in nonclinical safety assessment.
Introduction
The drug development industry has undergone extraordinary growth in the last few decades resulting in an increase in the number of new drugs approved. The 5-year average of the Food and Drug Administration (FDA) drug approvals is more than double what it was a decade ago. 1 The year 2020 also saw the second greatest number of drugs ever approved, with fifty-three new drugs, a number just shy of the all-time record of fifty-nine in 2018.1,2 While most new therapeutics are new chemical entities (NCE), biologics account for 27% of the drugs approved in the last 6 years. 2 This growth within the industry reflects significant advances in all aspects of drug development from basic science and understanding of molecular pathways to new technologies involved in nonclinical testing. Identification of molecular targets of diseases and new advances in human genome research have opened the door for significant advances in precision and personalized medicine in treatment of disease. This advance in personalized medicine was evident with the approval of chimeric antigen receptor (CAR) T-cells for treatment of certain blood cancers or viral vector delivery of therapeutic transgenes. Because of these advances, the pharmaceutical industry continues to invest large sums for the development of therapeutics for a broad range of human diseases.
The safety assessment of novel therapeutics intended for clinical trials is fundamentally based on the principles and recommendations of the International Council on Harmonization (ICH) as well as regulatory guidelines and guidance published by other regulatory authorities including the FDA, European Medicines Agency (EMA), and other national regulatory bodies. Pharmaceutical development scientists are continuously challenged to ensure nonclinical study designs reflect the standards of drug safety testing practice, but may also include novel endpoints that can determine the safety of a new therapeutic in clinical trials. The Predictive Safety Testing Consortium (PSTC), an independent group within the Critical Path Institute, has a focus on the identification and validation of new safety methods including biomarkers of toxicity with several biomarkers being included in nonclinical programs. In addition to the PSTC, research and development organizations within pharmaceutical companies and at academic research laboratories are key to the development of new methods. Moreover, the publication of data is an important vehicle for information transfer ultimately leading to improved testing as well as considerations for future testing. The year 2020 has also seen an unprecedent shift in collaboration within the industry, a direct result of the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) or COVID-19 pandemic and the need to develop new therapeutics quickly. This collaboration, in partnership with regulators has led to the development of new therapies and vaccines in a fraction of the time drug development historically took.
Benchmarking industry practices is important to enable critical evaluation along the path to evolve state-of-the-art pharmaceutical testing. The outcome of past industry surveys has had an impact on the regulatory guidance and the best practices in testing. Purves et al. 3 described industry practices for genotoxicity testing and criteria for determination of a positive response. This practice influenced the revision of ICH S2(R1) as well as the FDA guidance on determination of a weight-of-evidence approach for assessing genotoxicity responses. Subsequent reviews of industry practices have included testing with animal models of disease 4 and social housing of nonrodents for toxicity and safety pharmacology testing. 5 The results of several surveys have been published related to the field of safety pharmacology, sponsored by the Safety Pharmacology Society (SPS). Valentin et al. (2005) described the testing and evaluation of results for small molecules in the few years following implementation of ICH S7A. 6 This was followed by another survey a few years later examining CNS endpoints and inclusion of safety pharmacology endpoints, for example, cardiovascular, into routine general toxicology studies.7,8 Despite the impact these surveys can have on regulatory testing strategies and study design, there has been no systematic examination of toxicology study designs, endpoints being assessed or evaluation of datasets to document industry trends in nonclinical drug safety testing. This article attempts to identify current practices within the industry based on an extensive survey of safety pharmacologists, toxicologists, pathologists, and others involved in the conduct of nonclinical safety and toxicology studies.
Materials and Methods
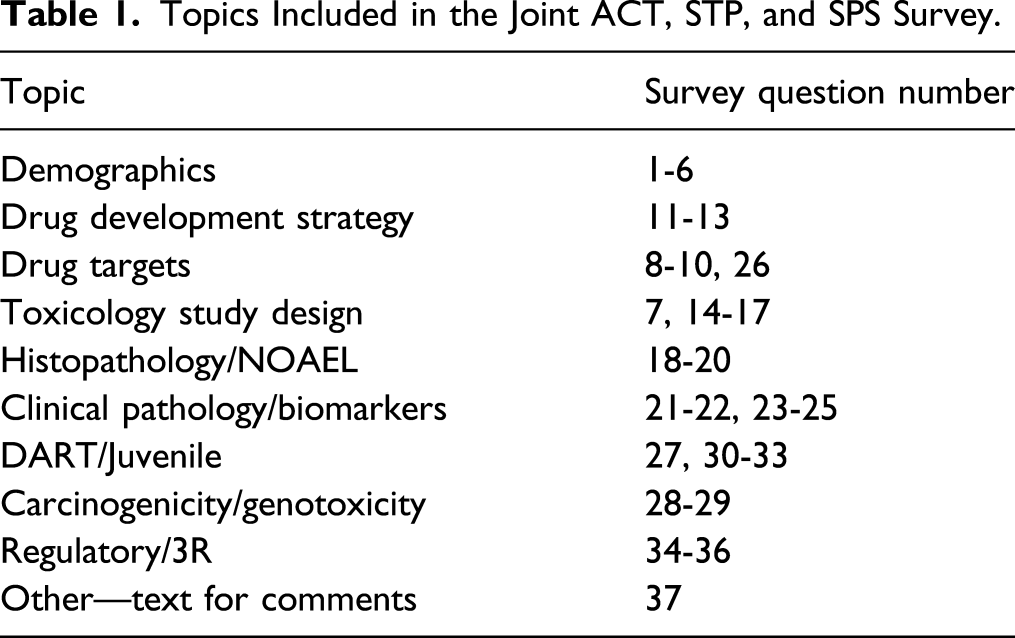
Topics Included in the Joint ACT, STP, and SPS Survey.
Statistical Analyses
The responses to many of the questions in the survey consisted of “Never,” “Rarely,” “Occasionally,” “Frequently,” “Always,” and “I Do Not Know.” For many of the questions, responses of “Never,” “Rarely,” and “I Do Not Know” were often less than 5% and usually less than 1%. Those responses are not included in the description of the data and as a result, not all responses will total to 100%. Quantitative data, when requested within a question are included in the description of the results, although at times, the data were combined to achieve greater clarity for instance when analyzing responses that were frequent for many questions. In questions inquiring about common practices, for example, drug therapeutics, toxicity outcomes, etc., the responses were limited to the last 5 years from initiation of the survey (2012-2017).
The independence of the frequency distributions between two variables were tested using the Chi-square test or the Fisher exact test. 9 Before conducting the independence tests, the categorical responses “Never” and “Rarely” were combined as “Never-Rarely” and the categorical responses “Frequently” and “Always” were combined as “Frequently-Always.” The categorical responses “I don’t know” and “N/A” were considered as missing responses. Statistical tests were performed to compare responses for certain questions, for example, drug development strategy, with demographic responses (e.g., company/institution size, job position).
Results
Demographics and Response Rate
There were 603 respondents accounting for a survey response rate of ∼24%. The survey was designed to exclude responders that are not intimately involved in the conduct of nonclinical toxicology studies, for example, academic graduate students. Likely due to the length of the survey, the item response rate declined as the survey questions progressed, such that the rate at Question 35 was only 9%. Excluding the last question which asked for toxicological issues not covered in the survey, all questions had 157 answers or more. The number of answers obtained from Question 11 to 31 and Question 34 and 35 was relatively stable between 213 and 246 answers. Despite the decrease in item response rate, the results of the survey are still considered valuable given the high number of participants to the survey.
The majority of respondents (∼76%) to the survey originated from the United States and Canada, while ∼17% were from Europe, including the UK, and the balance representing other geographic regions (Figure 1). Other respondents to the survey came from Nigeria, South Africa, Brazil, Denmark, Taiwan, and Israel, clearly representing an international response reflecting the global involvement in drug developmental modalities and in developing countries. Current trends of practices in nonclinical toxicology survey: geographical location. Responses (% of total; n=603) provided in the figure represent the proportion of participants that selected each category.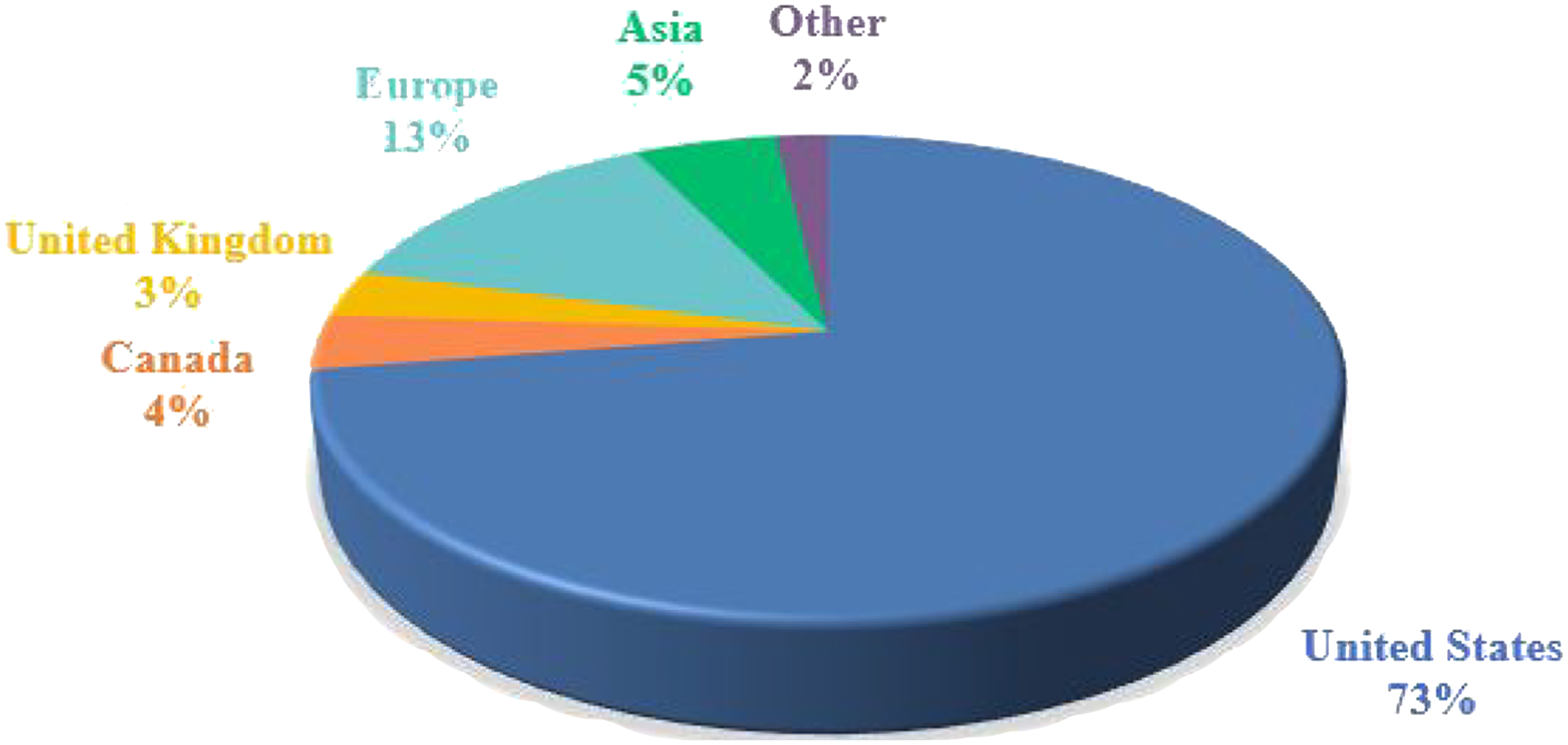
Nearly all responders (∼96%) had some role in nonclinical studies consisting of those involved with the design and execution of studies, monitoring of studies and generation of nonclinical data. Those that were not involved in the conduct of nonclinical studies were not offered to complete the survey. Of the remaining responders involved with nonclinical data, the majority were toxicologists (∼48%), pathologists (∼39%), or safety pharmacologists (∼17%) (Figure 2). Other responders included regulatory affairs specialists (∼4%), pharmacologists (∼6%), and other professionals including veterinarians, pharmaceutical scientists, and executives, making up the balance. About 45% of respondents had >20 years of service in the conduct of nonclinical study programs and only 10% had <5 years of service. Most respondents derived from the pharmaceutical and biotechnology industry (∼49%) followed by contract research organizations (∼22%) and consultants (∼17%). Additionally, there were respondents from academia (∼5%) and government/regulatory organizations (∼4%) (Figure 2). The respondents (∼38%) came primarily from large organizations (>5000 employees), and ∼26% of the responses were accounted for by smaller organizations comprised of less than 50 employees. Current trends of practices in nonclinical toxicology survey: demographics (i.e., profession, company type, years worked, and company size). Responses (% of total; n = 600 to n = 603) provided in the figure represent the proportion of participants that selected each category.
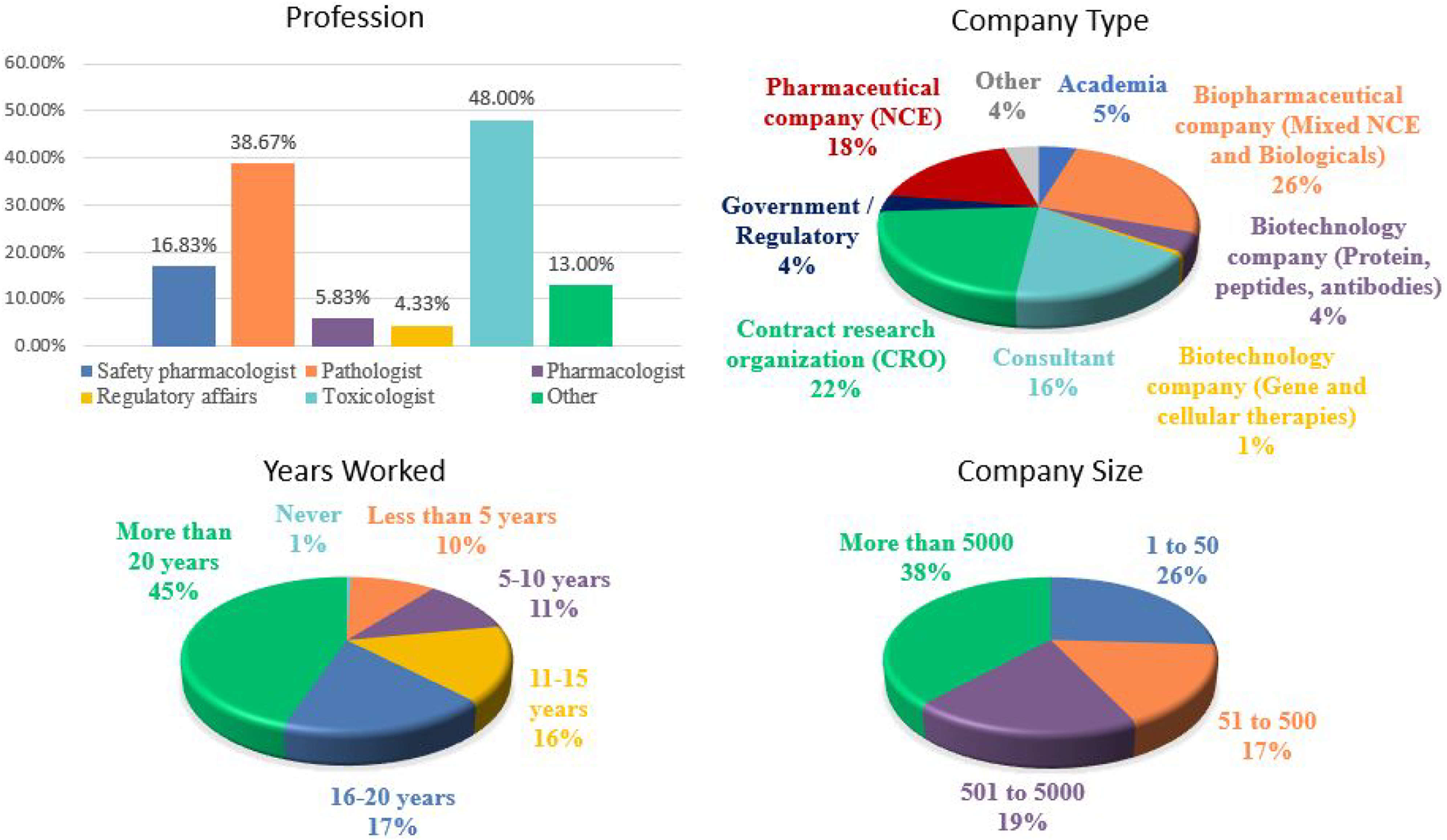
The responses obtained from this survey represent a diverse group of participants in different roles distributed across various organizations of various sizes and is thus likely representative of industry-wide practice in nonclinical toxicology testing.
Selection of a Molecule for Evaluation
Scientists involved in the drug development process are aware of the “drug development funnel,” a graphical representation of drug attrition rates where an estimated ten thousand compounds may enter the drug development screening process, but after over a decade of progressing through development, only a few molecules are granted marketing authorization.
10
While new chemical entities (NCEs), usually small chemical molecules, continue to make up the largest proportion of drug candidates screened (∼82%), a large percentage of respondents also indicated working with monoclonal antibodies (∼62%), peptides (∼53%), antibody drug conjugates (∼47%), and antibody-based therapies (∼45%) (Figure 3). This reflects an industry trend where there has been a large increase in the evaluation, and approval of, biotechnology-derived therapeutics within the last decade.
2
Other drug candidates included in the responses (∼20-35%) include cell and gene therapies, slow-release formulations, vaccines, antisense therapies, anti-viral therapies, and chemically modified RNA. Additionally, “Approved drugs” was a commonly specified response likely associated with the re-purposing of already approved drugs for another indication or route of administration. One advantage of this re-purposing approach is these drugs already have known safety profiles, and thus the time and financial resources and risk to develop these new indications is substantially less.
11
Although the survey was intended for those in the pharmaceutical industry, responses regarding food additives, environmental chemicals, and agrochemical were also included by responders. Overall, the responses to this survey encompassed a broad range of therapeutic classes indicative of the breadth of drug modalities in development the pharmaceutical industry as a whole. The types of drug candidates pursued for nonclinical testing from 2012-2017. Responses (% of total; n = 488) provided in the figure represent the proportion of participants that selected each category.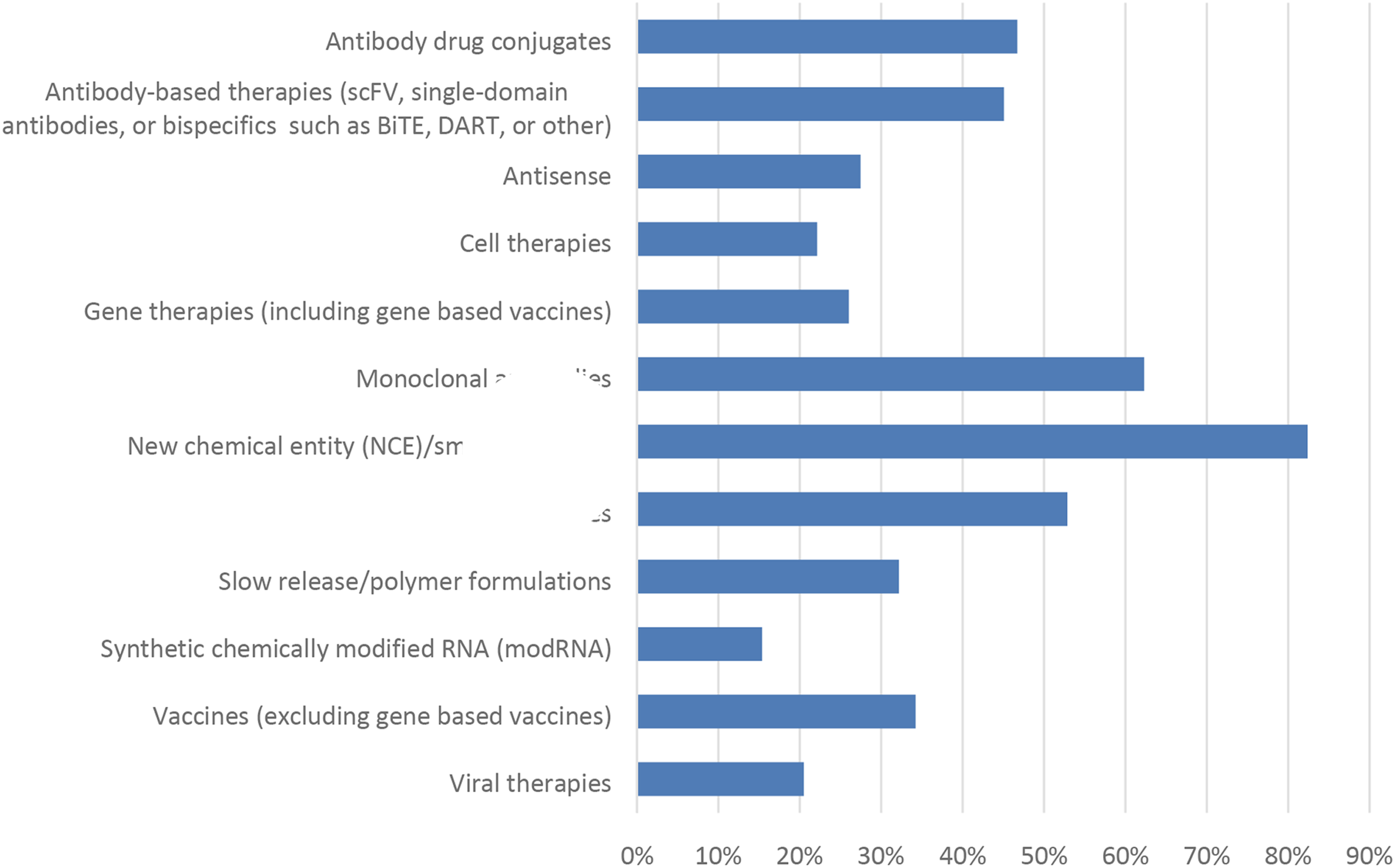
Similarly, the range of responses for drug indication was also quite broad (Figure 4). At the time of this survey (2017), developing drug candidates for oncology represented the greatest response (∼69%) followed by inflammation (∼55%). Cardiovascular and neurology also represented a large proportion of the responses (>40% each). These responses are consistent with the trend of drug approvals by the FDA where cancer products comprise the majority (34%) of approvals, followed by neurology products (15%).
1
The “Other” (∼13%) responses consisted of numerous indications including hematology, dermal, vaccines, otic, and autoimmune. Not specifically included in Figure 4 is the development of drug candidates for kidney disease which represented the largest proportion of responses in the “Other” category (∼2%). The types of indications pursued for nonclinical testing from 2012-2017. Responses (% of total; n = 485) provided in the figure represent the proportion of participants that selected each category.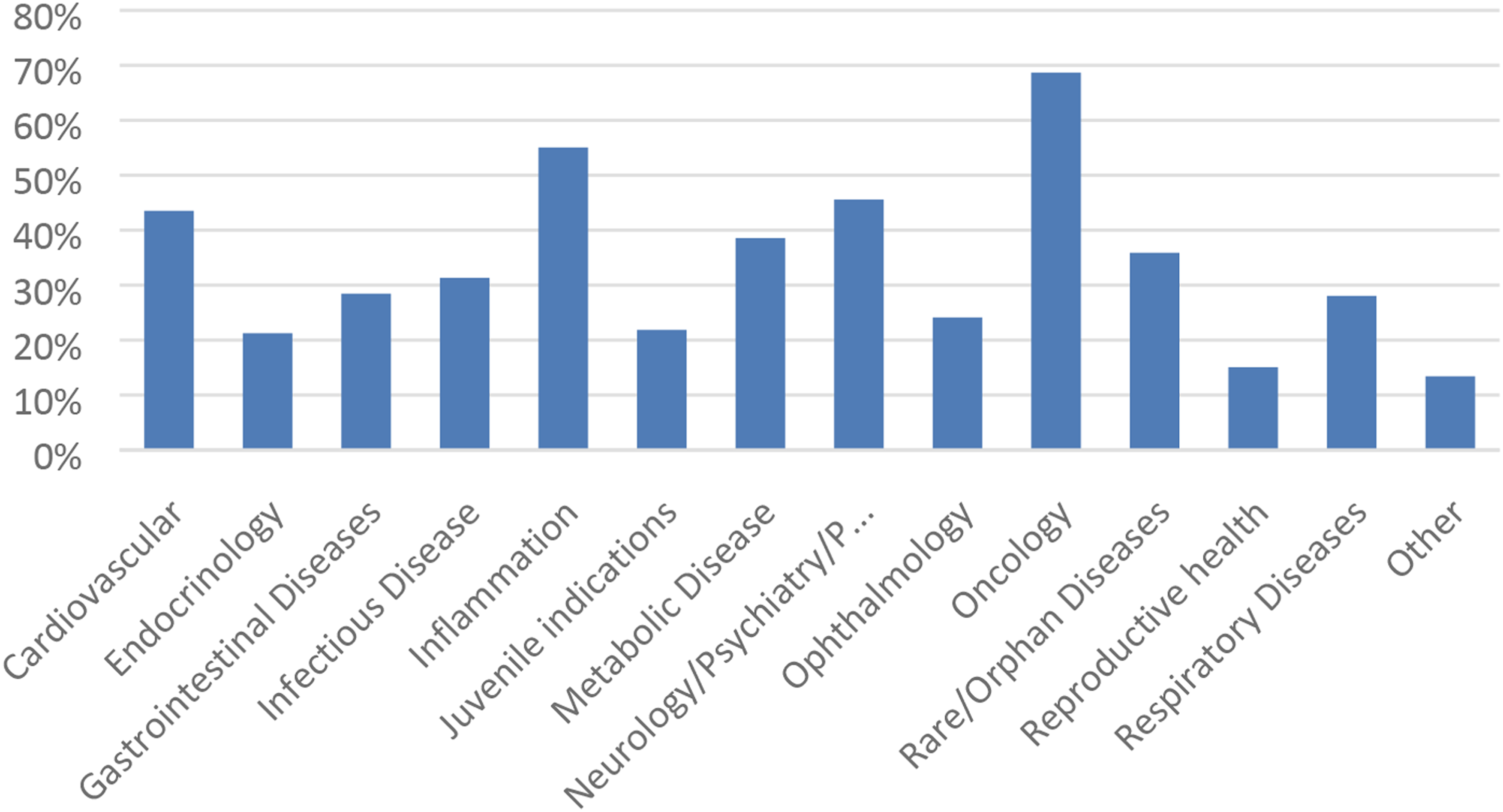
Safety Study Designs and Outcomes
As part of the typical drug development paradigm, several questions were asked in regard to early discovery toxicology and investigative toxicology regarding toxicology study designs. Questions related to early stage research revealed that ∼55% of the respondents utilized standard or screening studies to examine the initial toxicity of potential drug candidates. Other respondents (∼34%) used screening studies for issue resolution, that is, investigative toxicology studies to examine an effect observed in other studies (Figure 5). There was a significant correlation (Chi-square P < 0.01) between the size of the organization and early toxicology study design, where large organizations (>5000 employees) were more likely to use a standard battery of tests on all drug candidates followed by issue resolution studies, whereas small organizations (1-50 employees) were more likely to select tests for issue resolution but would not employ standard screening studies for all drug candidates. Early approaches to toxicology testing. Responses (% of total; n = 246) provided in the figure represent the proportion of participants that selected each category.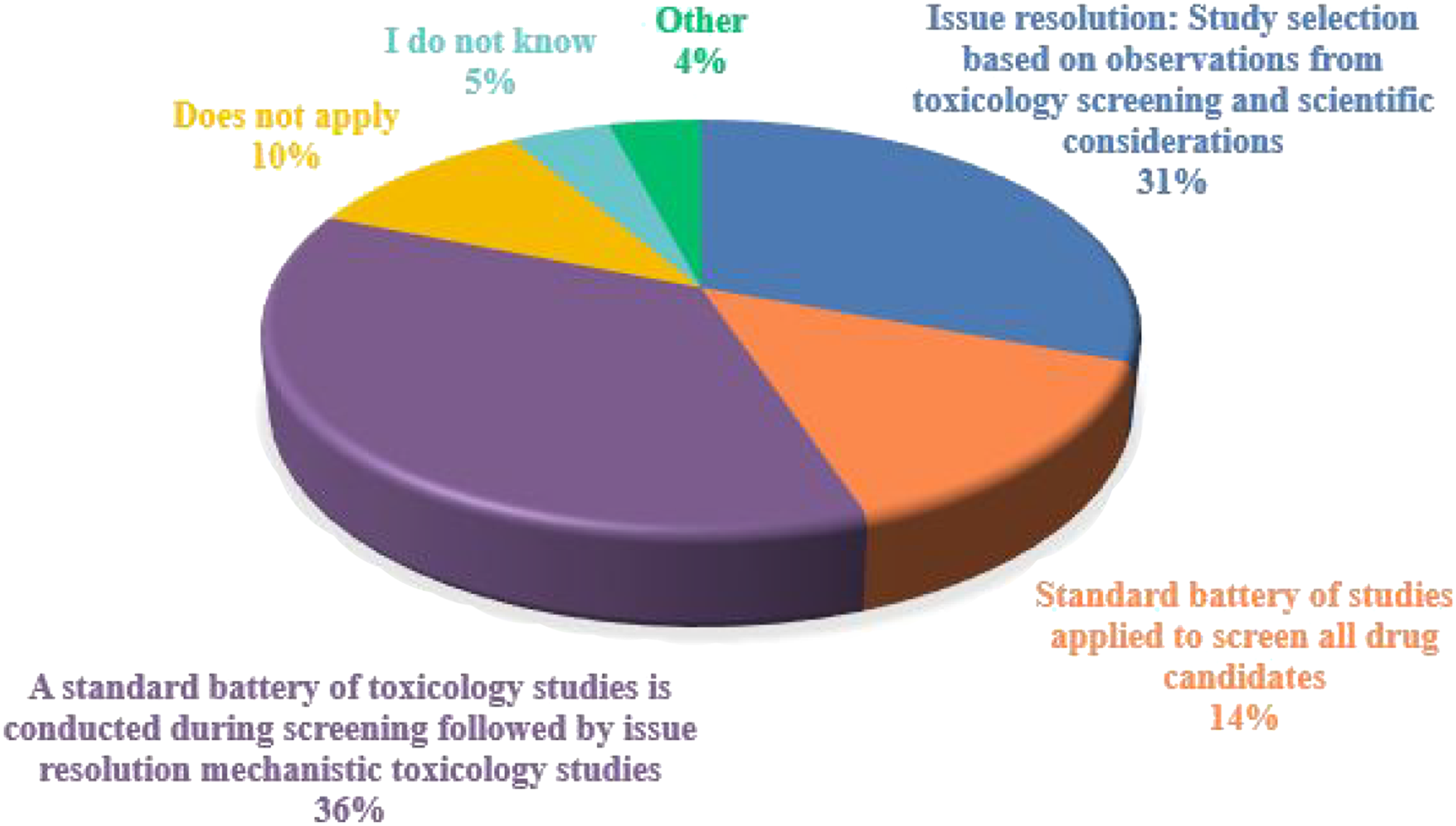
For general toxicology studies there are many parameters to consider when designing a study, including those related to animal selection and dose formulations. Approaches for species selection and general toxicology study design are described in regulatory documents and in a number of publications. When selecting a species of animal for use, rats were most commonly selected with ∼92% of respondents selecting “frequently” or “always.” (Figure 6) Dogs and nonhuman primates were the next most commonly selected with ∼65% selecting “frequently” or “always” for both species (Figure 6). Ferrets and hamsters were the least selected animal with ∼80% and ∼73% selecting “never,” respectively. Survey findings regarding the range of animal species selected and recovery animal inclusion in toxicology studies. Responses (% of total; n = 239) provided in the figure represent the proportion of participants that selected each category.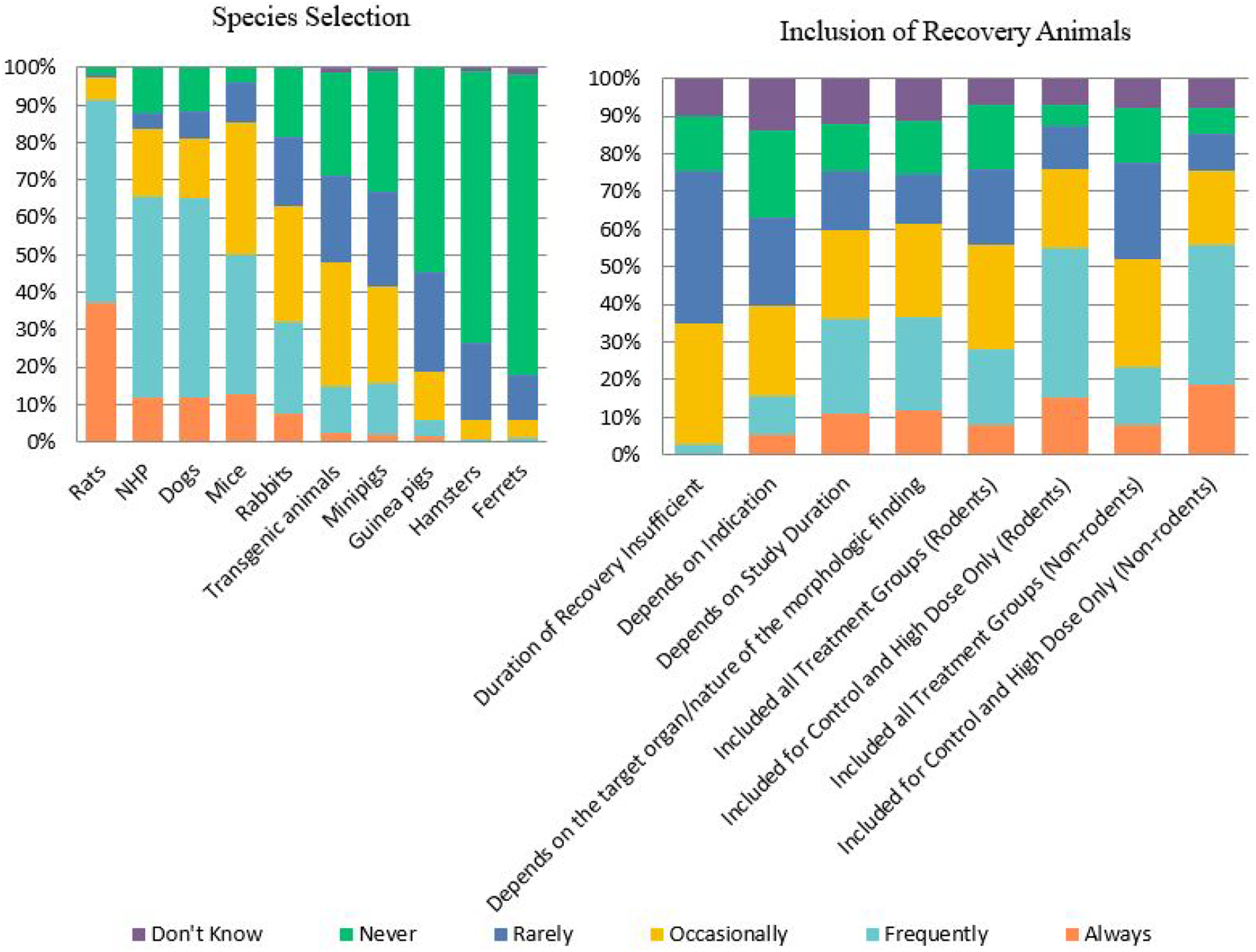
The use of recovery animals was highly variable, but recovery animals were generally included for control and high dose groups only, ∼40% of respondents “Frequently” included recovery animals in rodent studies and ∼37% in non-rodent studies (Figure 6). Approximately one-third of the respondents (32%) indicated that the duration of recovery was occasionally insufficient. The duration of the study and the target organ/nature of the morphologic finding identified were more frequently reported as factors that affected the duration of the recovery than the indication.
Drug administration was “Frequently” performed via oral gavage (59%) or intravenous bolus (∼46%) (Figure 7). Less common, but at least occasionally used by the majority of respondents, were subcutaneous, bolus, and continuous intravenous (IV) infusion methods. Intramuscular (IM) injection and dermal (topical) dose administration were at least occasionally used by almost half of the respondents. In addition, the majority of respondents had some experience over the past 5 years with intraperitoneal, intradermal, intranasal, inhalation, subcutaneous infusion (osmotic mini-pump), and oral (diet) routes. In contrast, over 60% of respondents reported never using epidural, intra-articular, intracerebral, intra-rectal, intrathecal, intravitreal/subretinal, ocular instillation (topical), and otic (ear) instillation as routes of administration. Additional routes indicated in the free text response included oral (via drinking water), subconjunctival (drug/device), implant (venous, IM, heart), intravaginal, and trans-tympanic/transbullar. Route(s) of drug administration used in toxicology studies. Responses (% of total; n = 240) provided in the figure represent the proportion of participants that selected each category.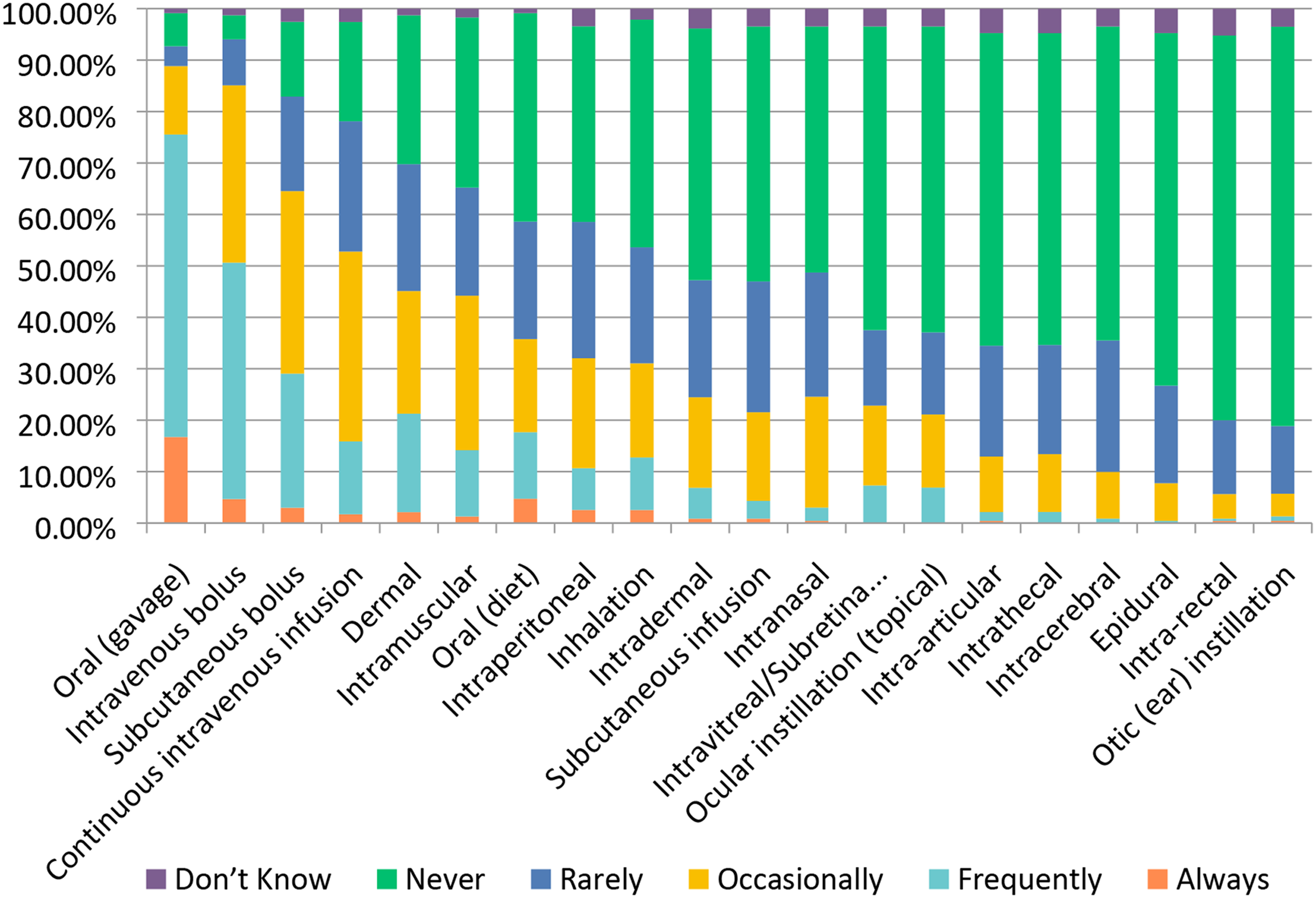
Dose level selection for pivotal GLP studies can include a number of factors, 5 of which were specifically surveyed. Dose range finding studies were often used with ∼56% of respondents selecting “Always” and ∼32% selecting “Frequently.” (Figure 8) The maximum tolerated dose and 10-fold safety margin based on projected human dose were also commonly selected for high doses with ∼45% and ∼39% selecting “Frequently,” respectively (Figure 8). Both the maximum feasible dose and the limit doses, as defined by ICH M3 (R2), are also utilized for high dose selection, but less consistently across responders. Other considerations indicated in the free-text response included an additional cushion over the anticipated targeted safety factor, a literature basis, and, for vaccine programs, dosing up to the clinical dose. Dose Selection for GLP Studies. Responses (% of total; n = 238) provided in the figure represent the proportion of participants that selected each category.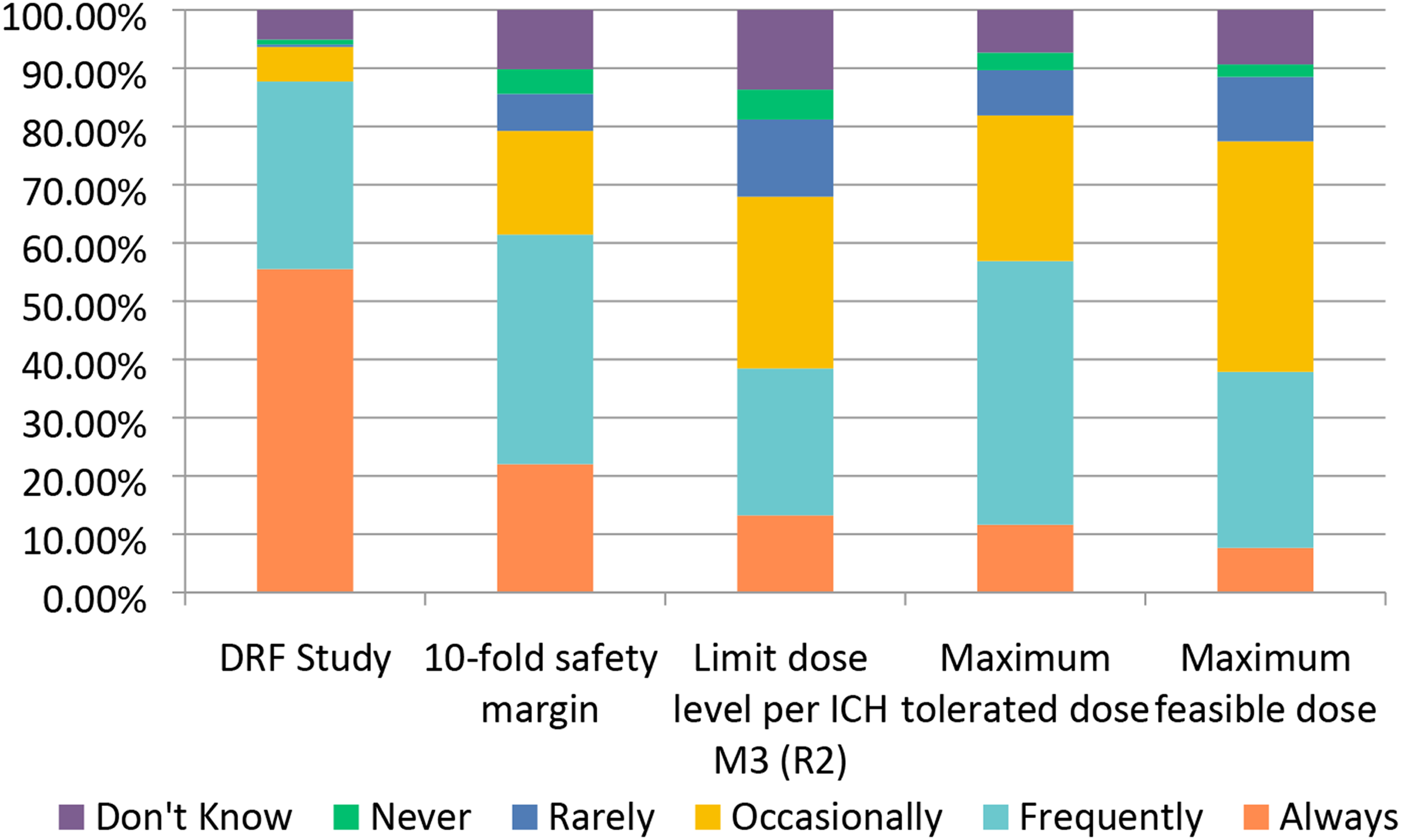
Histopathology is a key endpoint in nonclinical toxicology studies. There are multiple approaches to assessment of histopathology in toxicity studies, none of which are uniformly applied by the pool of respondents in this survey
12
(Figure 9). Some respondents indicated that they always use a 5-point grading scale, while others always use a 4-point grading scale, and the majority use a mixture of the two. Likewise, the use of pathology peer review was also variable with ∼49% of respondents “Always” used peer review for definitive (GLP or OECD) studies but only ∼12% selected “Always” for the use of peer review in exploratory toxicity studies (Figure 9). However, other queries demonstrated a clearly preferred response. For example, more than half of respondents indicated that histopathology findings “Frequently” or “Always” determine the NOAEL, “Frequently” or “Always” are categorized as adverse or non-adverse, and “Frequently” or “Always” are read to a no-observed effect level (NOEL). When it comes to reporting, approximately 53% of respondents selected “Always” or “Frequently” for the use of standardized SEND nomenclature for histopathology. Histopathology in Toxicology Studies. Responses (% of total; n = 239) provided in the figure represent the proportion of participants that selected each category.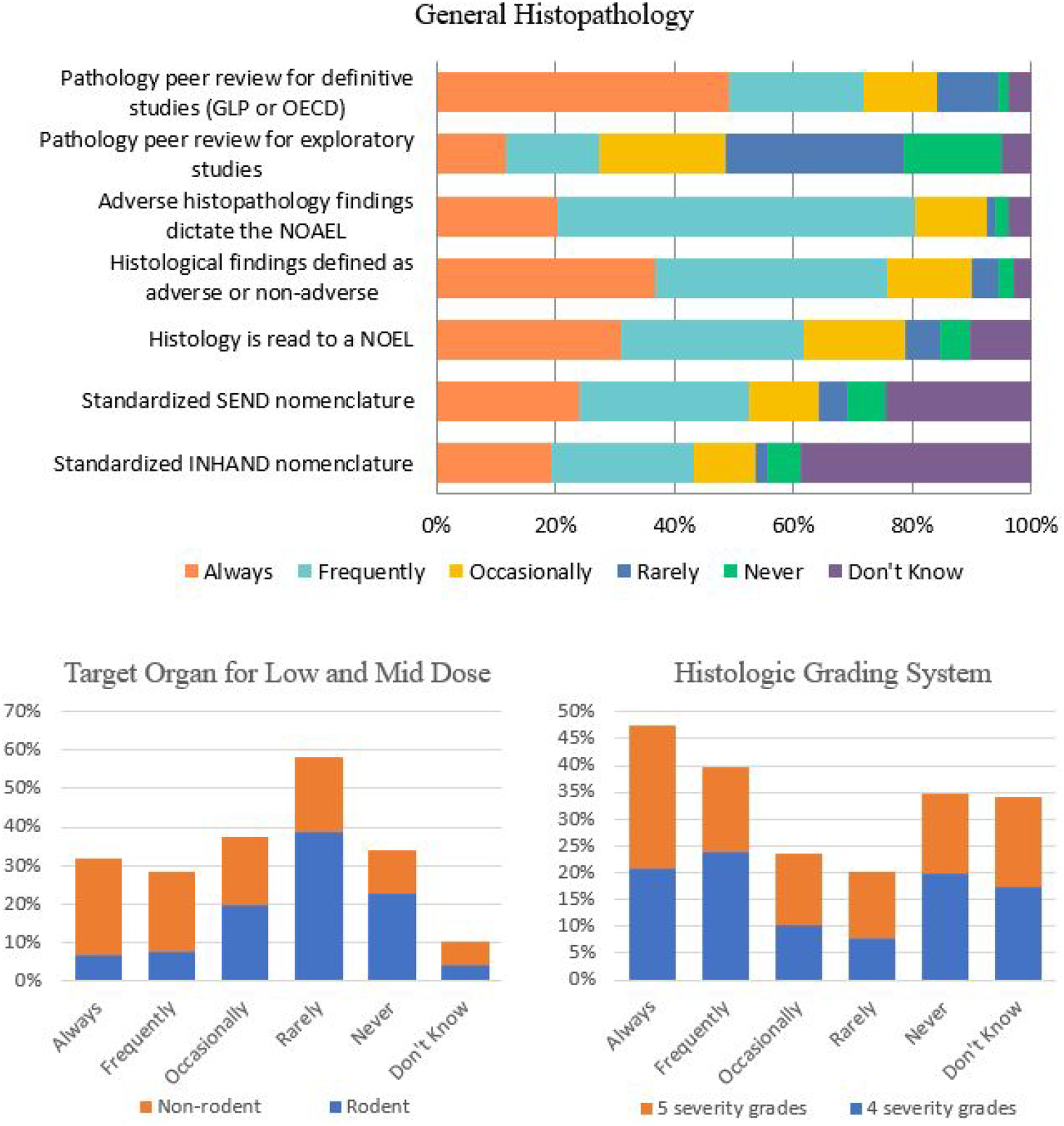
With the development of many new candidate molecules, there is an expectation of toxicity either related to the drug target (i.e., exaggerated pharmacology) or off-target effects. However, there are also examples of highly targeted and disease-specific candidate therapeutics that are not expected to demonstrate toxicity in healthy animals, or where there is no dose dependency of effects once pharmacologic effects are saturated. As shown in Figure 10, in this survey the toxicity most often reported by respondents was hepatotoxicity with ∼37% selecting “Frequently.” Hepatotoxicity is a toxicity that can be associated with either drug termination or market withdrawal. Gastrointestinal toxicity (∼69%), hematopoietic toxicity (∼67%), renal toxicity (∼61%), cardiac toxicity (∼53%), and neurotoxicity (∼45%) account for a large proportion of responses. Not surprisingly, immunotoxicity (∼61%) was also highly selected and likely reflects the shift in industry focus towards the development of oncology drugs that are comprised of biologics and antibody-based therapies.
13
Survey respondents reported that ototoxicity is rarely or never seen in part because it is not specifically evaluated in most study paradigms. Drug-Induced Toxicity Encountered in Testing (2012-2017). Responses (% of total; n = 482) provided in the figure represent the proportion of participants that selected each category.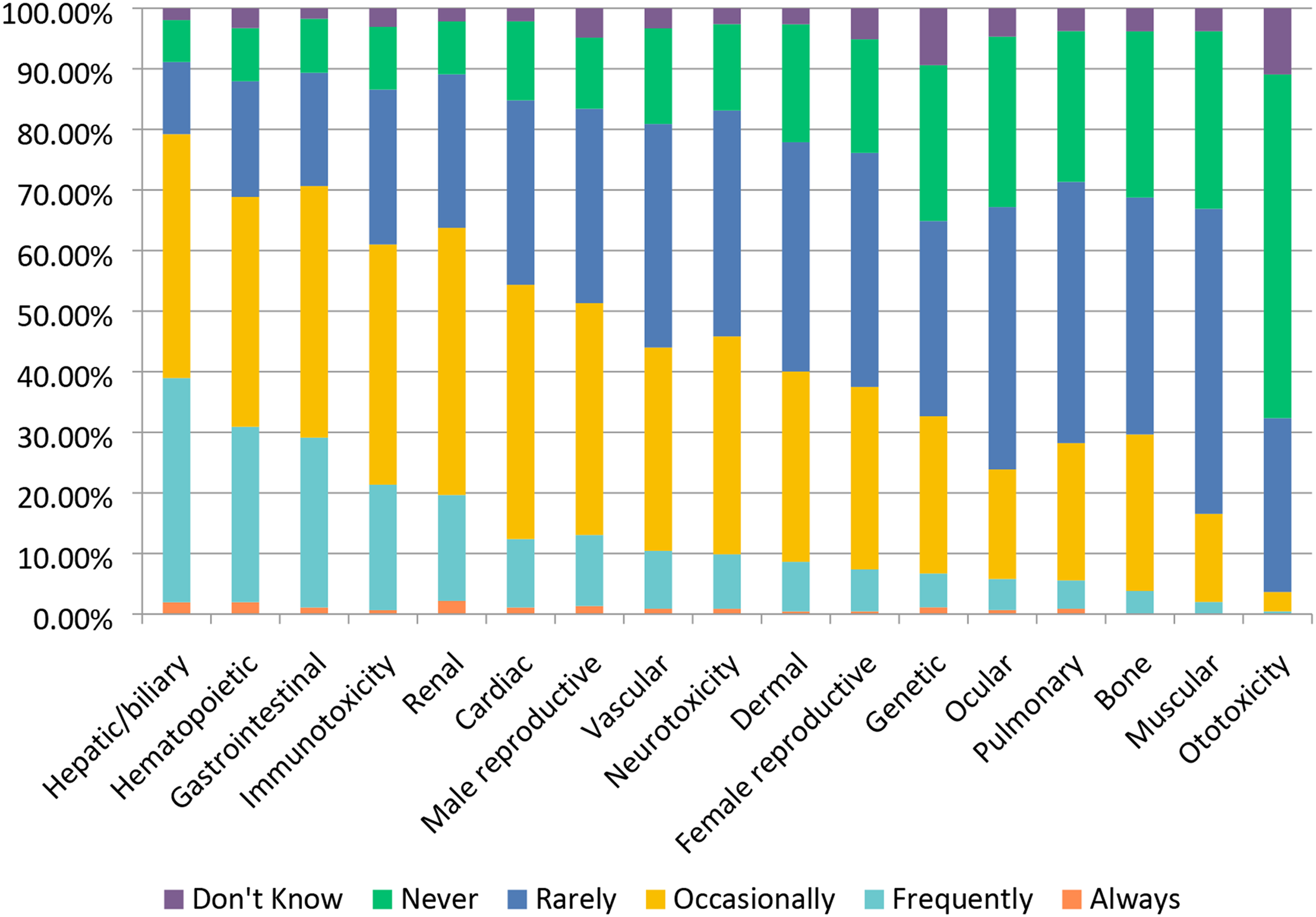
Specific assays and biomarkers are often used to measure toxicity in specific organ systems (Figure 11). The cardiovascular system, kidney, and liver all have distinct biomarkers that can be measured through various assays to determine toxicity. Common cardiovascular assays and biomarkers used by survey respondents included troponins ∼62%, the use of telemetry implants ∼54%, jacketed ECG ∼49%, and echocardiography ∼39%. Common renal biomarkers include creatine ∼84%, urinalysis ∼81%, albumin ∼81%, total protein ∼75%, and KIM-1 ∼47%. Commonly used liver biomarkers apart from standard clinical pathology parameters of ALT/AST, included bile acids (total or separated) ∼61%, glutamate dehydrogenase (GLDH) ∼39%, and sorbitol dehydrogenase ∼39%. An assessment of specific cardiovascular, renal and liver biomarkers of toxicity. Responses (% of total; n=226 for liver biomarkers, n = 231 for Renal biomarkers and n = 228 for Cardiac/vascular biomarkers) provided in the figure represent the proportion of participants that selected each category.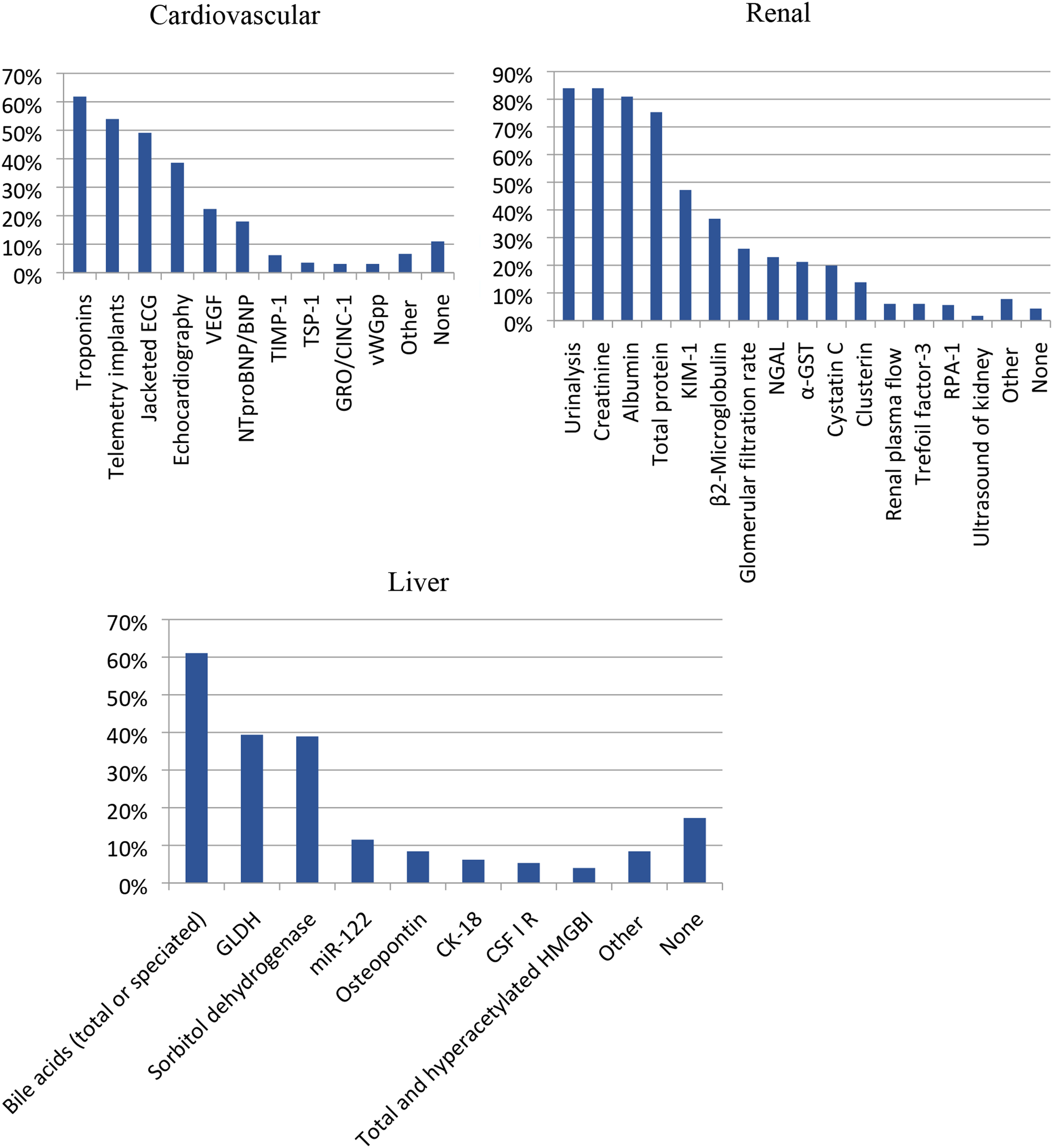
Specific Study Designs Used in Safety Evaluation
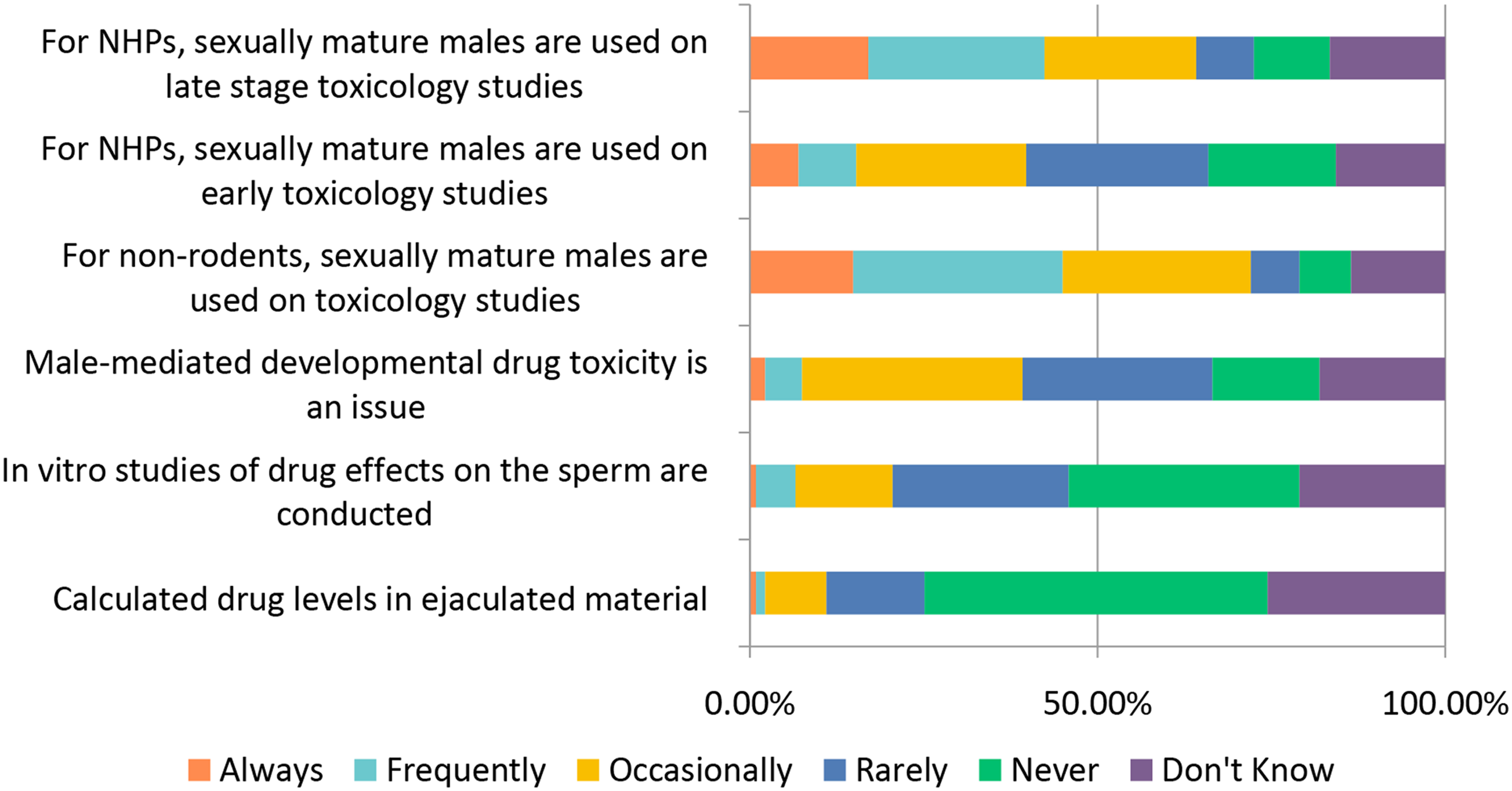
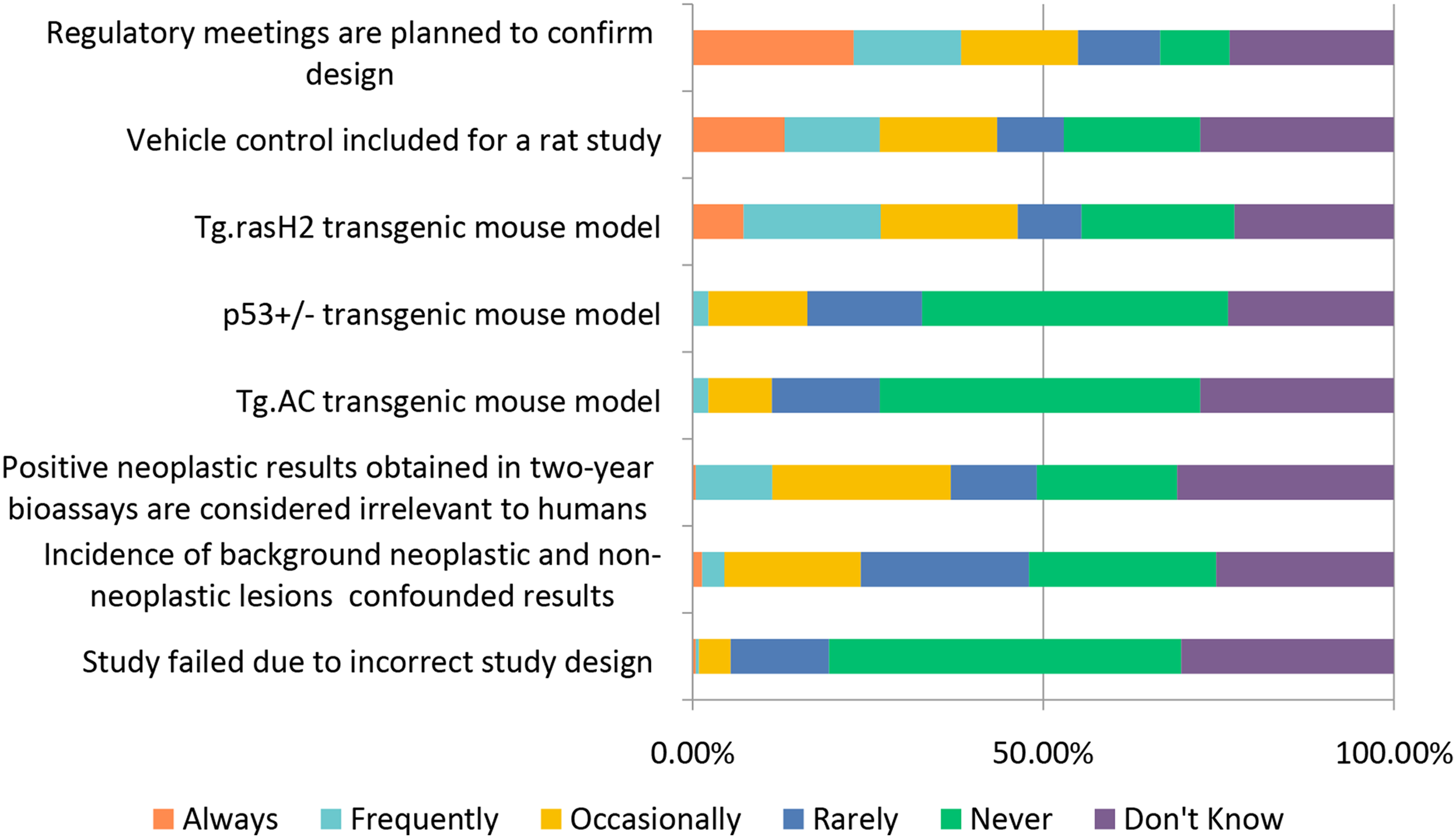
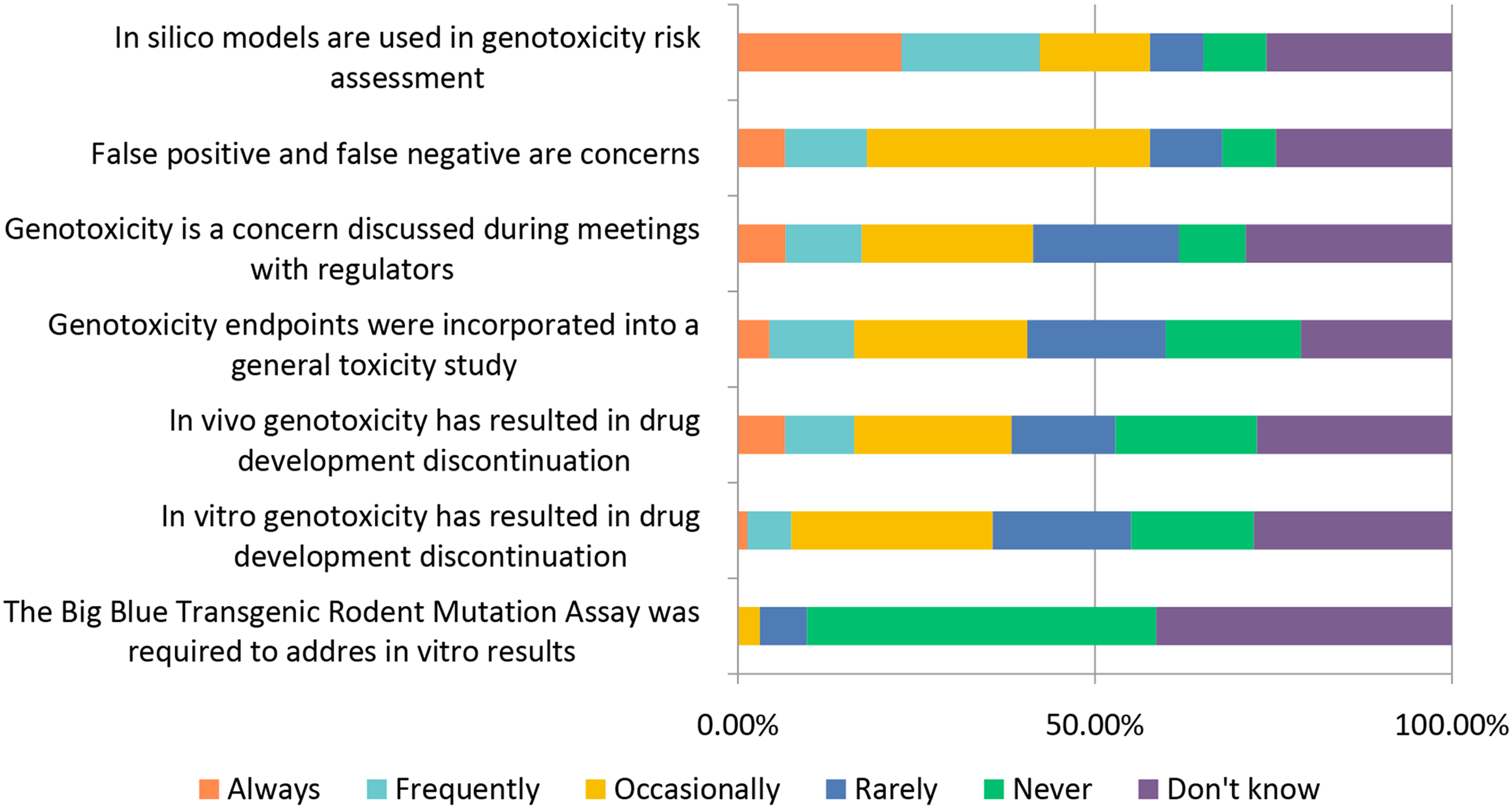
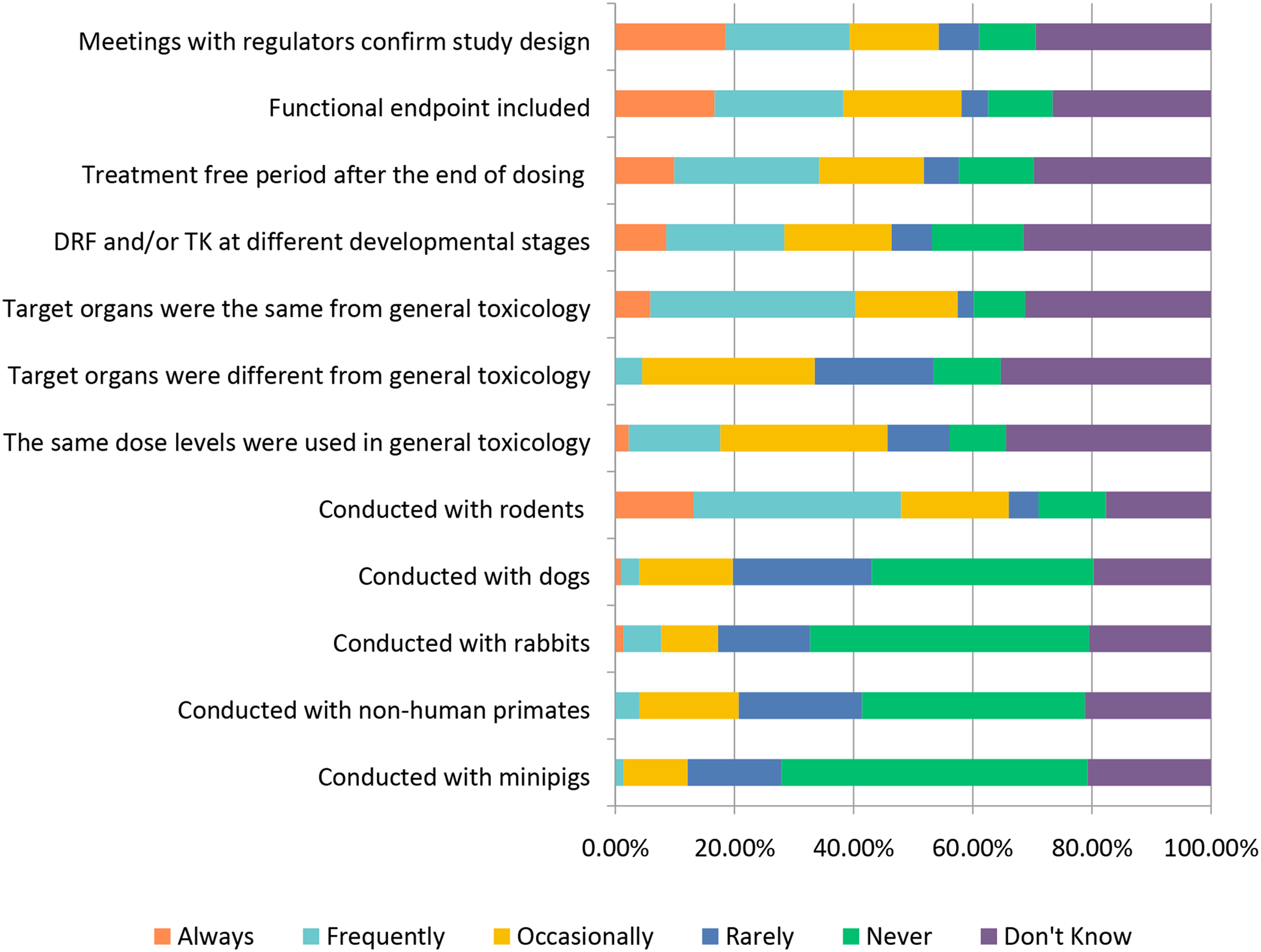
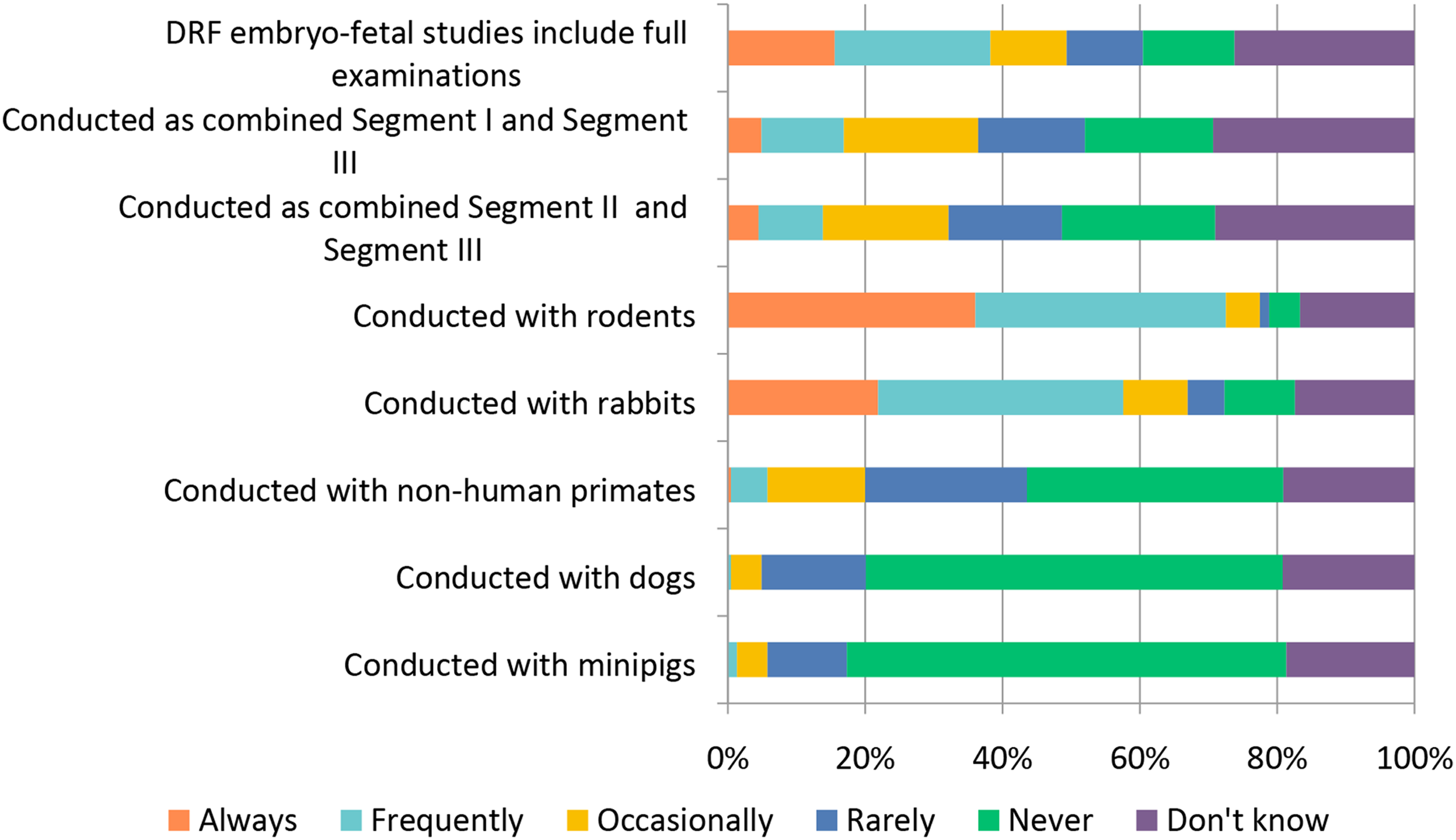
The general toxicology study designs covered above (selection of: species, recovery animals, route of administration, dose, histopathology, and biomarkers) are widely used in “general” toxicology studies to test new candidate molecules; however, there are also special study designs to consider when molecules are targeting a specific population (WOCBP, pediatric populations) or organ system (e.g., requiring more specialized interrogation). Studies requiring special design include endocrine, reproductive, developmental, juvenile, carcinogenicity, and genotoxicity (Figures 12–17). Study design characteristics for endocrine-related drug toxicity. Responses (% of total; n = 229) provided in the figure represent the proportion of participants that selected each category. Developmental drug toxicity in males. Responses (% of total; n = 231) provided in the figure represent the proportion of participants that selected each category. Study design characteristics for carcinogenicity studies. Responses (% of total; n = 222) provided in the figure represent the proportion of participants that selected each category. Study design characteristics for genotoxicity studies. Responses (% of total; n = 228) provided in the figure represent the proportion of participants that selected each category. Study design characteristics for juvenile toxicity studies. Responses (% of total; n = 223) provided in the figure represent the proportion of participants that selected each category. Study design characteristics for reproductive toxicity studies. Responses (% of total; n = 225) provided in the figure represent the proportion of participants that selected each category.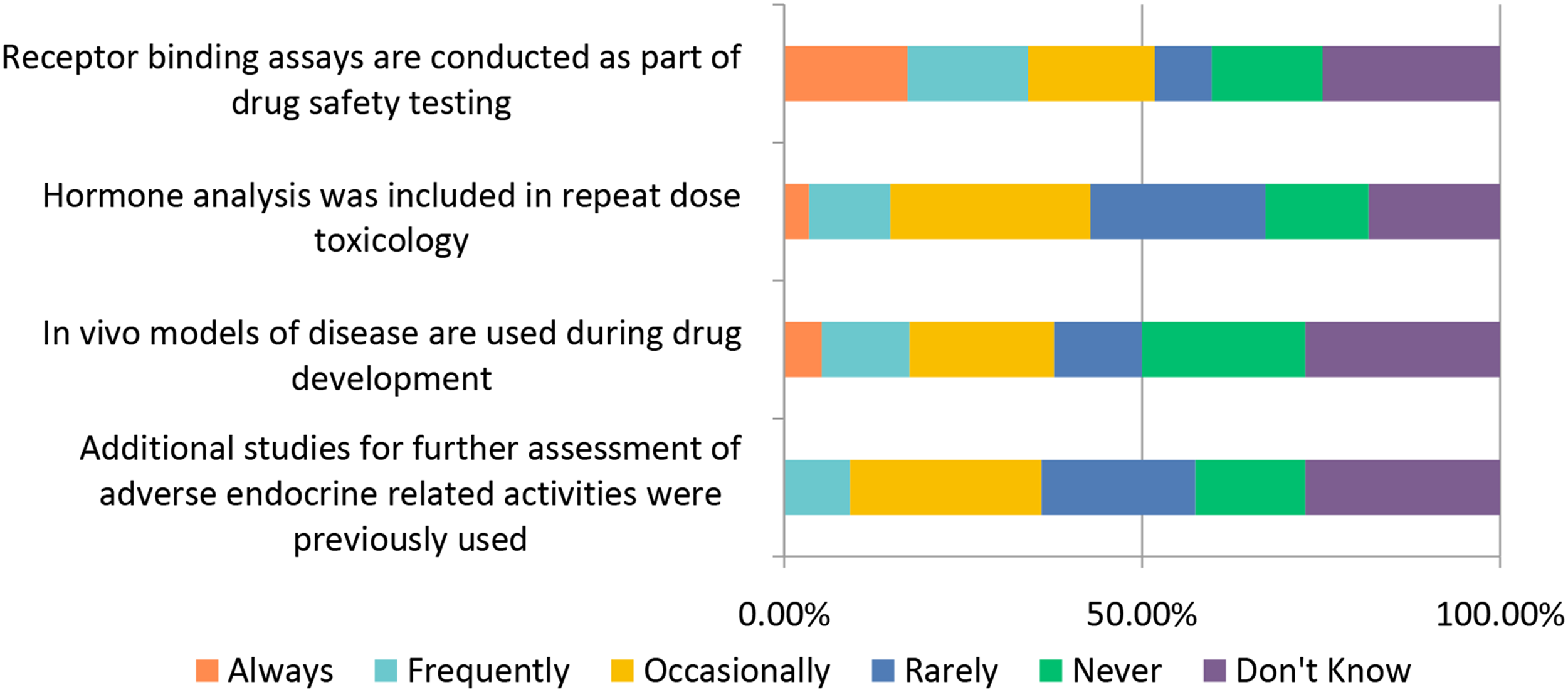
For endocrine system testing studies, respondents to the survey reported using receptor binding assays “Always” or “Frequently” ∼34% of the time and included hormone analysis in repeat dose toxicology “Occasionally” ∼28% of the time.
When testing male-mediated developmental drug toxicity, the majority of respondents indicated that male-mediated developmental drug toxicity is “Never” or “Rarely” an issue (∼42%) while 32% indicated the issue arose “Occasionally” (Figure 13). When conducting non-rodent toxicology studies, ∼45% of respondents indicated they “Frequently” or “Always” included sexually mature males. The responses were similar when asked whether sexually mature male non-human primates were used in late-phase toxicology studies. However, for early-phase toxicology studies, sexually mature male non-human primates were usually not used: ∼69% responded “Never/Rarely/Occasionally” and only ∼15% responded “Frequently” or “Always.” Other kinds of analyses (drug levels in ejaculate or in vitro analysis of sperm) are relatively infrequently conducted.
When carcinogenicity testing is warranted, the program usually includes a 2-year rat study as well as a 2-year wild type or 6-month transgenic mouse study. When planning carcinogenicity studies, ∼38% of respondents said they “Always” or “Frequently” meet with regulators to confirm the study design (Figure 14). When a transgenic mouse assay was conducted for carcinogenicity assessment, the Tg.RasH2 transgenic mouse model was the most frequently used
14
with approximately 39% of respondents “Frequently” or “Occasionally” using this mouse model while only ∼11% used the Tg.AC transgenic mouse model, and ∼16% used the p53+/− deficient mouse model. This is not surprising as the Tg.rasH2 transgene is more widely expressed than the Tg.AC transgene; the Tg.AC mouse model is more commonly used in dermal studies.
15
The Tg.rasH2 model also has a low rate of spontaneous tumors and is less prone to false positives.
16
All three transgenic mouse models can be acceptable for investigating carcinogenicity under the ICH S1B guidelines. Further, it was unusual for a carcinogenicity study to fail due to study design issues as 50% responded “Never,” 18% responded “Rarely” to “Occasional” and only 1% responded “Frequently” to “Always.”
When it comes to genotoxicity studies, ∼23% of respondents report “Always” using in silico modeling, a number slightly higher than what was reported for early stage toxicity studies for NCEs, at ∼20% (Figure 15). A similar percentage of respondents (∼40%) indicated that genotoxicity endpoints are “Occasionally” to “Always” incorporated into general toxicity studies as those responding “Never” to “Rarely.”
Juvenile toxicity studies are considered under the special study design topic due to the target (infant and/or pediatric) population; however, historically the study design has generally resembled that of general toxicity studies. Approximately 34% of respondents selected “Frequently” when asked if the target organs identified were the same as for adults (Figure 16). Similar to chronic toxicology studies, ∼39% of respondents selected “Frequently” or “Always” meeting with regulators when designing juvenile toxicity studies and these studies are most often conducted using rodents, with ∼35% selecting “Frequently.” The authors anticipate that approaches to juvenile animal studies are likely to shift somewhat with the recent completion of the ICH S11 Guideline on Nonclinical Safety Testing in Support of the Development of Pediatric Medicines (2020), which includes study design and endpoint recommendations17.
Reproductive toxicity studies are most commonly conducted in rodents with ∼79% selecting “Frequently” or “Always.” For those respondents providing a numerical response to the question, most respondents indicated that the number of females used in the conduct of rodent reproductive toxicity studies is in the range of 20-26 (Table 2). Rabbits are also commonly used with ∼57% selecting “Frequently” or “Always” (Figure 17). The combination of either Segment 1 (fertility and early embryonic development) or 2 (embryo-fetal development) studies with Segment 3 (peri- and postnatal development) studies was not common; only ∼14-16% responded this is done “Frequently” to “Always” while ∼35% indicated this was done “Rarely” to “Occasionally” and ∼19-22% selecting “Never.” The Number of Female Rodents Used for Reproductive Toxicity Studies (n = 158). aThe range of group sizes used in reproductive toxicology studies.

As noted above regarding juvenile toxicity, the practices for reproductive and developmental toxicity testing are likely to be influenced by the recent revision of the ICH S5 guideline.
Study Designs of NCEs vs Biologics
Historically, the bulk of drug development has been targeting NCEs, however, in recent years the development of biologics has increased progressively and now makes up ∼25% of new drugs approved in 2020. 2 While both NCEs and biologics have the potential to produce effective drug therapies, they work through different mechanisms of action and can elicit different responses within the organism. For example, due to the size and nature of the compound, biologics are much more likely to cause an immune reaction and induce the production of anti-drug antibodies (ADA). Because of these kinds of differences, the study design for testing toxicity of an NCE versus a biologic should be able to adequately characterize the toxicity associated with each compound. Specific guidance documents are also available to guide the safety assessment of biologics (ICH S6 for biotechnology derived pharmaceuticals).
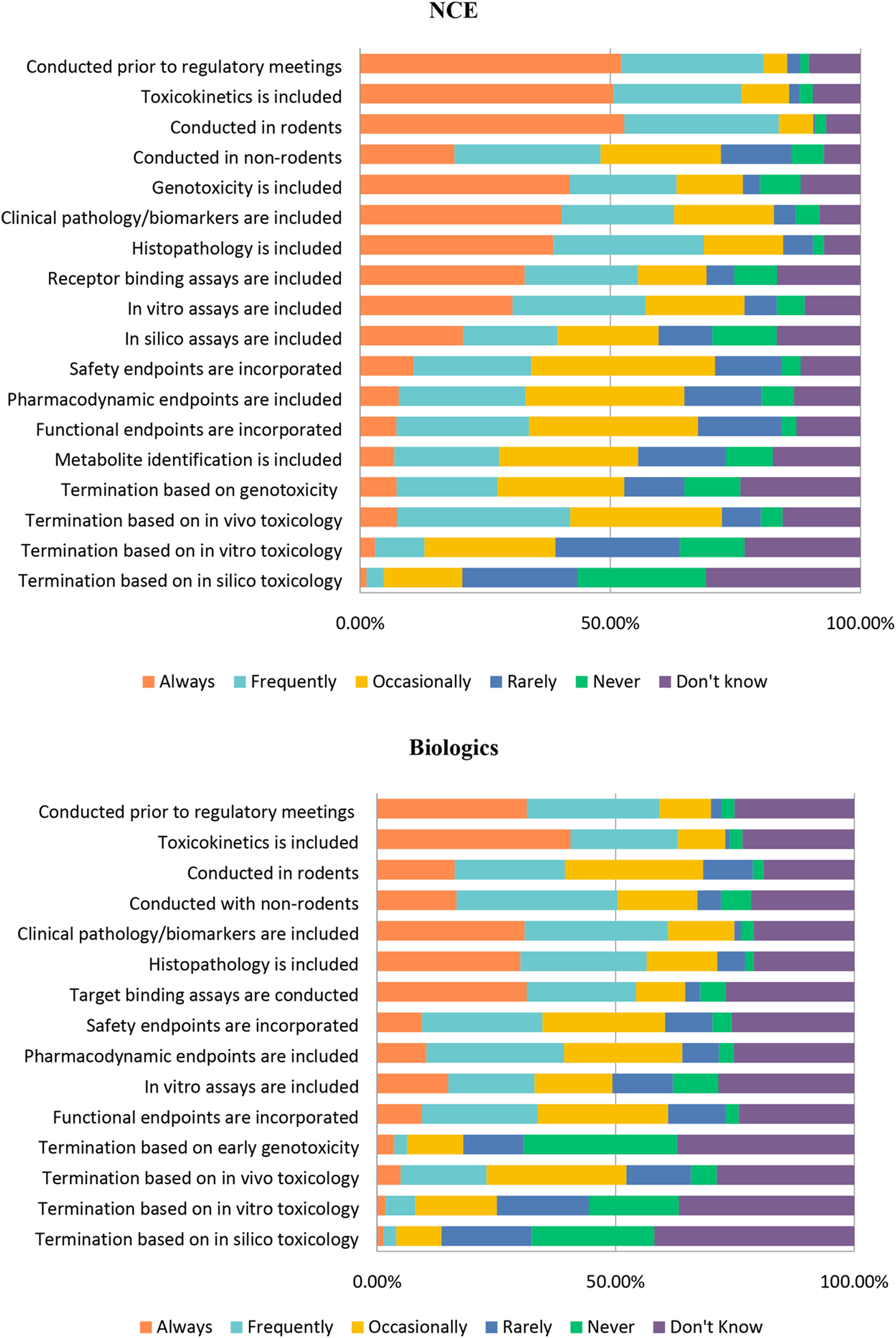
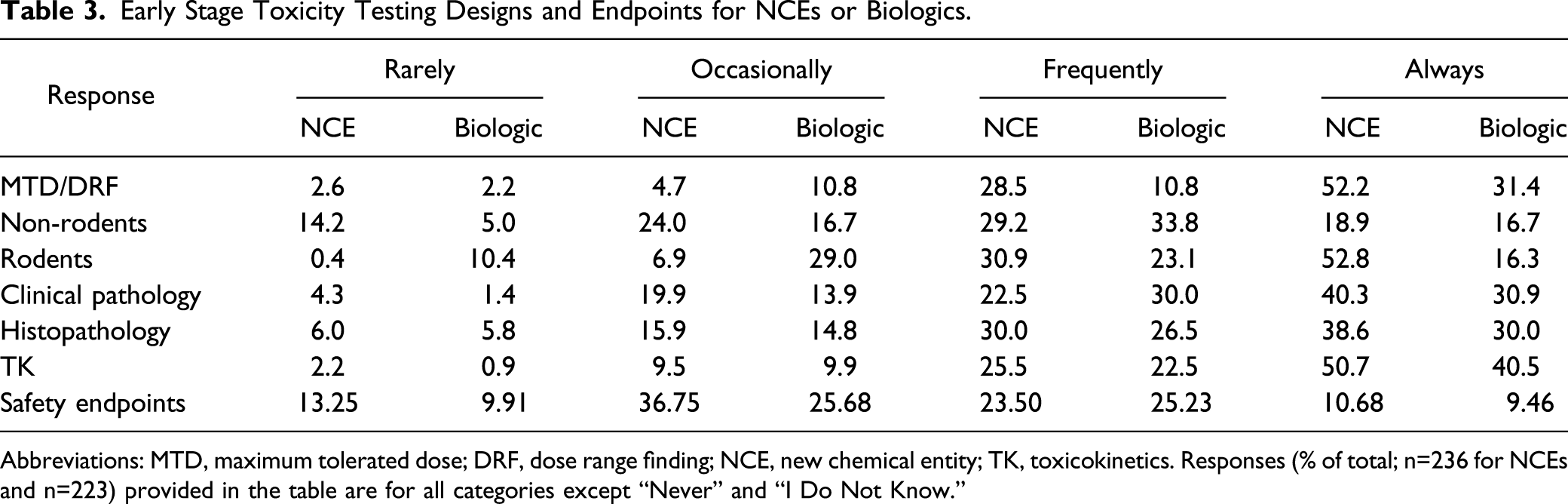
In order to parse out the similarities and difference between the two types of compounds, we asked a series of questions comparing various parameters of study design for NCEs and biologics (Figure 18). For early toxicology studies, whether for NCEs or biologics, respondents confirmed these studies were used for identifying a maximum tolerated dose (MTD) as part of range finding studies in both rodents and non-rodents. In addition, many of these early studies included histopathology, clinical pathology, and toxicokinetics (Table 3). There are many similarities between the responses for NCEs and biologics regarding the use of rodents and non-rodents; however, it is interesting to note that a much higher percentage of respondents selected “Always” for NCEs compared with biologics when it comes to the use of rodents (Figure 18). Importantly, whether using rodents or non-rodents, most respondents included clinical pathology, histopathology, toxicokinetics, and genotoxicity (NCEs only) in early toxicology studies. Safety endpoints were reported to be frequently or always included in efficacy studies by 34% of respondents. Early stage toxicity designs and study endpoints for NCEs or Biologics. Responses (% of total; n = 236 for NCEs and n = 223 for Biologics) provided in the figure represent the proportion of participants that selected each category. Early Stage Toxicity Testing Designs and Endpoints for NCEs or Biologics. Abbreviations: MTD, maximum tolerated dose; DRF, dose range finding; NCE, new chemical entity; TK, toxicokinetics. Responses (% of total; n=236 for NCEs and n=223) provided in the table are for all categories except “Never” and “I Do Not Know.”
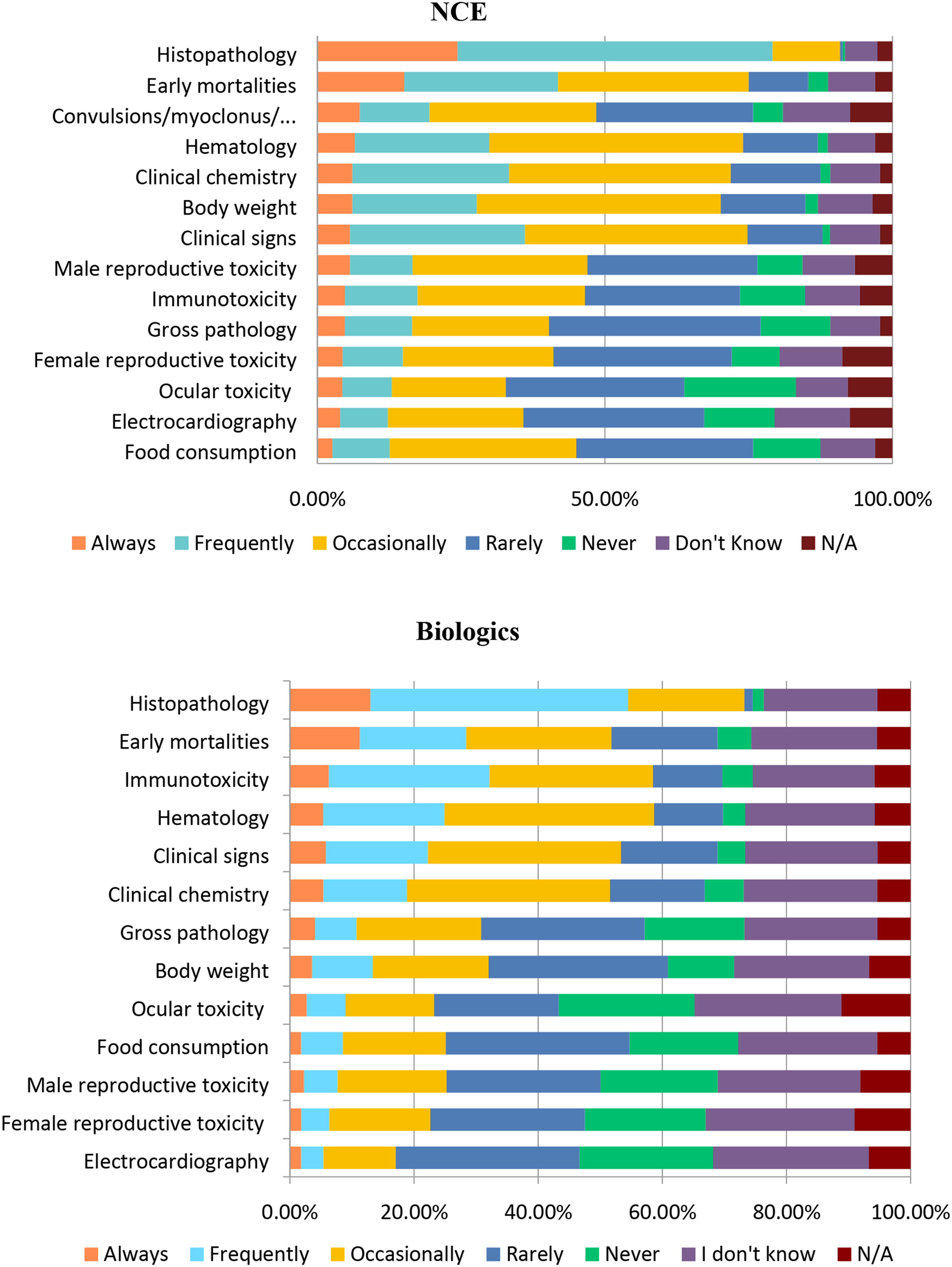
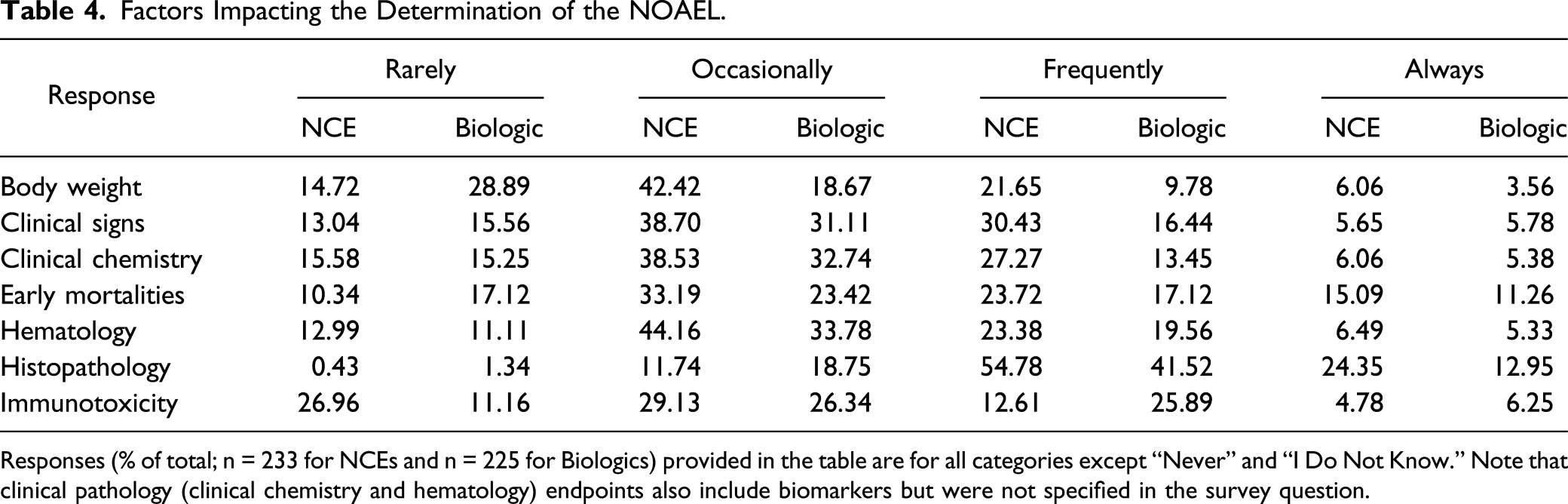
There are also similarities when it comes to determining the NOAEL for both NCEs and biologics (Figure 19). Histopathology was an important determinant of the NOAEL for both NCEs and biologics with ∼55% and ∼42%, respectively, selecting “Frequently” (Figure 19). An important difference to note is that respondents selected “Frequently” less often when it comes to using body weight, clinical signs, and clinical chemistry for determining the NOAEL for biologics. Not surprisingly, immunotoxicity was an important determinant of the NOAEL in many biologics, with approximately twice as many respondents selecting “Always” or “Frequently” compared to NCEs (32 vs 17%) and ∼27% of respondents reported “Rarely” using immunotoxicity for NCEs (Table 4). This result was expected as many biologics are immunomodulatory in nature. In addition, biologics administered in toxicity studies can often elicit an immune response to the xenoprotein, and therefore immunogenic potential and monitoring needs to be considered when designing the study. Factors that may impact determination of the NOAEL for NCEs or Biologics. Responses (% of total; n = 233 for NCE and n = 225 for Biologics) provided in the figure represent the proportion of participants that selected each category. Factors Impacting the Determination of the NOAEL. Responses (% of total; n = 233 for NCEs and n = 225 for Biologics) provided in the table are for all categories except “Never” and “I Do Not Know.” Note that clinical pathology (clinical chemistry and hematology) endpoints also include biomarkers but were not specified in the survey question.
Clinical Pathology Parameters Associated With Inflammation.
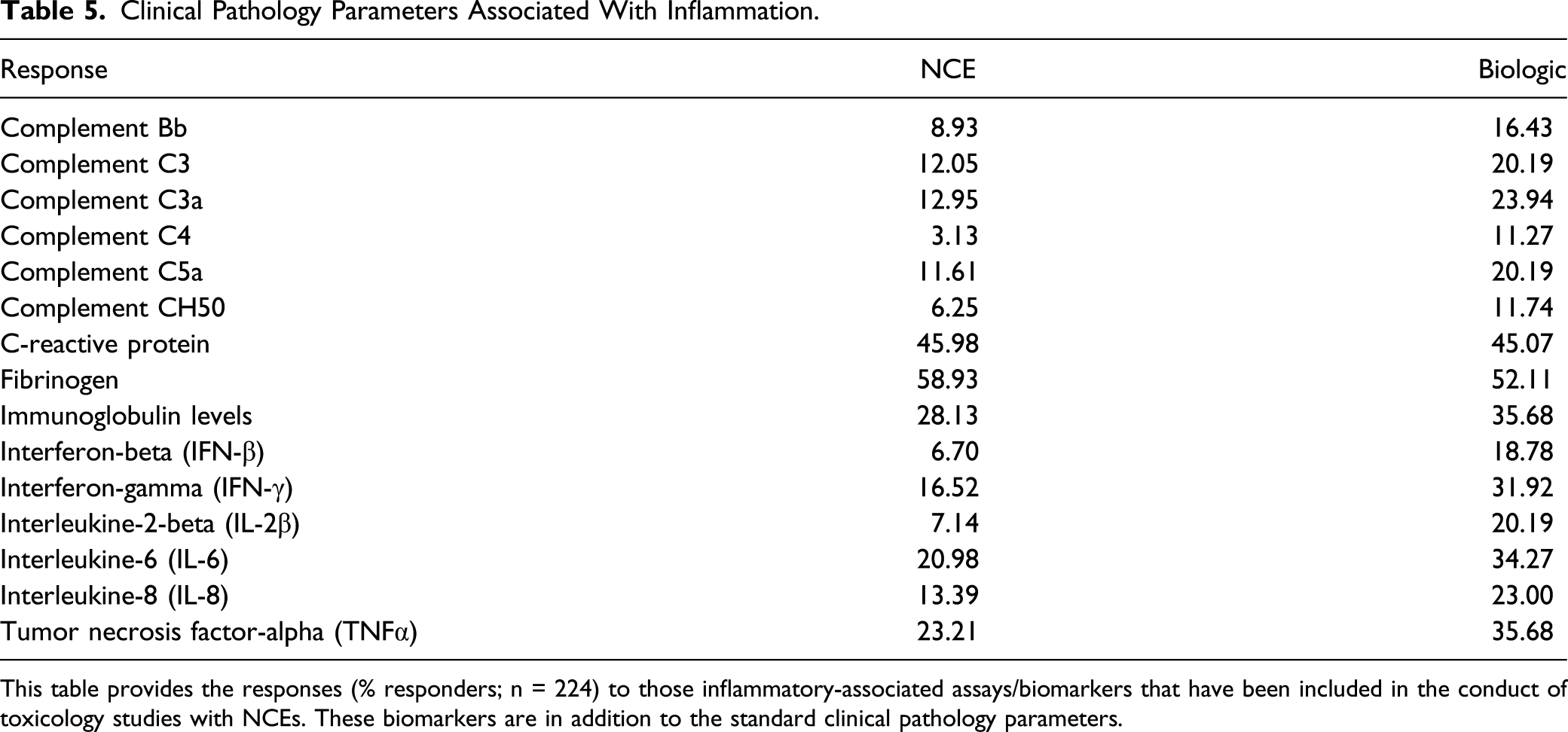
This table provides the responses (% responders; n = 224) to those inflammatory-associated assays/biomarkers that have been included in the conduct of toxicology studies with NCEs. These biomarkers are in addition to the standard clinical pathology parameters.
Challenges in Study Design
With selection of a lead compound, the challenges of drug development are just beginning. An initial challenge for toxicologists is delivering the drug to the experimental model at sufficiently high doses to induce toxicity. Accordingly, dose level selection was the most frequently encountered challenge, followed by compound solubility and availability in the appropriate vehicle (Figure 20). Stability of the compound within the dosing formulation was also selected “Occasionally” as a problem by ∼42% of respondents. Similarly, nearly half of respondents cited problems with the test article being the main cause for delays in toxicology programs. Additional reported challenges included the need for novel excipients for a proposed route of administration, a lack of stability for biologics, the need to deliver very low concentrations of protein, physical instability of suspensions, scheduling capacity limitations, and complexities in the development of pediatric (oral/liquid) formulations. Challenges encountered in toxicology study design. Responses (% of total; n = 491) provided in the figure represent the proportion of participants that selected each category.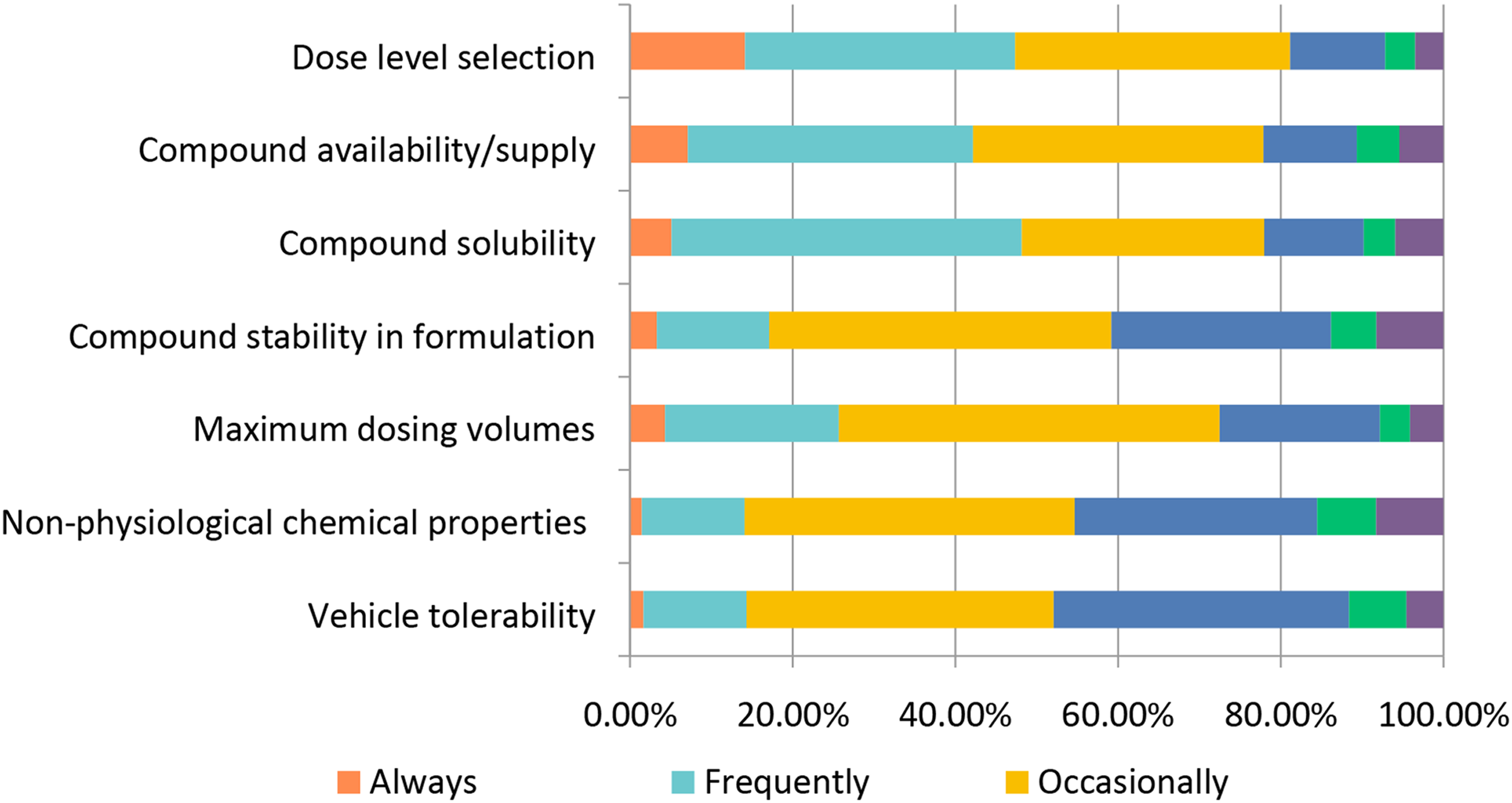
The process of taking a new drug from early discovery through ultimate regulatory approval requires considerable resources including both personnel and financial. From a nonclinical perspective, scientists are continuously challenged to limit the amount of testing necessary and, importantly, determine whether a therapeutic should be developed, that is, if a drug is to fail, that failure should be determined as early as possible. When asked if meetings with regulatory agencies were conducted prior to submission of a First in Human clinical protocol, ∼48% selected “Always” or “Frequently” regarding meetings being routinely conducted (Figure 21). The outcome of these meetings was more varied with ∼38% indicating that they “Occasionally” led to a change in study design and ∼32% selecting “Occasionally” there was conflicting regulatory advice. Regulatory meetings conducted prior to First in Human (FIH) submissions. Responses (% of total; n = 230) provided in the figure represent the proportion of participants that selected each category.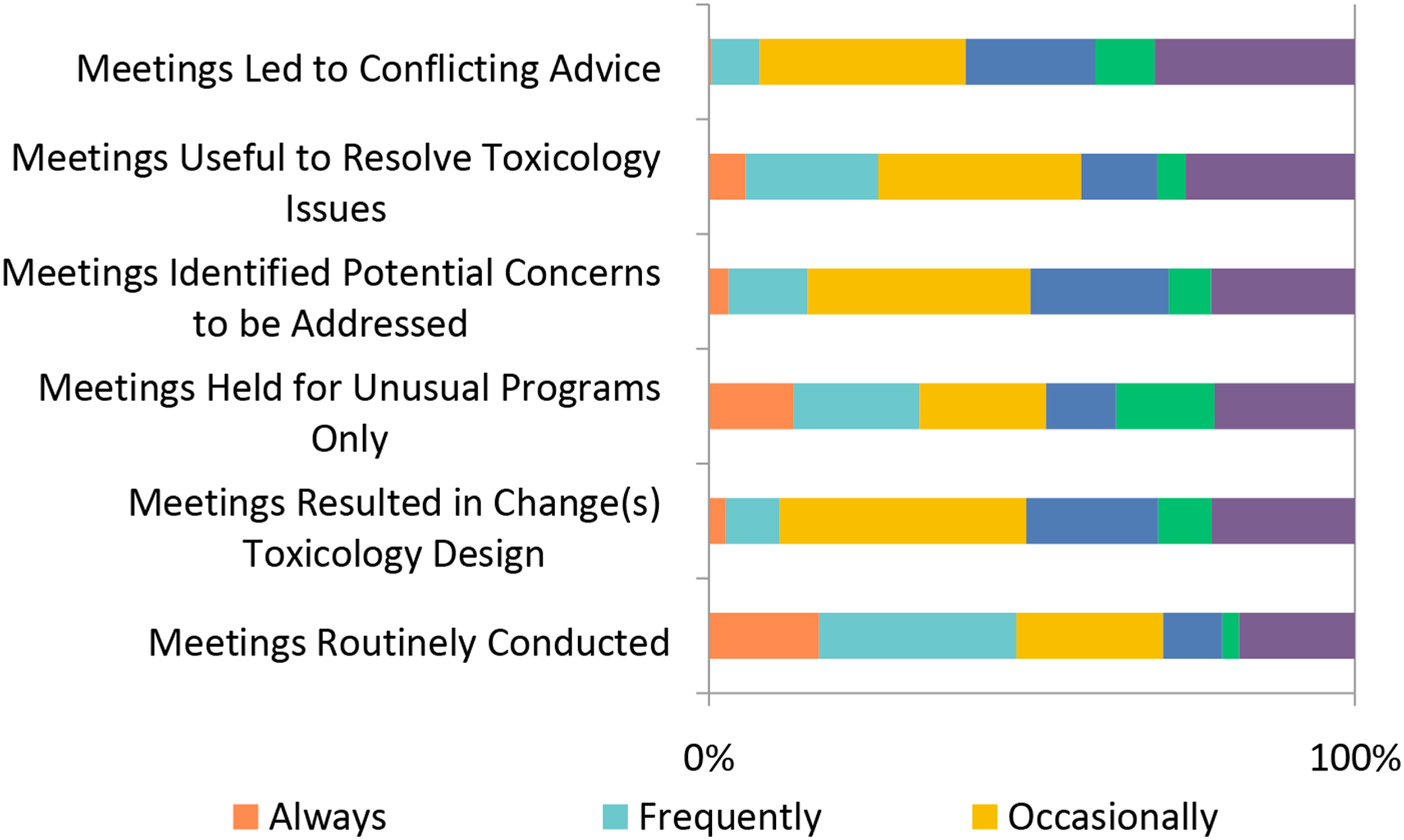
Additionally, the nonclinical industry invests resources into developing new methodologies to assist in the drug testing process. Along with the studies described above, the area of computational toxicology has seen a large expansion in terms of refining methods and alternative in silico models. In silico modeling for genotoxicity endpoints as well as other endpoints for small molecules has become relatively routine, particularly for the evaluation of genotoxic impurities. In this survey, the response to the use of in silico modeling was somewhat surprising. Only about 20% of the respondents “Frequently” or “Occasionally” used in silico models for early testing (Figure 18). The response of “Always” was about 20%. In line with the low number of respondents using in silico models, ∼ 27% of respondents said they only “Occasionally” consider alternatives to animal testing and ∼37% said they “Rarely” or “Never” consider alternatives to animals (Figure 22). The use of in silico models was significantly correlated (Chi-square P<0.001) to the size of the organization, where large organizations (>5000 employees) used in silico models more frequently. Since in silico methods are only one tool within a toolbox of other assays, termination of a compound from development was “Never” or “Rarely” (total ∼49%) based solely on an in silico assessment output. Similarly, a compound was “Rarely” or only “Occasionally” (total ∼37%) terminated based on the results of in vitro testing, for example, genotoxicity. However, based on correlating in vivo study findings, a compound may have been terminated “Frequently” (∼35%). Although a question of early termination based on the combined in vivo and in vitro results was not posed, it is plausible that a combined response would have been high. Although biologics are not usually subjected to in silico modeling, a similar response pattern was observed with in-vivo toxicology accounting for most program terminations for both biologics and small molecules. Collectively, these data demonstrate that drug development scientists take a holistic approach when deciding to pursue a drug for further development. Alternatives to the use of animals in toxicology studies. Responses (% of total; n = 234) provided in the figure represent the proportion of participants that selected each category.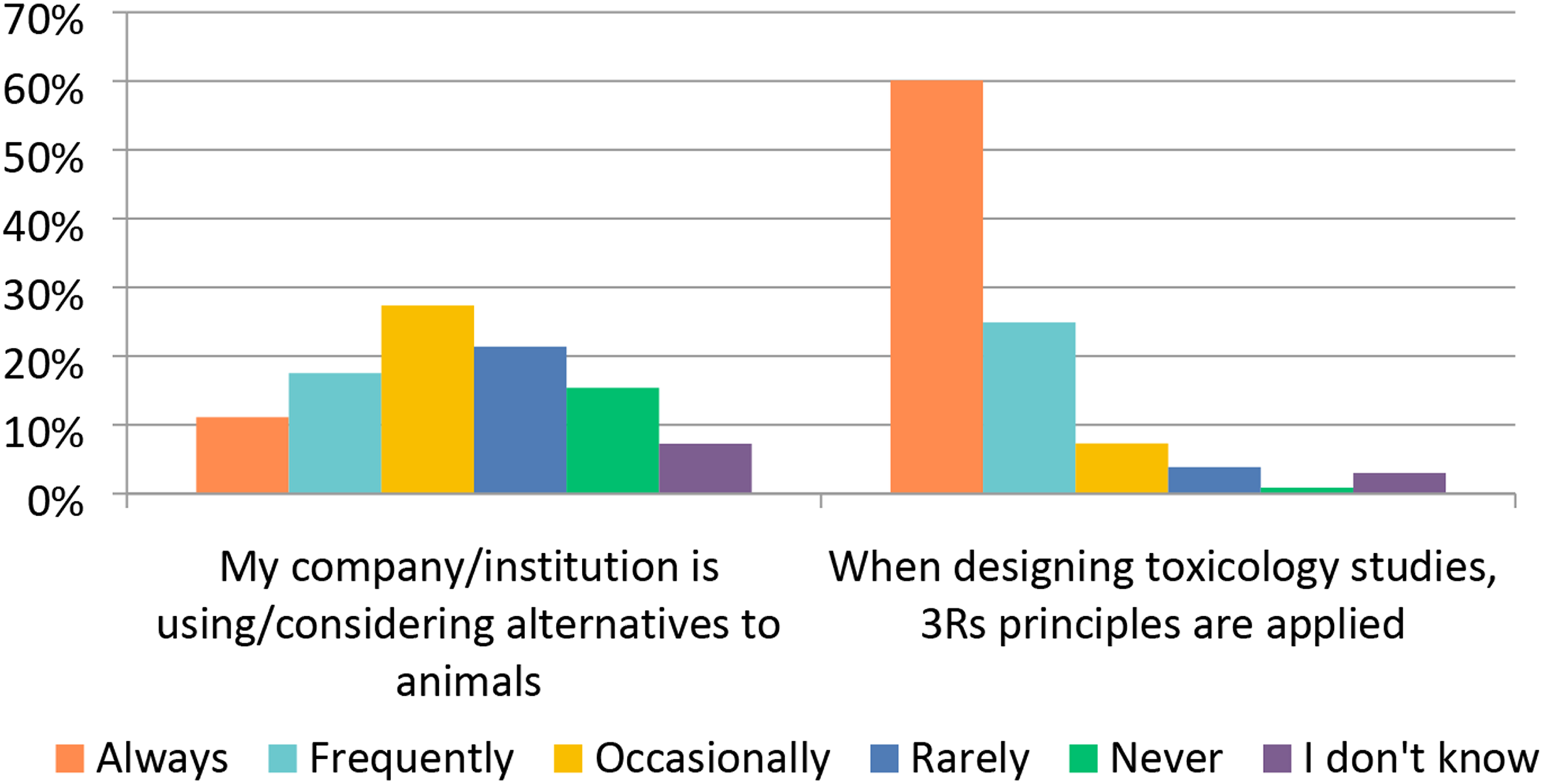
Conclusions
The current survey provides valuable data to help characterize industry practices as they relate to study design in drug safety testing. Strategies in drug safety testing are influenced by therapeutic indications but also by drug classes, regulatory guidelines, historical data, and experience. Sharing common practices amongst the non-clinical drug safety testing community offers the potential to evolve study design approaches and can also contribute to a healthy reflection on the path towards defining the drug safety testing strategies of tomorrow. Similar to previous industry surveys, results confirmed the important proportion of drugs in development for oncology with toxicology pillars including clinical pathology and histopathology that remain stable. The survey highlighted intrinsic differences but also similarities in the testing strategies and challenges that are encountered with NCE and biologics. New areas such as study design, development challenges, and regulatory interactions were explored in this survey and future industry practice examinations will be useful to better characterize drug development trends.
Footnotes
Acknowledgments
The authors wish to thank the Safety Pharmacology Society (SPS), American College of Toxicology (ACT) and Society for Toxicologic Pathology (STP) for support in the conduct of this survey.
Author Contributions
Simon Authier contributed to conception, contributed to acquisition, analysis, and interpretation, and drafted manuscript; William J Brock contributed to conception, contributed to acquisition, analysis, and interpretation, drafted manuscript, and critically revised manuscript; Wendy Halpern contributed to conception and design, contributed to acquisition, analysis, and interpretation, drafted manuscript, and critically revised manuscript; Stephanie N Harris contributed to analysis and interpretation, drafted manuscript, and critically revised manuscript; David Jones contributed to conception and design, contributed to acquisition, analysis, and interpretation, and critically revised manuscript; Timothy McGovern contributed to conception and design, contributed to acquisition, analysis, and interpretation, drafted manuscript, and critically revised manuscript; Pam D McGovern contributed to conception and design, contributed to acquisition, analysis, and interpretation, drafted manuscript, and critically revised manuscript; Michael K Pugsley contributed to conception and design, contributed to acquisition, analysis, and interpretation, drafted manuscript, and critically revised manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Disclaimer
This publication reflects the views of the authors and does not represent views or policies of any organization, including the USDA, US FDA and MHRA.