
Abstract
COVID-19 is a potentially fatal infection caused by the SARS-CoV-2 virus. The SARS-CoV-2 3CL protease (Mpro) is a viral enzyme essential for replication and is the target for nirmatrelvir. Paxlovid (nirmatrelvir co-administered with the pharmacokinetic enhancer ritonavir) showed efficacy in COVID-19 patients at high risk of progressing to hospitalization and/or death. Nonclinical safety studies with nirmatrelvir are essential in informing benefit-risk of Paxlovid and were conducted to support clinical development. In vivo safety pharmacology assessments included a nervous system/pulmonary study in rats and a cardiovascular study in telemetered monkeys. Potential toxicities were assessed in repeat dose studies of up to 1 month in rats and monkeys. Nirmatrelvir administration (1,000 mg/kg, p.o.) to male rats produced transient increases in locomotor activity and respiratory rate but did not affect behavioral endpoints in the functional observational battery. Cardiovascular effects in monkeys were limited to transient increases in blood pressure and decreases in heart rate, observed only at the highest dose tested (75 mg/kg per dose b.i.d; p.o.). Nirmatrelvir did not prolong QTc-interval or induce arrhythmias. There were no adverse findings in repeat dose toxicity studies up to 1 month in rats (up to 1,000 mg/kg daily, p.o.) or monkeys (up to 600 mg/kg daily, p.o.). Nonadverse, reversible clinical pathology findings without clinical or microscopic correlates included prolonged coagulation times at ≥60 mg/kg in rats and increases in transaminases at 600 mg/kg in monkeys. The safety pharmacology and nonclinical toxicity profiles of nirmatrelvir support clinical development and use of Paxlovid for treatment of COVID-19.
Introduction
Coronaviruses, like SARS-CoV-2, the causative agent of the COVID-19 pandemic, are dependent on the activity of the viral protease, Mpro (also known as 3CLpro) for viral replication. 1 The global uptake of prophylactic vaccines has been highly effective in limiting the spread and impact of the pandemic which, combined with hospital-based antibody therapeutics, has proven to be a highly effective disease mitigation strategy. However, despite the advances made in these areas, COVID-19 disease continues to be a global burden and additional therapeutic options, particularly oral antiviral agents, are needed and could be a key solution in addressing the possibility of vaccine resistant strains of SARS-CoV-2. Nirmatrelvir (PF-07321332) is being developed as an oral antiviral therapeutic for COVID-19 and is a potent and selective inhibitor of the SARS-CoV-2 protease, Mpro. 2 With its essential functional importance in virus replication together with the absence of closely related homologues in humans, 3 Mpro is an attractive antiviral drug target. Safety pharmacology and nonclinical toxicology studies are required per ICH M3(R2) to inform the benefit-risk of nirmatrelvir prior to administration to humans in clinical trials as well as in the real-world setting. Paxlovid is a therapeutic containing two molecular components, nirmatrelvir and the marketed drug, ritonavir. Ritonavir is inactive against SARS-CoV-2 Mpro but is co-administered as an exposure enhancer of nirmatrelvir. The use of ritonavir as a pharmacokinetic booster to enable high plasma concentrations of nirmatrelvir is well precedented in other antiviral therapies. 4 In humans, nirmatrelvir is rapidly metabolized by CYP3A, which is expected to lower its systemic exposure. Ritonavir inhibits the CYP3A-mediated metabolism of nirmatrelvir, thereby enabling increased plasma concentrations of nirmatrelvir and aiding to achieve and maintain therapeutically effective exposures. Ritonavir was not included in the nonclinical safety studies as (a) ritonavir is a marketed drug with well characterized nonclinical and clinical safety profile, 5 and no overlapping or additive toxicities between nirmatrelvir and ritonavir are expected; therefore, in accordance with ICH M3(R2) guidelines, 6 combination nonclinical safety studies are unlikely to provide additional information beyond the known individual toxicity profiles of nirmatrelvir and ritonavir; (b) the exposure boosting effects of ritonavir seen in humans do not translate completely in animals; 7 (c) high exposures of nirmatrelvir in nonclinical studies were achieved through use of solvate and formulations approaches; and (d) the duration of clinical therapy is short, ie, less than 10 days which mitigates the risk of duration-related cumulative effects.
We have characterized the nonclinical safety of nirmatrelvir in a series of safety pharmacology and repeat-dose toxicity studies in the rat and non-human primate. This data complements previously published nonclinical assessments of nirmatrelvir including genetic toxicity data. 2 We describe key nonclinical findings and provide context and discussion around the safety profile and justification for clinical use of this potentially life-saving medicine.
Methods
Experimental Standards and Guideline Compliance
All procedures were conducted in compliance with Good Laboratory Practice (GLP) for Nonclinical Laboratory Studies regulations as set forth in the Code of Federal Regulations (21 CFR Part 58). The study standards met acceptance criteria by regulatory agencies for the conduct of clinical trials and the marketing of drugs. Study designs and parameters evaluated in toxicity studies were consistent with accepted principles and practices as outlined in ICH and OECD guidelines as well as national regulations (US FDA, European Community Directives, and Japan regulations). The 3 Rs principles were considered in study designs, group sizes, and experimental repeats.
Drug and Formulation
Nirmatrelvir as a 1:1 methyl tert-butyl ether (MTBE) solvate was used for cardiovascular assessment in monkeys, neurofunctional and pulmonary assessment in rats, and the 2-week toxicity studies in rats and monkeys. Nirmatrelvir as a 50% spray dried dispersion (SDD) was used for the 1-month toxicity studies in rats and monkeys. Hydroxypropylmethylcellulose acetate succinate (HPMCAS) was used in SDD manufacturing. Control groups were administered either 2% Polysorbate 80 in 0.5% of methylcellulose in purified water, vehicle spiked with MTBE (at a concentration of 1.5% [w/v]), or HPMCAS (at a concentration of 50%) in 1% Soluplus® and 0.5% methylcellulose in purified water. Doses of nirmatrelvir were selected in accordance with applicable ICH guidelines (S7A, S7B or M3) and aimed to cover a range of plasma concentrations above the projected human plasma concentration in early clinical trials.
Animals
In vivo studies were conducted in accordance with the current guidelines for animal welfare (National Research Council Guide for the Care and Use of Laboratory Animals, 2011). The procedures used in these studies were reviewed and approved by the Institutional Animal Care and Use Committee.
Plasma Exposure Analysis
Plasma samples were analyzed for nirmatrelvir concentrations using a validated liquid chromatography tandem mass spectrometry (LC-MS/MS) method. In this assay, plasma samples and standards were subject to protein precipitation extraction and analyzed using LC-MS/MS. Analyst software (Version 1.7, Sciex, Framingham, MA) was used to measure peak areas, and peak area ratios of analyte to internal standard were calculated. A calibration curve was constructed from the peak area ratios of the standards by applying a weighted (1/×2) linear regression. All exposure (toxicokinetic) parameters were determined from group mean animal data using non-compartmental analysis in Watson LIMS (Version 7.6.1, Thermo Inc. Philadelphia, PA). The values for AUC24 were estimated using the linear trapezoidal rule.
Nervous System Assessment
This assessment included a functional observational battery (FOB) 8 followed immediately by a quantitative assessment of locomotor activity in naïve male Wistar Han (Crl:WI[Han]) rats (n = 6 per dose group; ∼300-350 g body weight). The number of animals included in each dose group is consistent with the recommended group size for similar rat neurofunctional assessments. 9 The FOB was started approximately 1 hour after vehicle administration, MTBE control, 60 mg/kg, or 1,000 mg/kg nirmatrelvir. The timing of the FOB relative to dose administration was selected based on the time to maximum plasma concentration (Tmax) observed in previous pharmacokinetic studies in rats (unpublished; data not shown).
The FOB was conducted over 3 consecutive experimental days and included assessments in the home cage, open field, and reflex testing. The timing of the FOB was balanced across dose groups and commenced at the same time (±30 minute) on each of the 3 experimental days. All observations were performed by trained observers who were blinded to the animals’ dose group. Quantitative locomotor activity (horizontal and vertical) was monitored over a 60 minute period for each animal using the automated Motor Monitor (Kinder Scientific Company, Chula Vista, CA). Locomotor activity monitoring began immediately following each animal’s FOB assessment. Each 60 minute session was divided into twelve 5 minute intervals and group means were calculated. Rats were euthanized via isoflurane followed by exsanguination upon completion of the locomotor activity assessment. To facilitate statistical analysis of the neurofunctional data, parameters were identified as continuous, discrete, or binary and the analysis was completed as follows: (1) For continuous parameters (defined as more than six distinct responses recorded across all groups), nirmatrelvir-administered groups were compared to the vehicle control using the following procedures: If Bartlett’s test for variance homogeneity
10
was not significant at the 1% level, parametric methods were applied to either the absolute or adjusted means if an analysis of covariance was performed. If the F1 approximate test for monotonicity of dose-response11,12 was not significant at the 1% level, Williams’ test for a monotonic trend was applied. If the F1 test was significant suggesting that the dose-response was not monotone, a Dunnett’s test13,14 was performed instead. If Bartlett’s test was significant at the 1% level, logarithmic and square-root transformations were employed. If Bartlett’s test was still significant, non-parametric methods were applied to mean ranks. If the H1 approximate test for monotonicity was not significant at the 1% level, Shirley’s test for a monotonic trend
15
was applied. If the H1 test was significant, Steel’s test
16
was performed instead. The MTBE control was compared to vehicle control using Wilcoxon rank sum test.
17
(2) For discrete parameters (defined as between three and six distinct responses recorded across all groups), nirmatrelvir-administered groups were compared to the vehicle control using the following procedures: if the Jonckheere-Terpstra test
18
was significant at the 5% level, the direction of the trend was established and one-tailed step-down testing in this direction was performed. If the Jonckheere-Terpstra test was not significant at the 5% level, the Kruskal-Wallis test19,20 was applied. If the Kruskal-Wallis test was significant at the 5% level, the nirmatrelvir-administered groups were compared to the vehicle control using exact Wilcoxon rank sum tests; otherwise, no further comparisons were made. The MTBE control was compared to vehicle control using exact Wilcoxon rank sum test. (3) For binary parameters (defined as two distinct responses recorded across all groups), nirmatrelvir-administered groups were compared to the vehicle control using the following procedures: if the Cochran-Armitage test
21
was significant at the 5% level, the direction of the trend was established and one-tailed step-down testing in this direction was performed. If the Cochran-Armitage test was not significant at the 5% level, the χ2 test
22
was applied. If the χ2 test was significant at the 5% level, the nirmatrelvir-administered groups were compared to the control using Fisher’s Exact tests;
23
otherwise, no further comparisons were made. The MTBE control was compared to vehicle control using Fisher’s Exact test.
Plasma concentrations were not measured in the neurofunctional assessment; however, exposures were extrapolated from the concentrations achieved in male rats on Day 1 of the 14-day toxicity study.
Pulmonary Assessment
Pulmonary assessments were conducted over 3 consecutive experimental days via whole body, unrestrained plethysmography in naïve male Wistar Han (Crl:WI[Han]) rats (n = 6 per dose group; ∼300-350 g body weight) using the BioSystem XA software package (Version 2.9.4.1; Buxco Electronics Inc, Wilmington, NC). The time of the pulmonary assessments commenced at the same time (±30 minute) on each of the 3 experimental days. Prior to dosing, animals were habituated to the plethysmography chambers on at least three occurrences for up to 60 minute each. On each day of dosing, pulmonary data was assessed predose for 20 minute (following 40 minute habituation). Animals were administered vehicle, MTBE control, 60 mg/kg, or 1,000 mg/kg nirmatrelvir and pulmonary parameters were collected continuously for 6 hour. Rats were euthanized via isoflurane followed by exsanguination upon completion of the pulmonary assessment. Respiratory rate, tidal volume, and minute volume were analyzed in 20 minute intervals from predose to 6 hour postdose. Separate analyses of MTBE control or nirmatrelvir effects for each 20 minute time interval, and for each animal’s average response over the course of the entire assessment, were performed using an analysis of covariance suitable for the randomized block design, with day of exposure as the blocking factor. The average response in the last 20 minute interval of predose baseline data was used as the covariate in the model and used to calculate the adjusted means. Comparisons of each dose group and the MTBE control to the vehicle control group were performed using t-tests based on the analysis of covariance model. The group adjusted means, differences from vehicle control, and 95% confidence intervals (95% CI) for the differences were calculated.
Plasma concentrations were not measured in the pulmonary assessment; however, exposures were extrapolated from the concentrations achieved in male rats on Day 1 of the 14-day toxicity study.
Cardiovascular Assessment
Male Mauritian cynomolgus monkeys (n = 8; 3-7 years of age at study start) underwent a surgical procedure for the placement of a telemetry device capable of transmitting arterial blood pressure, left ventricular pressure (LVP), body temperature, electrocardiogram (ECG), and activity data. One pressure catheter was placed in the femoral artery and advanced to the descending aorta and a second pressure catheter was placed into the left ventricle through the apex of the heart. The ECG leads were secured in a lead II configuration with the negative electrode attached to the pericardium near the right atria, and the positive electrode secured to the epicardium at the apex of the left ventricle. The body of the transmitter was implanted in an intramuscular pocket on the left flank of the animal. Telemetry ECG, arterial blood pressure, LVP, body temperature, and activity data were transmitted via the telemetry device to transceivers located near to the animals. The acquired signals were passed through a communication link controller to the computer-based data acquisition system. Telemetered data were continuously recorded from all animals for a minimum of 45 minute prior to dosing and continuing through at least 22 hour postdose (HPD). Ponemah Review/ECGpro (P3 Version 5.2, Data Sciences International, St. Paul, MN) was used for post-acquisition analysis of telemetered signals. Measurements derived from these signals included systolic (SBP), diastolic (DBP), and mean (MBP) blood pressure, heart rate (HR), ECG intervals (RR, PR, QRS, and QT), left ventricular systolic pressure (LVSP), and left ventricular end diastolic pressure (LVEDP), left ventricular LV + dP/dt max (an index of cardiac contractility), body temperature, and activity.
During the cardiovascular (CV) phase, animals were dosed in a Latin square crossover design and telemetry data were collected. On each day of dosing, animals received a dose of vehicle, MTBE control, or nirmatrelvir (20 or 75 mg/kg per dose, b.i.d.) via oral gavage to exceed projected clinical drug concentrations and characterize a dose/exposure response for any CV changes observed. There were at least 60 hour allotted between dosing days which is greater than 5 half-lives of nirmatrelvir to allow for test article clearance.
During the pharmacokinetic (PK) phase, all animals were dosed with nirmatrelvir at 75 mg/kg per dose (b.i.d.) for the provision of a full PK profile; blood samples were collected predose and approximately 0.5, 1, 2, 4, 6 (prior to second daily dose), 7 and 24 HPD. No telemetry data were obtained during the PK phase of this study.
The CV data were initially reduced to 1 minute means and further processed using Data Sciences International Reporting (DSIR) software (Version 2.3.0.0; Data Sciences International, St. Paul, MN) to provide 15 minute contiguous mean values from which appropriate tabular and graphical data summaries were generated. The 15 minute mean data were subsequently averaged by binning into four postdose periods as follows: Period 1 (0.75-5.5 hours post dose [HPD]), Period 2 (7.25-9.00 HPD), Period 3 (9.25-16.00 HPD), and Period 4 (16.25-20.5 HPD). These periods were selected to omit time points where room entries by veterinary staff for feeding and blood draws and the transition to lights out which may confound the CV signal quality. Summary data for these time periods were used for statistical analysis. An individual animal correction factor (IACF) was generated for each animal as a method of normalizing QT-interval over a range of RR-intervals thus providing the heart rate corrected QT-interval (QTc). IACF is the slope of the linear regression generated by relating RR-interval (RR) with QT-interval, generated from the vehicle data acquired for each animal.
Statistical analysis was performed by analyzing each response separately for each postdose period using the analysis of variance (ANOVA) to investigate differences due to treatment (vehicle and nirmatrelvir) while accounting for variation due to animal and study period (different days). Comparisons between nirmatrelvir and vehicle were made for each animal. Specifically, the model was: Parameter = Animal + Treatment + Period. Mean values and 95% confidence intervals (CI) are provided in the results section. If the 95% CI did not span zero, that indicates statistical significance at the 5% level (ie, P < 0.05). A Latin square crossover design with n = 8 monkeys has demonstrated sensitivity, to detect CV changes of approximately 5 mmHg (SBP), 10 bpm (HR), and 10 msec (QTc); based on power analysis of internal studies in monkeys using ANOVA statistical analysis and data averaged into “super-interval” postdose periods (unpublished data).
Nonclinical Toxicity Assessment
Wistar Han (Crl:WI[Han]) rats (n = 15 of each sex per group, 8-9 weeks of age at study start) were administered nirmatrelvir by oral gavage for a period of 2 weeks or 1 month (60, 200, or 1,000 mg/kg, q.d.) in two separate studies. Reversibility was evaluated in all dose groups following a 2-week recovery phase in both studies. In addition, Mauritian cynomolgus monkeys (n = 3-5 of each sex per group, >2.5 years of age at study start) were administered nirmatrelvir by oral gavage for a period of 2 week or 1 month (20, 50, or 300 mg/kg per dose, b.i.d.). The reversibility of effects was evaluated in control and high dose groups following a 2-week recovery phase in the 1-month study. Doses of nirmatrelvir were selected in accordance with ICH M3 (R2), based on the previous dose range finding studies in rats and monkeys (unpublished; data not shown), and aimed to cover a range of plasma concentrations above the projected human plasma concentration in early clinical trials.
All animals were observed daily for clinical signs of toxicity. Body weight and food consumption were recorded weekly for rat and daily for monkeys. Other observations and measurements included electrocardiograms (monkey only) and ophthalmoscopic examinations. For clinical pathology assessments, blood was collected from caudal vena cava (rats) or femoral vein (monkeys), and urine was collected following overnight collection (rats) or via cystocentesis (monkeys). Standard hematology, clinical chemistry, and coagulation parameters were measured using a Siemens Advia 2120 Hematology analyzer (Siemens Healthcare Diagnostics, Tarrytown, NY), a Siemens Advia 1800 Chemistry analyzer (Siemens Healthcare Diagnostics, Tarrytown, NY), and an STA Compact Hemostasis analyzer (Diagnostica Stago, Asnieres-sur-Seine, France), respectively. Urine was analyzed on a Siemens Novus urine analyzer (Siemens Healthcare Diagnostics, Tarrytown, NY).
Descriptive statistics were generated for each parameter and group at each scheduled sampling time or each time interval. Statistical tests were conducted at the 5 and 1% significance levels. A nonparametric (rank-transform) one way analysis of variance on all groups was conducted, with two-sided trend tests and two-sided pairwise comparisons of each group to the reference group using Dunnett’s test. Statistical tests were not done if n was less than 3 animals. Clinical pathology results were reported as ratio of the nirmatrelvir-related finding relative to the most recent baseline values for monkeys and to group mean values for rats. Conclusions regarding relationship of clinical pathology results to nirmatrelvir were made using a weight-of-evidence approach as previously described. 24 Rats were euthanized with isoflurane and exsanguination and monkeys with pentobarbital and exsanguination.
Plasma drug concentrations were measured. At the end of the dosing (10 rats per sex per group or 3 monkey per sex per group) and recovery (5 rats per sex per group or 2 monkeys per sex from control and 300 mg/kg per dose, b.i.d. groups) phase animals were euthanized and necropsied. After gross examination select organs were weighed and a comprehensive set of tissues was collected and processed for microscopic examination from all animals. Adversity assessments and considerations for setting the no-observed-adverse-effect-levels (NOAELs) included integrated evaluation of the incidence and severity of clinical and pathology findings, and their potential impacts on functional capacity to maintain homeostasis or compensate for an additional challenge. 25
Results
Safety Pharmacology
In the nervous system assessment, there were no effects of nirmatrelvir on FOB parameters at any dose tested. Compared with vehicle control, rats administered 1,000 mg/kg nirmatrelvir had up to 36% lower vertical activity when first placed in the activity chamber (beam breaks (mean ± SEM) at interval 1: vehicle = 45.5 ± 2.7; 1,000 mg/kg = 29.2 ± 2.5, P < 0.001) and up to 806% higher horizontal activity levels during later intervals of the assessment (beam breaks (mean ± SEM) at interval 7: vehicle = 21.0 ± 9.0; 1000 mg/kg = 190.2 ± 69.3, P = 0.021). Locomotor activity returned to vehicle control levels by the end of the assessment, which was approximately 2-2.5 HPD (Figures 1A and B). Changes in quantitative locomotor activity and pulmonary endpoints in rats following a single acute dose of nirmatrelvir. (A,B) Group means ± SEM (n = 6 males/group) are presented for quantitative locomotor activity recorded for 60 min at approximately 1-1.5 HPD (immediately following the FOB assessment at 1 h postdose). (C-E) Group means ± SEM (n = 6 males/group) are presented for pulmonary function parameters assessed continuously for 6 h postdose. The test article contained approximately 15% MTBE; therefore, the amount of MTBE administered to the MTBE control group (150 mg/kg) was approximately the amount of MTBE present in the high dose of 1,000 mg/kg nirmatrelvir. Statistically significant differences are highlighted between 1,000 mg/kg and vehicle (*P < 0.05), between 60 mg/kg and vehicle (#P < 0.05), and between MTBE Control and vehicle (^P < 0.05).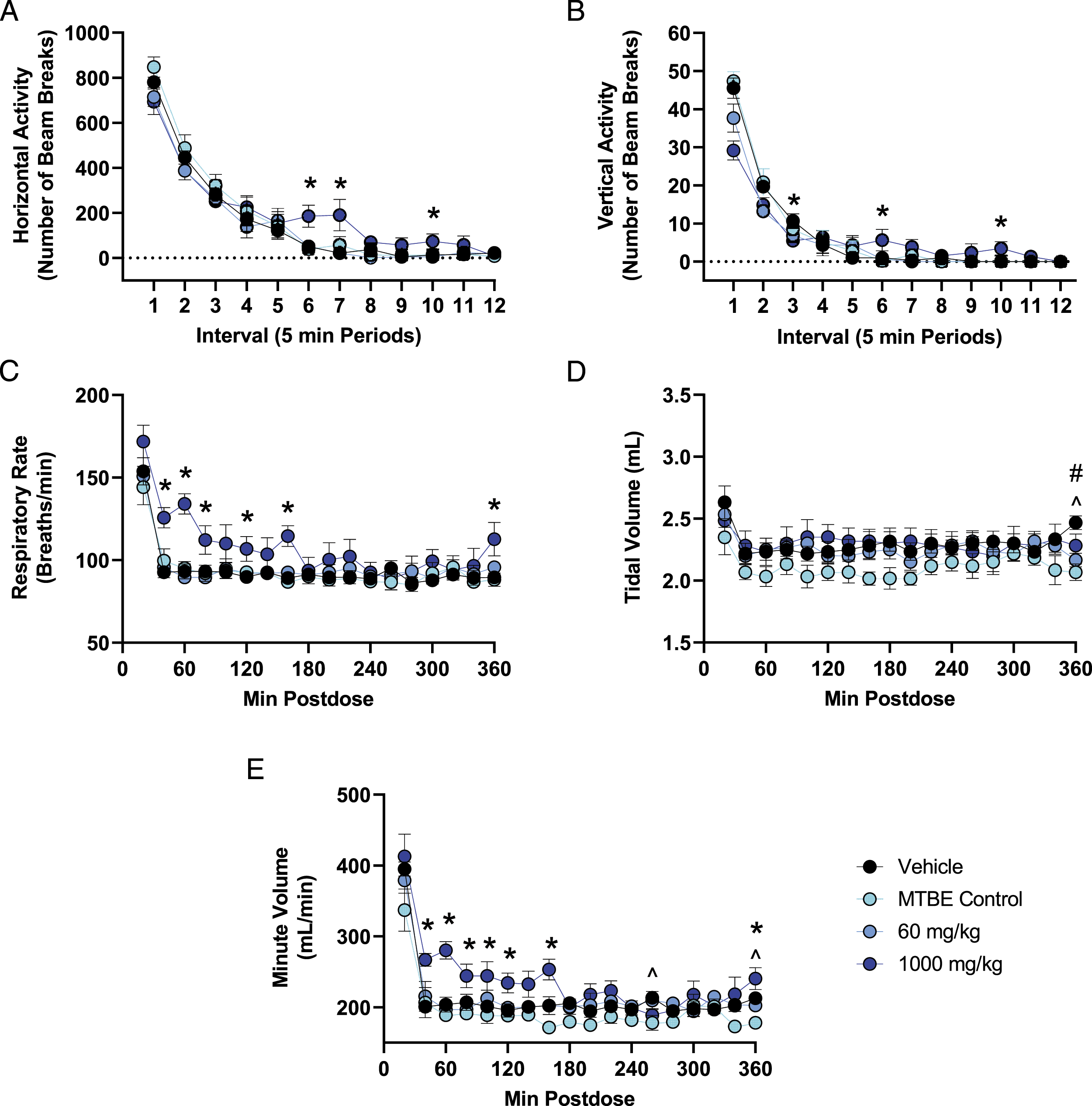
In the pulmonary function assessment, administration of 1,000 mg/kg nirmatrelvir resulted in up to 44% higher respiratory rate (breaths/min (mean ± SEM) at 60 minute postdose: vehicle = 92.8 ± 4.3; 1,000 mg/kg = 132.0 ± 4.4, P < 0.001) and up to 38% higher minute volume (mL/min (mean ± SEM) at 60 minute postdose: vehicle = 203.2 ± 7.5; 1,000 mg/kg = 280.1 ± 7.5, P < 0.001) compared with vehicle control. The increase in respiratory rate and minute volume was observed from approximately 40-160 minute postdose. Minute volume is a function of respiratory rate and tidal volume; therefore, the increase in minute volume was due to the increased rate of respiration with no change in tidal volume. Pulmonary parameters in animals administered 1,000 mg/kg nirmatrelvir returned to vehicle control levels by 180 minute postdose (Figures 1C-E). Following administration of a single dose of 60 and 1,000 mg/kg (q.d.) the mean Cmax (±SD) was 11.0 ± 1.73 and 72.4 ± 21.5 μg/mL, respectively, and the mean Tmax was 0.5 and 4.0 hours postdose, respectively. At 1 hour postdose (which is the time at which the neurofunctional assessment began), mean plasma concentrations (±SD) were 10.6 ± 0.475 and 52.1 ± 18.1 μg/mL for the 60 and 1,000 mg/kg (q.d.) dose, respectively.
In the monkey cardiovascular study, oral administration of nirmatrelvir at 20 mg/kg per dose (b.i.d.) produced no changes in any parameter measured (Figure 2). Nirmatrelvir at 75 mg/kg per dose (b.i.d.) produced a decrease (mean ± 95% CI) in HR (−8 ± 6 to −14 ± 5 beats/min) and increases in SBP, DBP and MBP (+3 ± 2 to +5 ± 4 mmHg). The RR-interval was increased (+37 ± 19 to +52 ± 17 ms), consistent with the decrease in HR during this same time. Increases in both the PR-interval (+3 ± 2 ms) and QT-interval (+11 ± 8 to +13 ± 5 ms) were observed, although both effects were considered secondary to the decrease in HR. When the QT-interval was corrected for HR (QTc), there was a nirmatrelvir -related decrease (−5 ± 4 to −7 ± 4 ms). Nirmatrelvir at 75 mg/kg per dose (b.i.d.) also produced decreases in LV + dP/dt max (−306 ± 222 to −364 ± 265 mmHg/s). There were no changes in QRS-interval at any dose and all measured changes returned to vehicle control levels within 24 HPD (see Figure 2). There were no quantifiable concentrations of nirmatrelvir in animals administered vehicle or in samples collected prior to dosing during the CV or PK phases indicating a sufficient washout between doses. The mean (±SD) Cmax following administration of the 75 mg/kg per dose (b.i.d.) was 14.7 ± 9.24 μg/mL. Detailed 15 minute averaged CV data are presented in Supplemental Figure 1. .Summary of effects of nirmatrelvir cardiovascular parameters in cynomolgus monkeys. Each individual animal (n = 8) was administered each treatment using a latin square cross-over design with sufficient washout period between dosing days to ensure clearance of nirmatrelvir. Following continuous collection, data were initially reduced to 1-minute means and further processed to provide 15 min mean values which were binned into four postdose periods. The data are represented as the change in each endpoint (mean ± 95% confidence interval) compared with vehicle control values for systolic blood pressure (SBP), diastolic blood pressure (DBP), heart rate (HR), and QTc interval, respectively, which are available in Supplemental Table 1. Statistical differences due to treatment were determined using ANOVA analysis. *P < 0.05 indicates statistically significant difference compared to vehicle.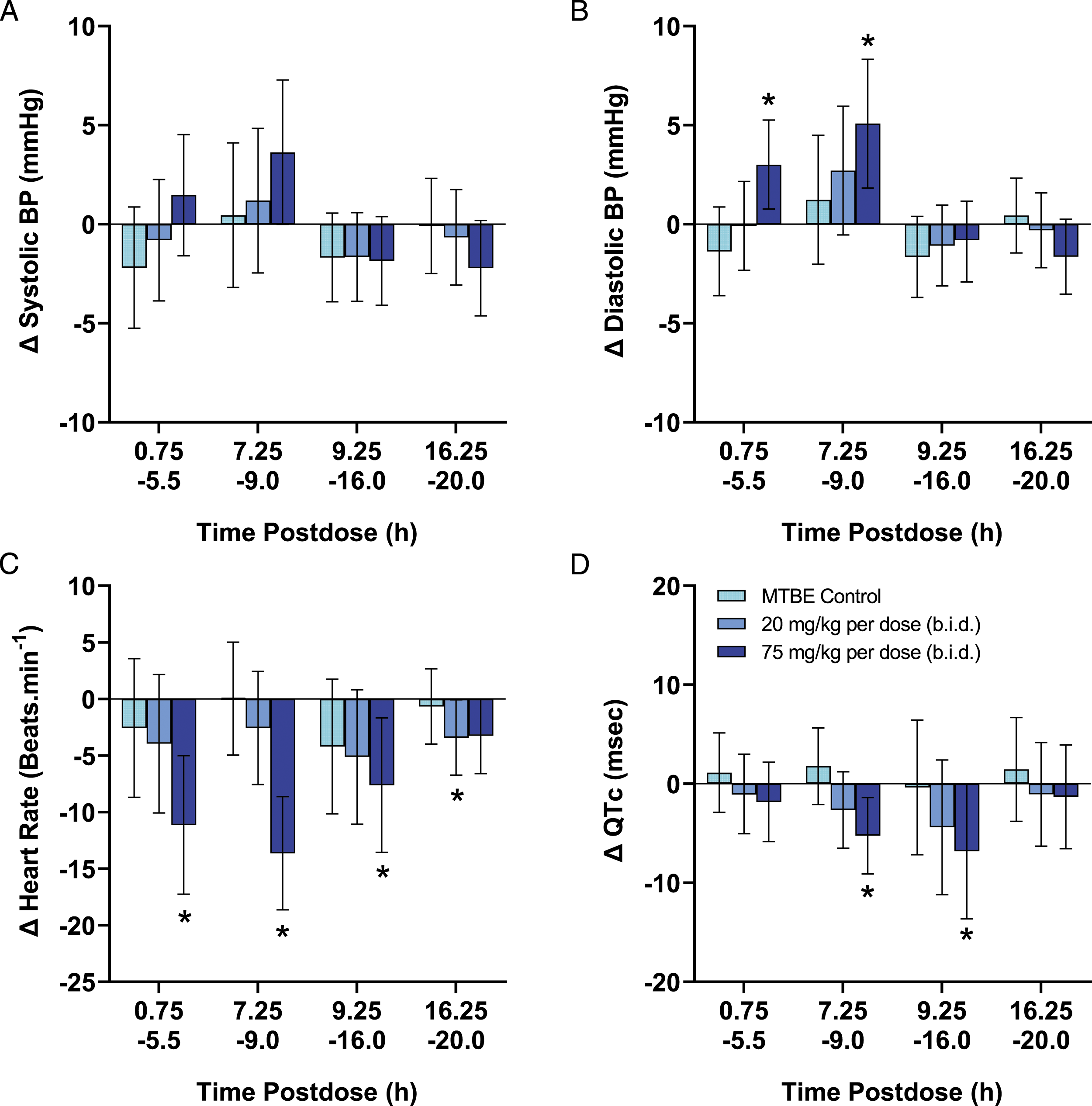
Mean Plasma Concentrations and Exposure Margins Relative to Predicted Human Clinical Exposures Across in vivo Studies.
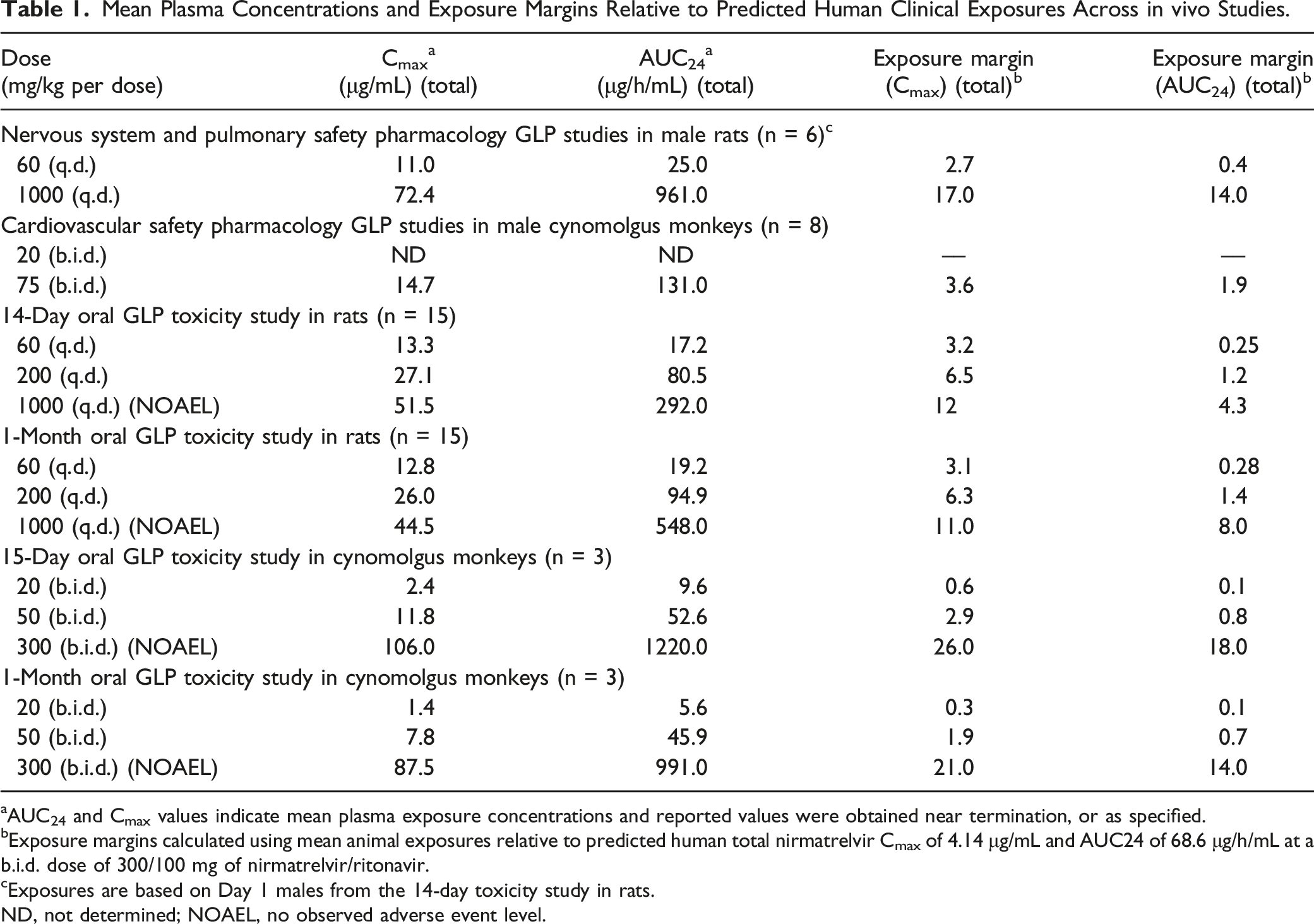
aAUC24 and Cmax values indicate mean plasma exposure concentrations and reported values were obtained near termination, or as specified.
bExposure margins calculated using mean animal exposures relative to predicted human total nirmatrelvir Cmax of 4.14 μg/mL and AUC24 of 68.6 μg/h/mL at a b.i.d. dose of 300/100 mg of nirmatrelvir/ritonavir.
cExposures are based on Day 1 males from the 14-day toxicity study in rats.
ND, not determined; NOAEL, no observed adverse event level.
Rat Toxicity Studies
Nirmatrelvir-Related Clinical Pathology Findings in 1-Month Rat Toxicity Study.
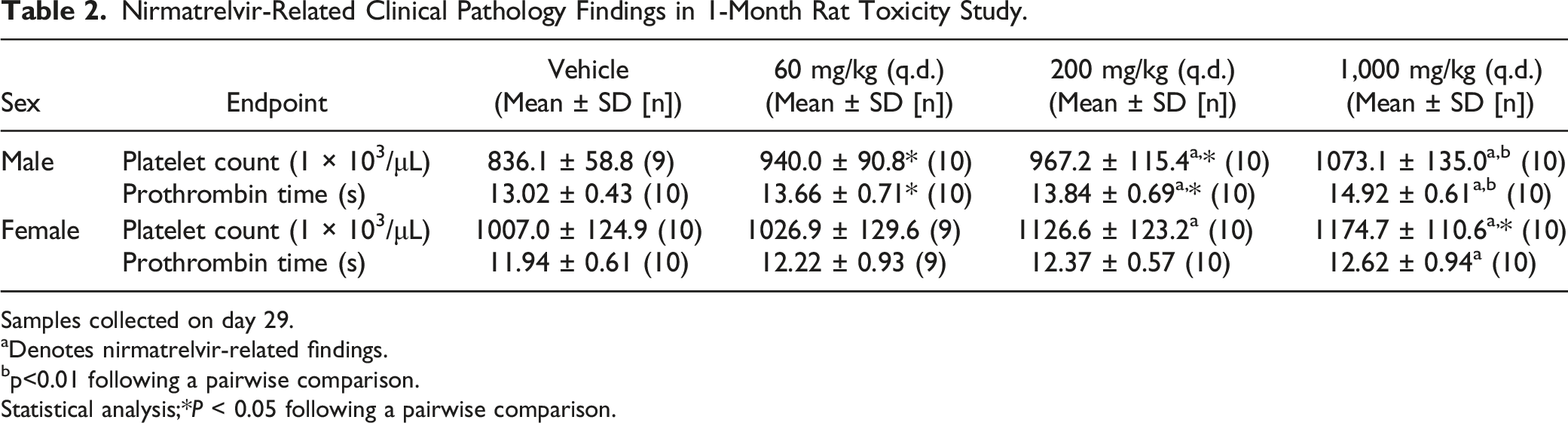
Samples collected on day 29.
aDenotes nirmatrelvir-related findings.
bp<0.01 following a pairwise comparison.
Statistical analysis;*P < 0.05 following a pairwise comparison.
Effect of Nirmatrelvir on Body, Liver, and Heart Weight in 2-Week and 1-Month Rat Toxicity Studies.

SD, Standard Deviation; n, number of individual animals.
ap<0.01 following a pairwise comparison.
bp<0.05 denotes a statistically significant trend.
cp<0.01 denotes a statistically significant trend.
ddenotes and ascending trend.
edenotes a descending trend.
Statistical analysis;*P < 0.05 following a pairwise comparison.
Group Incidences and Severities of Nirmatrelvir-Related Microscopic Findings in a 2-Week Rat Toxicity Study.
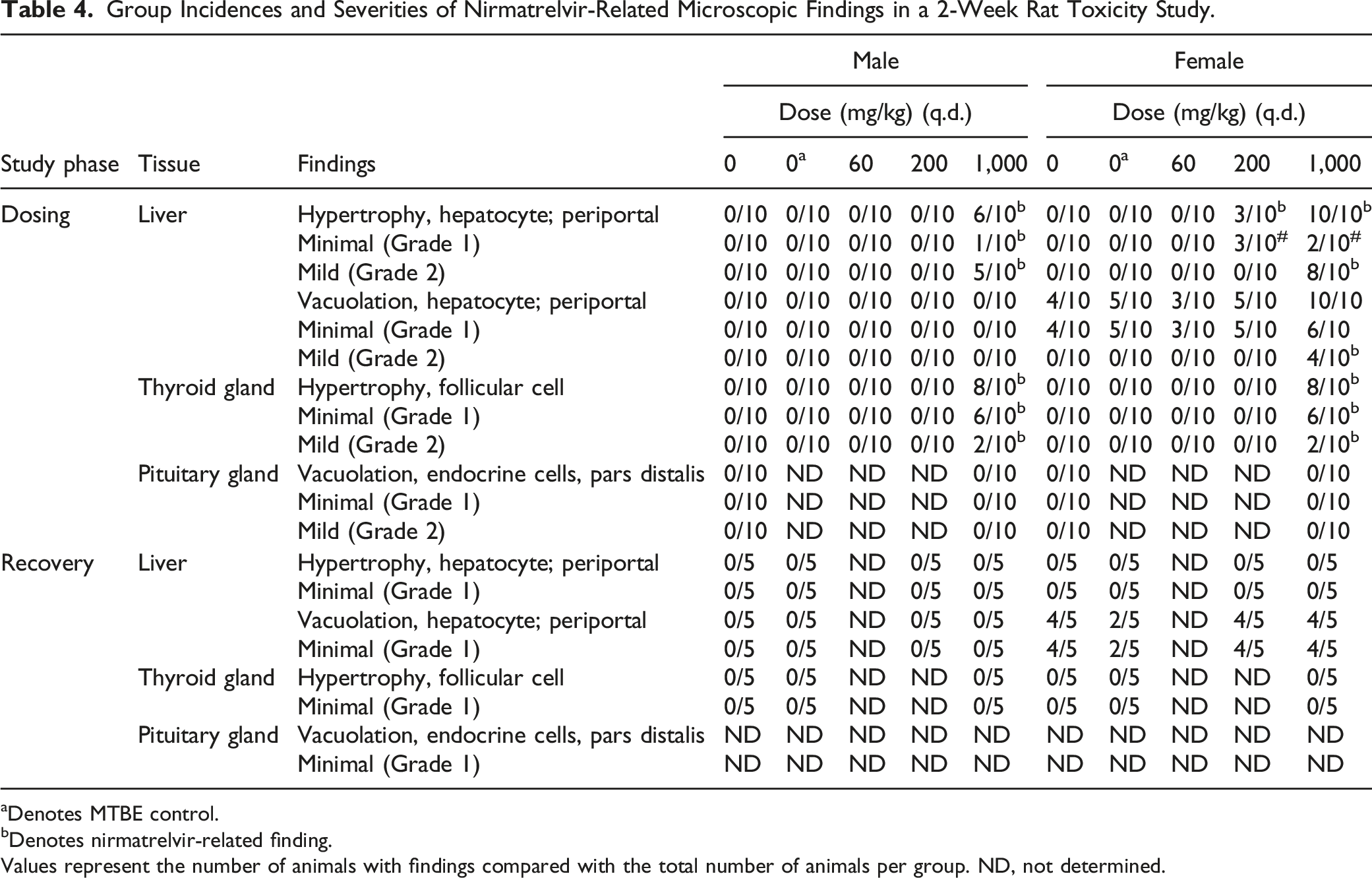
aDenotes MTBE control.
bDenotes nirmatrelvir-related finding.
Values represent the number of animals with findings compared with the total number of animals per group. ND, not determined.
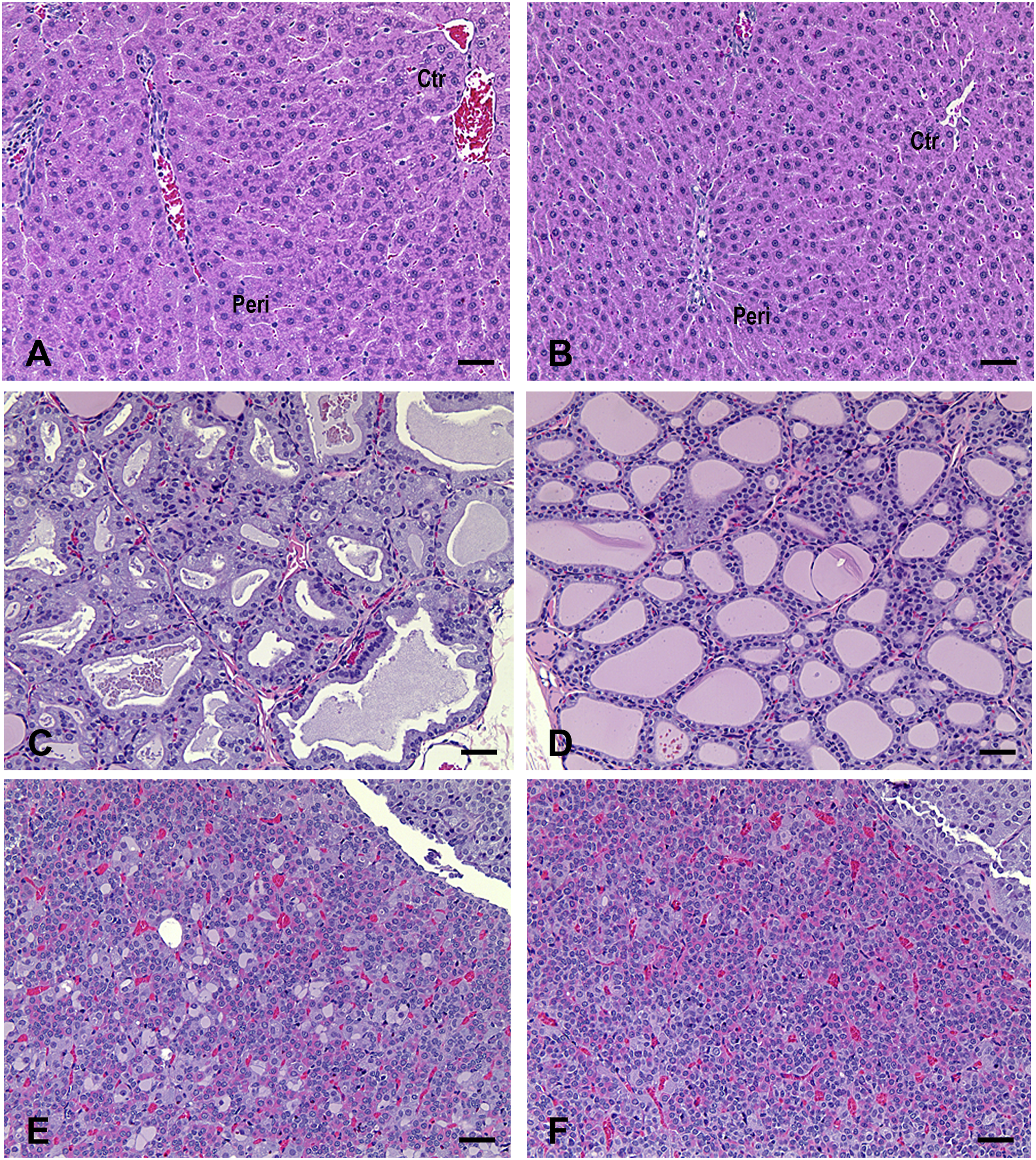
Representative microscopic changes in the liver, thyroid gland, and pituitary gland of rats following administration of nirmatrelvir (1,000 mg/kg) in a 1-month toxicity study. (A) Liver: periportal hepatocellular hypertrophy characterized by slight enlargement of periportal hepatocytes with abundant finely granular eosinophilic cytoplasm and sinusoidal compression. Peri: periportal area. Ctr: centrilobular area. Two out of 10 males and 2/10 females in the 200 mg/kg group, and 9/10 males and 10/10 females in the 1,000 mg/kg group had periportal hepatocellular hypertrophy. (B) Liver: normal. (C) Thyroid gland: follicular cell hypertrophy characterized by increased size and height of follicular cells with more amphophilic to vacuolated cytoplasm and decreased colloid in the follicle lumens with variable staining intensity. One out of 10 males in the 60 mg/kg group, 4/10 males and 2/10 females in the 200 mg/kg group, and 10/10 males and 10/10 females in the 1000 mg/kg group had thyroid follicular cell hypertrophy. (D) Thyroid gland: normal. (E) Pituitary gland: vacuolated cells (basophils or chromophobes) within pars distalis are slightly enlarged and have a large single or several cytoplasmic vacuoles with displaced nuclei. Two out of 10 males in the 60 mg/kg group, 2/10 males in the 200 mg/kg group, and 5/10 males in the 1,000 mg/kg group exhibited vacuolated cells in the pituitary gland. (F) Pituitary gland: normal. Scale bar = 50 μm. H&E staining.
Group Incidences and Severities of Nirmatrelvir-Related Microscopic Findings in a 1-Month Rat Toxicity Study.
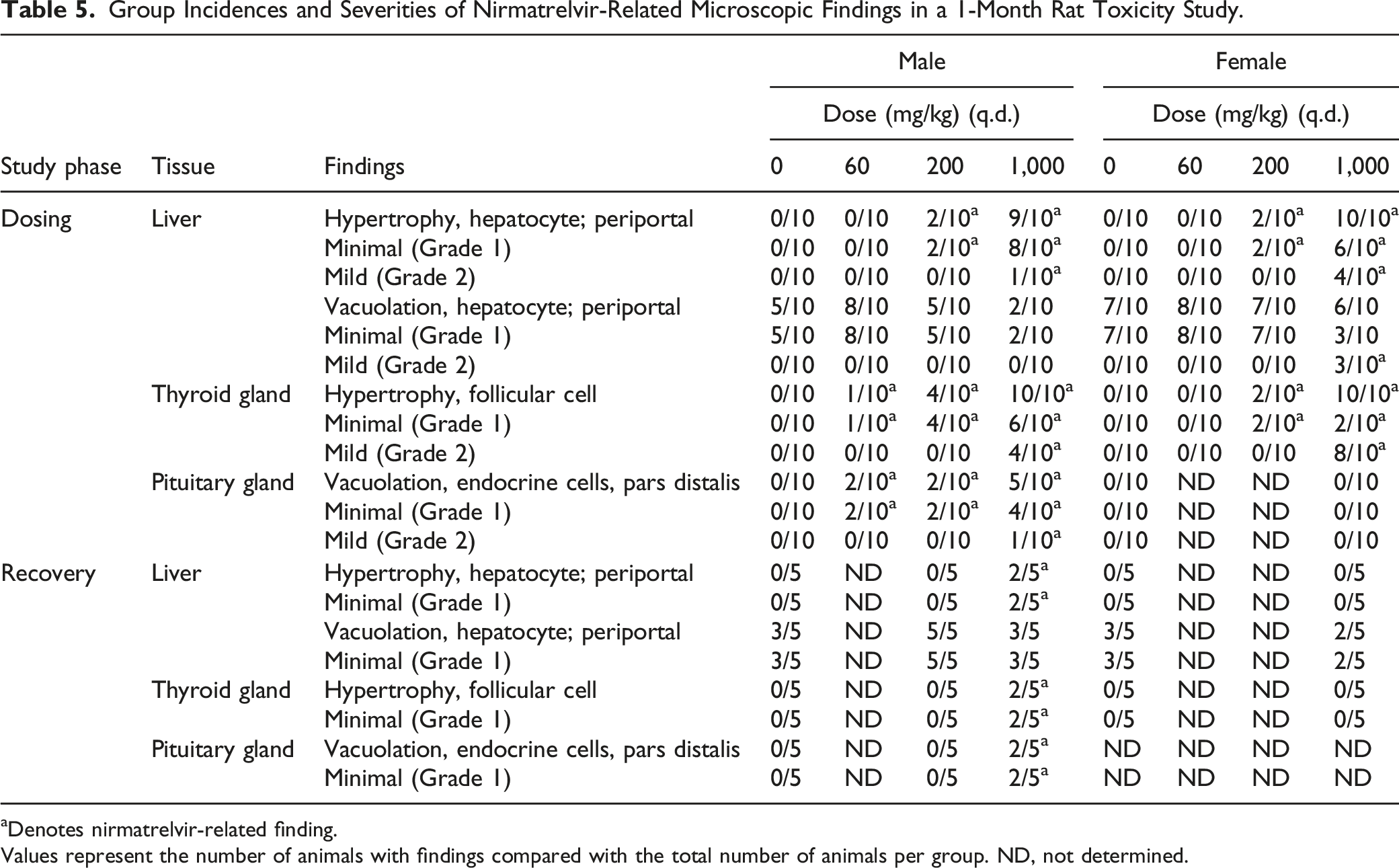
aDenotes nirmatrelvir-related finding.
Values represent the number of animals with findings compared with the total number of animals per group. ND, not determined.
Monkey Toxicity Studies
Nirmatrelvir-Related Clinical Pathology Findings in 1-Month Monkey Toxicity Study.
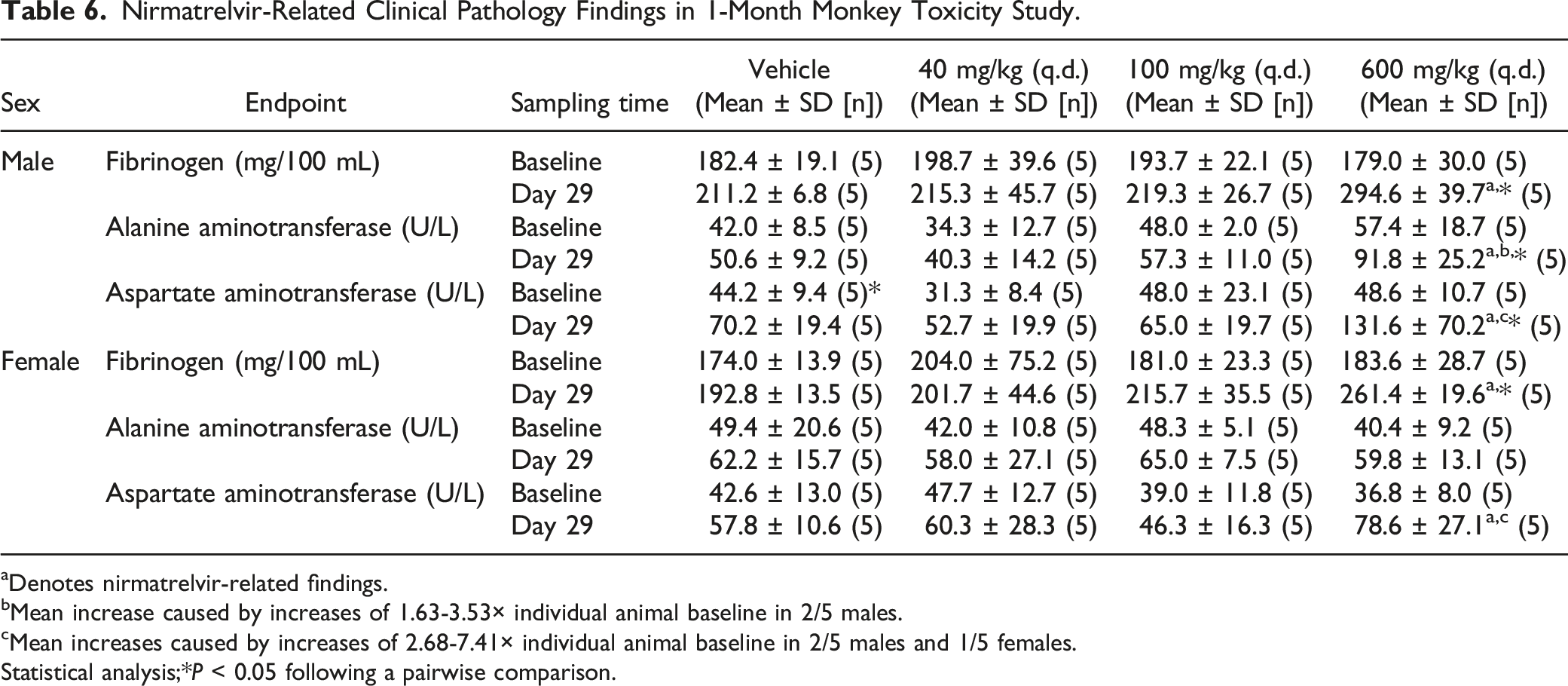
aDenotes nirmatrelvir-related findings.
bMean increase caused by increases of 1.63-3.53× individual animal baseline in 2/5 males.
cMean increases caused by increases of 2.68-7.41× individual animal baseline in 2/5 males and 1/5 females.
Statistical analysis;*P < 0.05 following a pairwise comparison.
Discussion
An oral antiviral agent for COVID-19 with a favorable safety profile that could support administration to a broad patient population is expected to be a valuable tool in mitigating the severity and duration of this disease in affected subjects. Previous studies assessing the in vitro -off target pharmacology of nirmatrelvir against a broad panel of secondary pharmacology targets have shown it to be highly selective with respect to its primary pharmacology, and no genetic toxicity risks were observed. 2 Here, we provide an expanded preclinical in vivo data safety package for nirmatrelvir covering safety pharmacology and toxicology studies in rats and monkeys that highlight the lack of associated adverse effects across these studies.
In the quantitative locomotor activity assessment, a typical pattern of activity is a high level of exploratory activity that wanes to normal low (sleeping) daytime activity levels. The higher activity observed during the later intervals of the locomotor activity assessment occurred at a similar time postdose when higher respiratory rates were observed in the pulmonary function assessment. Hence, it is likely that higher activity levels could have impacted respiratory rates in rats administered the highest dose of nirmatrelvir (1,000 mg/kg; 11× the predicted human total nirmatrelvir Cmax at a dose of 300/100 mg per dose (b.i.d.) nirmatrelvir/ritonavir; Table 1). Inclusion of positive control compounds is not required for well characterized in vivo models (eg, FOB) per ICH S7A; however, in some instances, positive control compounds are incorporated into study designs to contextualize the findings for a novel drug relative to known pharmacological mechanism and/or human effects. Although a positive control compound was not incorporated in this study, the neurofunctional profile of nirmatrelvir is divergent from that of known pharmacological mechanisms that increase locomotor activity and respiratory rate (eg, amphetamine). Relatively low doses of amphetamine (3 mg/kg) increase spontaneous activity and affect a number of FOB endpoints in the autonomic, neuromuscular, sensorimotor, and behavioral domains in rats. 30 Nirmatrelvir did not affect any FOB endpoints up to 1,000 mg/kg. Although plasma exposures were not directly measured in this study, safety margins were calculated using the maximum plasma concentrations achieved on Day 1 of the corresponding 14-day toxicity study in rats, which was conducted at the same dose levels. At 1-hour postdose (which is the time the neurofunctional assessment was conducted), plasma exposures were within 1.4-fold of the Day 1 Cmax, which provides confidence in the exposure-response relationship and safety margins achieved (Table 1). The translation of changes in neurofunctional endpoints in rodents to human adverse events (AEs) is not clear. On one hand, rodent data for some reference drugs with known CNS activity have been shown to be reflective of human effects, including both central nervous system stimulant (eg, amphetamine) and sedative (eg, chlorpromazine) effects. 31 On the other hand, in an evaluation of novel chemical entities, Mead et al 32 found no predictive value of nonclinical neurofunctional endpoints to the 5 most common central nervous system AEs observed in phase 1 clinical trials. Decreased horizontal and/or vertical locomotor activity was suggested as a plausible nonclinical correlate of dizziness, somnolence/fatigue and/or pain in humans; however, the nonclinical locomotor activity data was not predictive of those human AEs. However, there could be translation to other human AEs that were not included in the analysis. While the reason for changes in locomotor activity following administration of nirmatrelvir is unknown, it is unlikely to be due to direct effects on central nervous system function as (1) the intended pharmacological target is not present in mammals; (2) a comprehensive secondary pharmacology screen for off-target effects indicated no binding and/or functional activity at any target at relevant concentrations, 2 and (3) compared to the peripheral compartment, nirmatrelvir is expected to have limited distribution into the brain because its structure mimics a peptide (peptidomimetic) and in vitro transporter studies (unpublished; data not shown) indicate it is a substrate for MDR1 p-glycoprotein, which is a transporter protein located on the blood-brain-barrier that limits the distribution of substances into the brain.33,34 In addition, there were no changes in telemetry-based activity in the monkey cardiovascular study (up to 3.6× the predicted human total nirmatrelvir Cmax at a dose of 300/100 mg per dose (b.i.d.) nirmatrelvir/ritonavir; Table 1) and no related clinical observations in repeat-dose toxicity studies in rats and monkeys (up to 12× and 26× the predicted human total nirmatrelvir Cmax at a dose of 300/100 mg per dose (b.i.d.) nirmatrelvir/ritonavir, respectively; Table 1).
The cardiovascular profile of nirmatrelvir in cynomolgus monkeys was relatively mild with no CV effects detected with a 20 mg/kg per dose (b.i.d.) and transient increases in blood pressure with decreases in HR, LV + dP/dt max and QTc detected at 75 mg/kg per dose (b.i.d.). This mild CV profile combined with a relatively short clinical dosing regimen (a 5-day treatment period) suggest there is unlikely to be a significant risk to human subjects. Furthermore, no effects on ECG parameters were observed in the human Phase 1 studies up to projected supratherapeutic exposures. 27 The lack of any QTc prolongation in monkeys is consistent with the lack of activity in the human Ether-à-go-go-Related Gene (hERG) patch clamp assay (IC50 >300 μM) 2 and helps support the principle of a nonclinical “double negative” (ie, low risk for hERG block and in vivo QTc prolongation) as discussed by Vargas et al 35 and described in the recent draft S7B/E14 Q&A document 36 used to inform regulatory decisions on conduct of clinical thorough QT/QTc (TQT) studies.
Based on the repeat dose toxicity studies conducted in nonclinical species, the liver, thyroid, and pituitary were identified as target organs due to microscopic findings in the rat studies. Liver findings in rats included minimal to mild periportal hepatocellular hypertrophy with occasional vacuolation of periportal hepatocytes in females. These microscopic findings in the liver were associated with higher mean liver weights and the macroscopic finding of enlarged liver size at high dose. The hepatocellular hypertrophy and hepatocyte vacuolation were consistent with microsomal enzyme induction. 37 Thyroid gland follicular cell hypertrophy occurred and correlated with the higher liver weights and hepatocellular hypertrophy. Finding in the pituitary gland consisted of cytoplasmic vacuolation of the endocrine cells in the pars distalis possibly reflecting increased secretory activity in subpopulations of the hormone secreting cells (thyrotropin-secreting cells). The pattern of linked findings in the liver, thyroid and pituitary glands are consistent with a rat specific response to hepatic enzyme induction resulting in increased thyroid hormone catabolism, raised serum thyroid stimulating hormone and thyroid follicular cell hypertrophy and anterior pituitary vacuolation.28,38,39 This mechanism is usually considered to have little to no relevance to humans mostly because of the marked differences in plasma half-life of thyroid hormones and in binding to transport proteins between rodents and humans.28,29 None of the microscopic findings were considered adverse based on their limited severity or magnitude, lack of microscopic evidence of associated tissue damage (ie, necrosis or inflammation), or adverse clinical correlate. Reversible, slightly lower heart weights without microscopic correlates noted in female rats administered 1,000 mg/kg (q.d.) in the 2-week study lacked reproducibility in the 1-month study at an equivalent dose (exposure) level. Thus, this finding was deemed to be of no toxicological significance.
Alterations in coagulation parameters and/or inflammatory markers (ie, increased fibrinogen) were considered nonadverse based on their small magnitude and absence of clinical or microscopic correlates. Prolongations in APTT and/or PT were only seen in rats and not in monkeys and have not been observed in clinical studies. The mechanism for these effects is unclear. Other viral protease inhibitors, eg, HIV protease inhibitors such as tipranavir and darunavir, have also been reported to cause prolongations in APTT and/or PT in nonclinical species,40,41 but these prolongations have also not translated to humans. 42
Sporadic increases in ALT and AST were noted in the monkey 1-month repeat dose study only, without correlating clinical, macroscopic, or microscopic findings. These changes are monitorable in patients and have not been observed in clinical studies. 26
Collectively, these assessments demonstrate the favorable toxicity profile of nirmatrelvir with a sufficient therapeutic window based on exposure margins using the NOAEL in the repeated dose toxicity studies of 11×/8.0× (Cmax/AUC24) and 21×/14× (Cmax/AUC24) for rats and monkeys, respectively, over predicted human therapeutic exposures. The reproductive safety of nirmatrelvir has been demonstrated in dedicated nonclinical developmental and reproductive studies, 43 and it is worth noting that nirmatrelvir did not negatively impact the reproductive organs based on macroscopic and microscopic evaluations in the rat and monkey toxicology studies.
In conclusion, the suite of nonclinical safety pharmacology and toxicology studies described here support the favorable clinical benefit-risk profile that has been observed in clinical studies with Paxlovid (nirmatrelvir co-administered with the pharmacokinetic enhancer, ritonavir). The combination of robust efficacy in COVID-19 patients at high risk of progressing to hospitalization and/or death (Paxlovid Phase 2-3 data)26,44 and the excellent preclinical and clinical safety profiles further support the potential of this therapy in treating the COVID-19 patient population, who until now had limited treatment options. Recognition of this potential is highlighted by the fact that, following a thorough review of clinical and nonclinical data, emergency use authorizations for Paxlovid have been granted in the US, UK, Canada, Australia, and several other countries.45-48
Supplemental Material
Supplemental Material - Comprehensive Nonclinical Safety Assessment of Nirmatrelvir Supporting Timely Development of the SARS-COV-2 Antiviral Therapeutic, Paxlovid™
Supplementary Material for Comprehensive Nonclinical Safety Assessment of Nirmatrelvir Supporting Timely Development of the SARS-COV-2 Antiviral Therapeutic, Paxlovid™ by Jean G. Sathish, Siddhartha Bhatt, Jamie K. DaSilva, Declan Flynn, Stephen Jenkinson, Amit S. Kalgutkar, Maggie Liu, Balasubramanian Manickam, Jason Pinkstaff, William J. Reagan, Norimitsu Shirai, Ahmed M. Shoieb, Madhu Sirivelu, Saurabh Vispute, Allison Vitsky, Karen Walters, Todd A. Wisialowski, and Lawrence W. Updyke in International Journal of Toxicology
Supplemental Material
Supplemental Material - Comprehensive Nonclinical Safety Assessment of Nirmatrelvir Supporting Timely Development of the SARS-COV-2 Antiviral Therapeutic, Paxlovid™
Supplementary Material for Comprehensive Nonclinical Safety Assessment of Nirmatrelvir Supporting Timely Development of the SARS-COV-2 Antiviral Therapeutic, Paxlovid™ by Jean G. Sathish, Siddhartha Bhatt, Jamie K. DaSilva, Declan Flynn, Stephen Jenkinson, Amit S. Kalgutkar, Maggie Liu, Balasubramanian Manickam, Jason Pinkstaff, William J. Reagan, Norimitsu Shirai, Ahmed M. Shoieb, Madhu Sirivelu, Saurabh Vispute, Allison Vitsky, Karen Walters, Todd A. Wisialowski, and Lawrence W. Updyke in International Journal of Toxicology
Footnotes
Author Contributions
Jean G. Sathish contributed to conception, contributed to interpretation, drafted manuscript, and critically revised manuscript; Siddhartha Bhatt contributed to acquisition and analysis, drafted manuscript, and critically revised manuscript; Jamie K. DaSilva contributed to acquisition and analysis, drafted manuscript, and critically revised manuscript; Declan Flynn contributed to acquisition and analysis, drafted manuscript, and critically revised manuscript; Stephen Jenkinson contributed to analysis and interpretation, drafted manuscript, and critically revised manuscript; Amit S. Kalgutka contributed to analysis and interpretation, drafted manuscript, and critically revised manuscript; Maggie Liu contributed to analysis and interpretation, drafted manuscript, and critically revised manuscript; Balasubramanian Manickam contributed to analysis and interpretation, drafted manuscript, and critically revised manuscript; Jason Pinkstaff contributed to interpretation, drafted manuscript, and critically revised manuscript; William J. Reagan contributed to acquisition and analysis, drafted manuscript, and critically revised manuscript; Norimitsu Shirai contributed to analysis and interpretation, drafted manuscript, and critically revised manuscript; Ahmed M. Shoieb contributed to analysis and interpretation, drafted manuscript, and critically revised manuscript; Saurabh Vispute contributed to acquisition and analysis, drafted manuscript, and critically revised manuscript; Allison Vitsky contributed to acquisition and analysis, drafted manuscript, and critically revised manuscript; Karen Walters contributed to acquisition and analysis and critically revised manuscript; Todd A. Wisialowski contributed to acquisition, analysis, and interpretation, drafted manuscript, and critically revised manuscript; Madhu Sirivelu contributed to analysis and interpretation, drafted manuscript, and critically revised manuscript; Lawrence W. Updyke contributed to analysis and interpretation, drafted manuscript, and critically revised manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors are employees of Pfizer Inc. Several authors are stockholders in Pfizer Inc.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Pfizer Inc.
Institutional Review Board Statement
In vivo studies were conducted in accordance with the current guidelines for animal welfare (National Research Council Guide for the Care and Use of Laboratory Animals, 2011). The procedures used in these studies were reviewed and approved by the Institutional Animal Care and Use Committee.
Data Availability
All data supporting the results described are provided within the manuscript and in the online supplemental data.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.