
Abstract
Sotorasib is a first-in-class KRASG12C covalent inhibitor in clinical development for the treatment of tumors with the KRAS p.G12C mutation. A comprehensive nonclinical safety assessment package, including secondary/safety pharmacology and toxicology studies, was conducted to support the marketing application for sotorasib. Sotorasib was negative in a battery of genotoxicity assays and negative in an in vitro phototoxicity assay. Based on in vitro assays, sotorasib had no off-target effects against various receptors, enzymes (including numerous kinases), ion channels, or transporters. Consistent with the tumor-specific target distribution (ie, KRASG12C), there were no primary pharmacology-related on-target effects identified. The kidney was identified as a target organ in the rat but not the dog. Renal toxicity in the rat was characterized by tubular degeneration and necrosis restricted to a specific region suggesting that the toxicity was attributed to the local formation of a putative toxic reactive metabolite. In the 3-month dog study, adaptive changes of hepatocellular hypertrophy due to drug metabolizing enzyme induction were observed in the liver that was associated with secondary effects in the pituitary and thyroid gland. Sotorasib was not teratogenic and had no direct effect on embryo-fetal development in the rat or rabbit. Human, dog, and rat circulating metabolites, M24, M10, and M18, raised no clinically relevant safety concerns based on the general toxicology studies, primary/secondary pharmacology screening, an in vitro human ether-à-go-go-related gene assay, or mutagenicity assessment. Overall, the results of the nonclinical safety program support a high benefit/risk ratio of sotorasib for the treatment of patients with KRAS p.G12C-mutated tumors.
Introduction
Sotorasib is a novel, first-in-class, potent, and highly selective small molecule inhibitor that covalently binds to the KRASG12C (Kirsten rat sarcoma viral oncogene homolog with a G12C amino acid substitution) oncoprotein and impairs downstream oncogenic signaling exclusively in KRAS p.G12C (Kirsten rat sarcoma viral oncogene homolog gene with a mutation resulting in a G12C amino acid substitution) tumor cells. 1 The covalent binding and inhibition of KRASG12C by sotorasib requires the interaction with the thiol group of Cys12, resulting in a precise interaction that is specific for KRASG12C. 2 The inhibitor contains a thiol reactive, acrylamide moiety that covalently modifies the cysteine residue and locks KRASG12C in the inactive guanosine diphosphate-bound conformation. This blocks the interaction of Kirsten rat sarcoma viral oncogene homolog (KRAS) with effectors such as RAF, thereby preventing downstream proliferation and survival signaling, including the phosphorylation of extracellular signal-regulated kinase (ERK).1,3-5
In vitro and in vivo pharmacology studies confirmed the predicted, specific pharmacologic mechanism of sotorasib. 1 In the in vitro assays, sotorasib potently inhibited recombinant KRASG12C but had minimal effect on wild type (WT) KRAS. Sotorasib inhibited KRASG12C cellular signaling and viability only in KRAS p.G12C cell lines and not in lines with WT KRAS or with other mutations in KRAS. Inhibition of KRASG12C by sotorasib blocked multiple nodes in downstream proliferation and survival pathways and induced markers of apoptosis. In the in vivo studies, sotorasib covalently modified KRASG12C and significantly inhibited ERK1/2 phosphorylation (p-ERK) at doses as low as 3 mg/kg. In xenograft tumor efficacy studies, sotorasib significantly inhibited tumor growth at 3 mg/kg, and at 100 mg/kg achieved almost complete regression in numerous KRAS p.G12C cell line-derived xenograft and patient-derived xenograft models. Sotorasib had no effect on KRAS p.G12V or p.G12D models and did not affect body weight in any study. In immune competent mice, treatment with sotorasib resulted in a proinflammatory tumor microenvironment and produced durable cures alone as well as in combination with immune checkpoint inhibitors. 1 Overall, these data suggest that sotorasib should be an effective antitumor agent for treatment of KRAS p.G12C-mutated advanced cancer.
Sotorasib has been evaluated as monotherapy and in combination with various other anticancer agents for the treatment of adult patients with KRAS p.G12C mutated locally advanced or metastatic non-small cell lung cancer, colorectal cancer, and other solid tumors.6-9 Sotorasib was administered as multiple doses of 180 to 960 mg once daily (QD). Recently, the US Food and Drug Administration granted Breakthrough Therapy designation for sotorasib for the treatment of patients with locally advanced or metastatic non-small cell lung cancer with the KRAS p.G12C mutation.
The KRAS p.G12C mutation has only been reported in tumor tissue and is not present in normal tissue.10-13 Cysteine proteome profiling with sotorasib for potential “off target” cellular proteins demonstrated that the Cys12 peptide from KRASG12C was the only peptide that met the criteria for covalent target engagement among 6,451 unique cysteine-containing peptides. 1 These data suggest that the primary pharmacology-related on-target effects and closely related off-target effects of sotorasib will be minimal in normal “nontumor bearing” animals or normal tissues/cells in patients with cancer. However, small molecule covalent inhibitors may have a risk to cause metabolite-mediated toxicity.14,15
A comprehensive nonclinical safety assessment of sotorasib was performed for the marketing authorization application. The assessment consisted of secondary and safety pharmacology studies, exploratory and Good Laboratory Practice (GLP) compliant repeat-dose toxicology studies in the rat and dog, genotoxicity studies, rat and rabbit embryo-fetal development toxicology studies, and other studies including in vitro/in vivo mechanistic studies, studies on metabolites, and an in vitro phototoxicity study. Here, we report the results of the nonclinical safety program and the uniquely favorable safety profile of sotorasib.
Methods
Test and Control Articles
Sotorasib (AMG 510) was synthesized at Amgen, Inc, California.1,2 The vehicle control articles were 2% (wt/vol) hydroxypropyl methylcellulose (HPMC) 1% (wt/vol) Tween 80 (Polysorbate 80) in reverse osmosis deionized water for the rat and dog 28-day repeat-dose studies and dog cardiovascular safety pharmacology study; 20% Captisol, pH 2.2 in reverse osmosis deionized water for the 3-month rat study and 7-day mechanistic study, and rat embryo-fetal development study; 2% HPMC 1% Tween 80 pH 2.4 for the 3-month study in the dog and rabbit embryo-fetal development study; and 30% (wt/vol) Captisol in ultrapure water for the rat combined in vivo micronucleus test and comet assay. Sotorasib was suspended with the vehicle control at appropriate concentrations, and dose formulation analysis was routinely performed to confirm expected test article concentration and homogeneity.
Animals
The Sprague Dawley (Crl: CD[SD]) rats and beagle dogs were selected for toxicology assessment based on the similarity of in vitro metabolic profiles with humans and a large background database for nonclinical toxicology assessment. New Zealand White (Hra:[NZW]SPF) female rabbits were used for the assessment of potential effects on embryo-fetal development. All animals were housed at Association for Assessment and Accreditation of Laboratory Animal Care international-accredited facilities. All animal studies were approved by the local Institutional Animal Care and Use Committee. All in vivo nonclinical safety studies summarized here, except for the 7-day mechanistic study in the rat and maternal tolerability study in the rabbit, were performed in accordance with GLP regulations (US Code of Federal Regulations, Title 21, Part 58: GLP for Nonclinical Laboratory Studies).
Rats were socially housed by sex (up to 3 animals/sex/cage) in solid bottom cages with bedding. Animals had ad libitum access to water and pelleted feed, except for an overnight food fast prior to blood collection at scheduled necropsy.
Beagle dogs were socially housed (up to 3 animals/sex/cage) in stainless steel cages equipped with an automatic watering valve. PMI Nutrition International Certified Canine Chow No. 5007 was provided daily.
Pregnant female rabbits were individually housed in stainless steel cages. Animals had ad libitum access to water and PMI Nutrition International Certified Rabbit Chow No. 5322 was provided daily.
All animals were maintained on a 12:12-hour light: dark cycle in rooms with appropriate temperature (approximately 16-26 °C), humidity (26%-70%), and ventilation (10 or more air changes per hour) controls. At study completion, animals were euthanized under deep anesthesia induced by isoflurane or carbon dioxide inhalation for the rat, or sodium pentobarbital injection for the dog, pregnant female rabbit, and live rat, and rabbit fetuses, followed by complete exsanguination.
In Vitro Secondary Pharmacology Screening to Evaluate Off-Target Activities
The secondary pharmacology of sotorasib was assessed using in vitro radioligand displacement, enzyme activity, and transporter uptake assays against various off targets including receptors, enzymes, ion channels, and transporters (Eurofins CEREP SA). The first set of panels includes 41 targets (Supplementary Table 1) and the second set of panels includes 105 targets (Supplementary Table 2). Compound binding or uptake was calculated as a percentage inhibition of the binding of a radioactively labeled ligand specific for each target. Compound enzyme inhibition effect was calculated as a percentage inhibition of control enzyme activity. Sotorasib was also evaluated in a kinase screening panel (KINOME scanMAXSM, Eurofins DiscoverX) that utilizes competition binding assays16,17 to probe the activities of the test compound against a total number of 468 kinases (Supplementary Table 3). Binding data are calculated as a % of control values.
Phototoxicity Studies
Potential phototoxicity of sotorasib was evaluated in the in vitro 3T3 NRU phototoxicity test in line with Organization for Economic Co-operation and Development Guideline 18 432 and International Council on Harmonization (ICH) S10 for photosafety evaluation of pharmaceuticals. 19
Genotoxicity
The potential genotoxicity of sotorasib was assessed in the genetic toxicology battery including the GLP Ames test and the GLP combined in vivo mammalian erythrocyte micronucleus test and comet assay in the rat.
In the GLP Ames test, Salmonella typhimurium strains TA1535, TA1537, TA98, TA100, and Escherichia coli strain WP2 uvrA were exposed to sotorasib at a range of concentrations from 1.58 to 5,000 μg/plate (the standard limit dose for this assay), in the presence and absence of a supplemented rat liver fraction (S9 mix), using the plate incorporation version of the bacterial mutation test.
In the GLP-combined in vivo mammalian erythrocyte micronucleus test and comet assay, female SD rats (5/group) received sotorasib by oral gavage at 0, 200, 600, or 2,000 mg/kg QD for 4 days. Blood for micronucleus evaluation and liver samples for the comet assay were collected 3 hours following the last administration, and frequency of micronucleated reticulocytes and comet tail DNA was evaluated.
In Vitro Dog Hepatocyte Assay
An in vitro enzyme induction assay in cultured dog hepatocytes was performed to evaluate the ability of sotorasib and metabolite M24 (a major circulating metabolite in the dog) to induce cytochrome P450 (CYP) and uridine diphosphate glucuronosyltransferase (UGT). The cultured dog hepatocytes were treated with sotorasib (2-200 µM) or M24 (1-30 µM) for 3 consecutive days. Messenger RNA (mRNA) expression of CYP1A1, CYP1A2, CYP2B11, CYP3A12, UGT1A6, and UGT2B31 by quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR) and in vitro enzyme activity of CYP1A1/2, CYP2B11, CYP3A12, UGT1A, and UGT2B were assessed. The prototypical inducers 3-methylcholanthrene, β naphthoflavone, phenobarbital, dexamethasone, pregnenolone 16α carbonitrile, and rifampin were used as positive controls.
Cardiovascular Safety Pharmacology
The in vitro effects of sotorasib on human ether-à-go-go-related gene (hERG) potassium channel function were examined in a GLP study. The concentration–response relationship of the effect of sotorasib (10, 30, 100, and 300 μM) on the hERG potassium channel current (a surrogate for IKr, the rapidly activating delayed rectifier cardiac potassium current) was evaluated at near physiological temperature in stably transfected mammalian (HEK 293) cells that express the hERG gene. 20 The vehicle control was a 4 (2-hydroxyethyl) 1 piperazineethanesulfonic acid buffered physiological saline solution and supplemented with 0.3% dimethyl sulfoxide (DMSO). The positive control was terfenadine (0.3% vol/vol in DMSO).
Sotorasib was also evaluated in a telemetry dog safety pharmacology study. Dogs were implanted at least 2 weeks prior to dosing with intravenous/diaphragmatic leads and the body of the transmitter, with the negative electrocardiogram (ECG) lead placed in the jugular vein advanced to a position cranial to the right atrium and the positive ECG lead placed on the diaphragm close to the apex of the heart. Using a double Latin square dosing design, where each animal is administered all dose levels during the study with all dose levels being represented on each dosing day, sotorasib was administered by oral gavage to beagle dogs at 0, 30, 100, or 300 mg/kg on days 1, 4, 8, and 11. Telemetry data were collected continuously from at least 90 minutes predose to 25 hours postdose on every dosing day.
Repeat-Dose Toxicology Studies
In the 28-day rat study, rats received sotorasib QD at 0, 30, 100, or 200 mg/kg (10/sex/group for terminal necropsy, 5/sex/group at 0 and 200 mg/kg for the 28-day recovery necropsy). Separate satellite groups (4/sex/group) were used for toxicokinetic assessment. In the 3-month study, rats received sotorasib QD at 0, 60, 180, or 750 mg/kg (10/sex/group for terminal necropsy, 5/sex/group for the 2-month recovery necropsy). Separate satellite groups (5/sex/group) were used for toxicokinetic assessment.
In the 28-day dog study, beagle dogs received sotorasib QD at 0, 30, 100, or 300 mg/kg (3/sex/group). In the 3-month study, dogs (3/sex/group) received sotorasib at 0, 200, or 1,000 mg/kg (divided twice daily [BID]).
The dose levels in the initial 28 day studies in both rats and dogs were determined based on the previously completed exploratory dose range finding studies; however, the systemic exposures to sotorasib were not as high as expected because of changes in sotorasib crystal form. Thus, higher dose levels were selected in the 3-month studies in both species to characterize further safety profile under higher sotorasib systemic exposure.
Study end points included toxicokinetics, clinical observations, body weight, food consumption, ophthalmic examinations, electrocardiographic evaluations (dogs only), clinical pathology, organ weights, macroscopic observations, and light microscopic evaluation of a full set of tissues.
Rat Renal Mechanistic Study
A mechanistic investigation in the rat was conducted to better elucidate the mechanism of renal toxicity. 21 The study was designed to characterize a time course of sotorasib-related renal tubular degeneration/necrosis in the male rat over a 7-day period and to correlate with blood and urine biomarkers of kidney toxicity. In addition, sotorasib and its metabolites were analyzed in plasma, urine, kidney, and liver using high resolution mass spectrometry and/or matrix-assisted laser desorption/ionization (MALDI). Rats received oral doses of sotorasib at 0, 60, or 750 mg/kg.
Embryo-Fetal Development Toxicology Studies
Potential effects of sotorasib on pregnant animals and embryo-fetal development were evaluated in the SD rat and New Zealand White rabbit following the administration of sotorasib to the dam from implantation to closure of the hard palate.
In the rat study, pregnant animals (20/group in the main study and 3/group in the toxicokinetic phase) received sotorasib at 0, 60, 180, or 540 mg/kg by oral gavage QD beginning on gestation day (GD) 7 and continuing through GD 17. Blood samples were collected from pregnant females on GD 12 and 21 and from fetuses on GD 21 to determine the concentration of sotorasib in the plasma. All rats assigned to the toxicokinetic phase were euthanized on GD 13 and those assigned to the main study were euthanized on GD 21.
In the rabbit study, pregnant animals (20/group) received 0, 10, 30, or 100 mg/kg sotorasib by oral gavage from GDs 7 through 19. The dose levels were selected based on nontolerability at 300 and 1,000 mg/kg in the preliminary dose range-finding study in the pregnant rabbit (data not shown). Rabbits were euthanized on GD 29. Blood samples were collected on GD 11 to determine the concentration of sotorasib in the plasma.
The study end points included maternal viability, clinical signs, body weight, food consumption, macroscopic observations, ovarian and uterine contents (including number and distribution of corpora lutea, implantations, live and dead fetuses, and early and late resorptions), gravid uterine weight, and fetal parameters (including sex, body weight, and external, visceral, and skeletal abnormalities).
Metabolite Safety Assessment
Screening assessments including potential primary or secondary (off target) pharmacology effects and effects on in vitro hERG potassium channel and mutagenicity were performed for 3 circulating metabolites, M24 (AMG3368167), M10 (AMG3375854), and M18 (AMG3413829), identified in humans, rats, and dogs (Supplementary Figure 1). The screening assessments including primary pharmacology were assessed in the same way as for sotorasib, including coupled nucleotide exchange assay, p-ERK assay, and cell viability assay. 1 The secondary pharmacology (potential off-target effects) was assessed in the same way as for sotorasib described above. Potential effects on hERG channel were assessed in the non-GLP hERG binding assay. 22 Potential mutagenicity was assessed in either non-GLP micro Ames test (for M24) or in silico mutagenicity assessment using CASE Ultra software (for M10 and M18).23,24
Results
In Vitro Secondary Pharmacology Screening to Evaluate Off-Target Activities
In the secondary pharmacology screening, sotorasib at a 10 μM concentration did not inhibit or stimulate any off targets with greater than 50% of control activity, which is the recommended cutoff value to qualify for a significant effect. The concentration of 10 μM is approximately 6.5-fold higher than the free fraction (approximately 1.5 μM) of the maximum observed concentration (Cmax) of 7,650 ng/mL sotorasib in human plasma at the clinical highest therapeutic dose (960 mg QD). In a kinase screening panel, sotorasib at 10 μM showed no binding to 468 different kinases with a percentage of control value (a value with inverse relationship to affinity) of 35 or less—the recommended cutoff to avoid false positives for kinase-inhibitor pairs. Overall, in vitro secondary pharmacology screening results suggest that sotorasib is highly selective for KRASG12C.
Phototoxicity Studies
Sotorasib at concentrations from 0.032 to 100 μg/mL was negative in an exploratory in vitro study using 3T3 fibroblasts.
Genotoxicity
In the preliminary genotoxicity studies, sotorasib was negative in an exploratory bacterial mutagenicity Ames assay, but positive in the exploratory in vitro micro-human peripheral blood lymphocytes micronucleus assay (data not shown). Therefore, following ICH S2(R1) guidance (2011), 25 an in vitro bacterial mutagenicity Ames assay and in vivo genotoxicity assessment with 2 different tissues (combined in vivo mammalian erythrocyte micronucleus test and the alkaline comet assay in the rat) were conducted and determined to be negative.
In Vitro Dog Hepatocyte Assay
The ability of sotorasib and metabolite M24 (a major circulating metabolite in the dog) to induce CYPs and UGT was evaluated in cultured dog hepatocytes by qRT-PCR and in vitro enzyme activity assays. Increased mRNA expressions of UGTs (UGT1A6: 2.37x and UGT2B31: 5.65x) as well as some CYP isozymes (CYP1A1: 4.42x, CYP2B11: 10.7x, and CYP3A12: 3.79x) with sotorasib compared to vehicle control were confirmed.
Cardiovascular Safety Pharmacology
In the in vitro hERG assay, sotorasib inhibited hERG current with an IC50 value of 54.8 μM. The IC50 value of 54.8 μM is approximately 36-fold higher than the free fraction (approximately 1.5 μM) of the Cmax of 7,650 ng/mL sotorasib in human plasma at the highest therapeutic dose (960 mg QD). Therefore, no clinically significant interaction with the hERG channel is expected over the proposed clinical dose range.
In the in vivo cardiovascular safety pharmacology study, there were no qualitative ECG effects or quantitative changes in ECG; hemodynamic parameters including heart rate, blood pressure, or cardiac contractility; or body temperature up to the highest dose tested (300 mg/kg). The lack of acute changes in cardiovascular parameters in the stand-alone study was consistent with the results of the 28-day and 3-month repeat-dose toxicology studies in dogs (see below).
A 28-Day Repeat-Dose Toxicology Study in the Rat
Sotorasib was well-tolerated in the 28-day rat study; the severely toxic dose in 10% of the animals was determined to be greater than the highest dose tested (> 200 mg/kg). Sotorasib-related changes included a minimal to mild decrease in red blood cell (RBC) mass (hemoglobin, RBC count, and hematocrit) associated with minimal reticulocyte response (increases in males and decreases in females), and an increase in platelets, a minimal to mild increase in white blood cells, and a minimal increase in spleen weight (Table 1 and Supplementary Table 5). Sotorasib-related light microscopic changes were confined to the kidney characterized by minimal to mild renal tubular epithelial degeneration/necrosis that was restricted to the proximal tubules in the outer stripe of the outer medulla (OSOM). In the recovery group, clinical pathology parameters were similar to control, the spleen weight was similar to control, and the renal tubular injury had partially reversed; however, a few tubules were surrounded by fibroplasia.
Summary of Repeat-Dose Toxicology Studies in the Rat.

Abbreviations: KIM-1, kidney injury molecule 1; RBC, red blood cell; STD10, severely toxic dose in 10% of animals; WBC, white blood cell.
A 3-Month Repeat-Dose Toxicology Study in the Rat
In the 3-month rat toxicology study, higher exposure levels were achieved which exceeded those in the clinic (Supplementary Table 4). Sotorasib-related changes included a minimal to moderate decrease in RBC mass at ≥ 60 mg/kg (Supplementary Table 5), a minimal to moderate increase in total bilirubin at ≥ 180 mg/kg and γ-glutamyltransferase at 750 mg/kg, a mild increase in cholesterol at ≥ 60 mg/kg, a mild to marked decrease in triglycerides at ≥ 180 mg/kg, and a minimal to moderate decrease in globulins at 750 mg/kg (Table 1).
Sotorasib-related light microscopic changes were confined to the kidney characterized by renal tubular degeneration/necrosis of the proximal tubules of the OSOM. As compared to the 28-day study, the severity (minimal to marked) and incidence of the renal changes had increased which was attributed to the longer study duration and higher systemic exposures to sotorasib (Figure 1). The renal changes were also associated with increased kidney weight, macroscopic observations of rough surface or dark discoloration, and alterations in clinical pathology parameters (eg, blood urea nitrogen [BUN] and creatinine; Table 1). In the recovery animals, there were no sotorasib-related changes with the exception of histopathological changes in the kidney. By light microscopy, there was evidence of tubular regeneration (eg, basophilic tubules) but was associated with interstitial fibrosis and glomerulosclerosis, which would not be expected to be reversible.
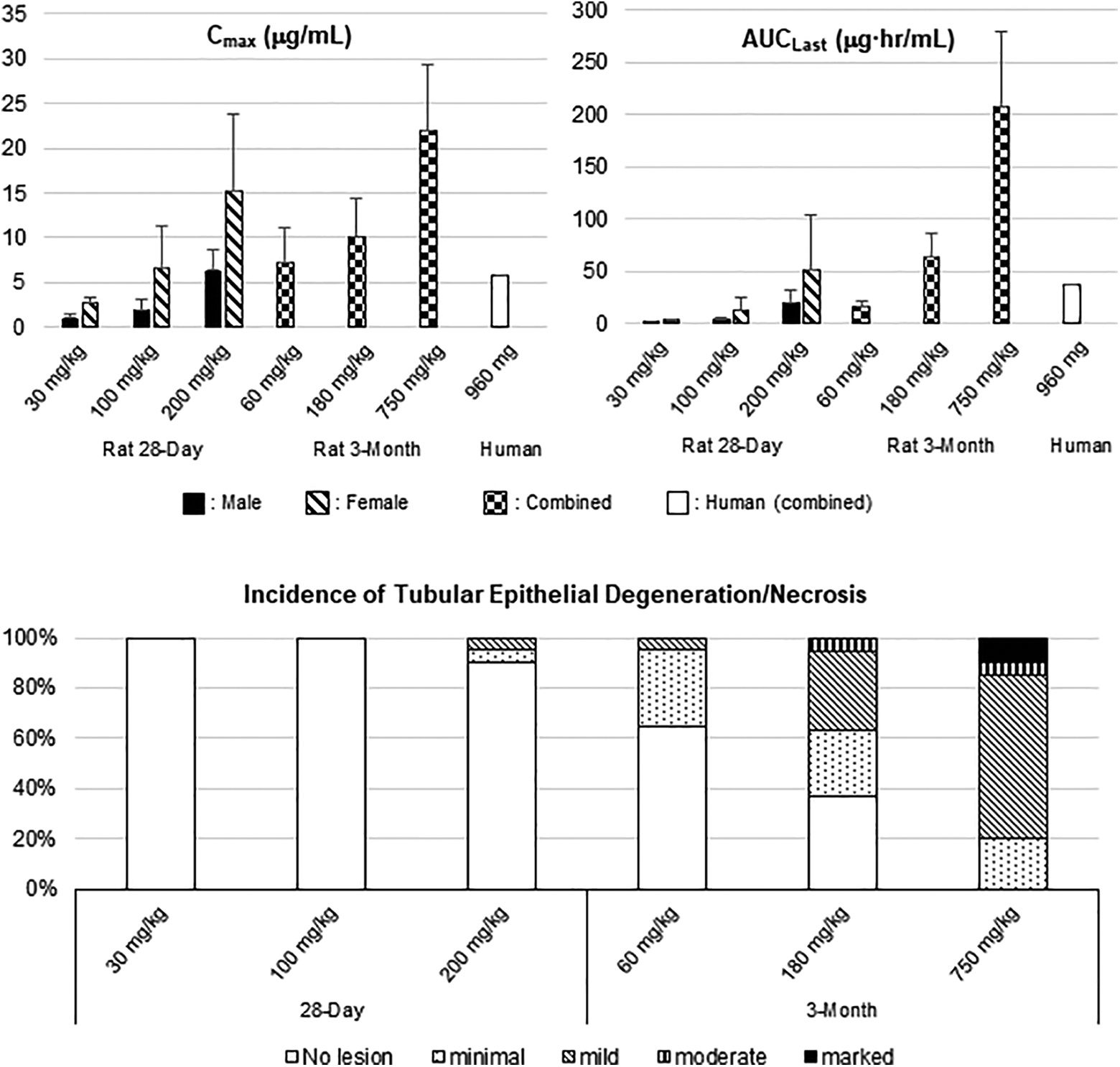
Comparison of sotorasib exposures and percentage incidence/severity of tubular epithelial degeneration/necrosis between the 28-day and 3-month rat repeat-dose toxicology studies. Top graphs: systemic exposures to sotorasib (Cmax and AUC) after the last dose. Bottom graph: percentage incidence/severity of renal tubular epithelial degeneration/necrosis. Human exposure (empty bar): Cmax (5.78 µg/mL) and AUC0-24 h (38.2 µg·h/mL) on day 8 at the highest clinical dose (960 mg). AUC indicates area under the concentration–time curve from time zero to the time of the last quantifiable concentration; Cmax, maximum observed drug concentration during a dosing interval.
A 28-Day Repeat-Dose Toxicology Studies in the Dog
Sotorasib was well-tolerated in the 28-day dog study; the highest nonseverely toxic dose was determined to be the highest dose tested (300 mg/kg). Key sotorasib-related changes in the dog consisted of a minimal to mild decrease in RBC mass associated with decreased reticulocytes (Table 2).
Summary of Repeat-Dose Toxicology Studies in the Dog.

Abbreviations: BID, twice daily; RBC, red blood cell.
A 3-Month Repeat-Dose Toxicology Studies in the Dog
In the 3-month dog study, higher dose levels were evaluated (200 and 1,000 mg/kg/d divided BID); however, even at the top dose, the area under the concentration–time curve (AUC) exposure was lower than those observed in the rat toxicology studies and in the clinic (Figure 2 and Supplementary Table 4).
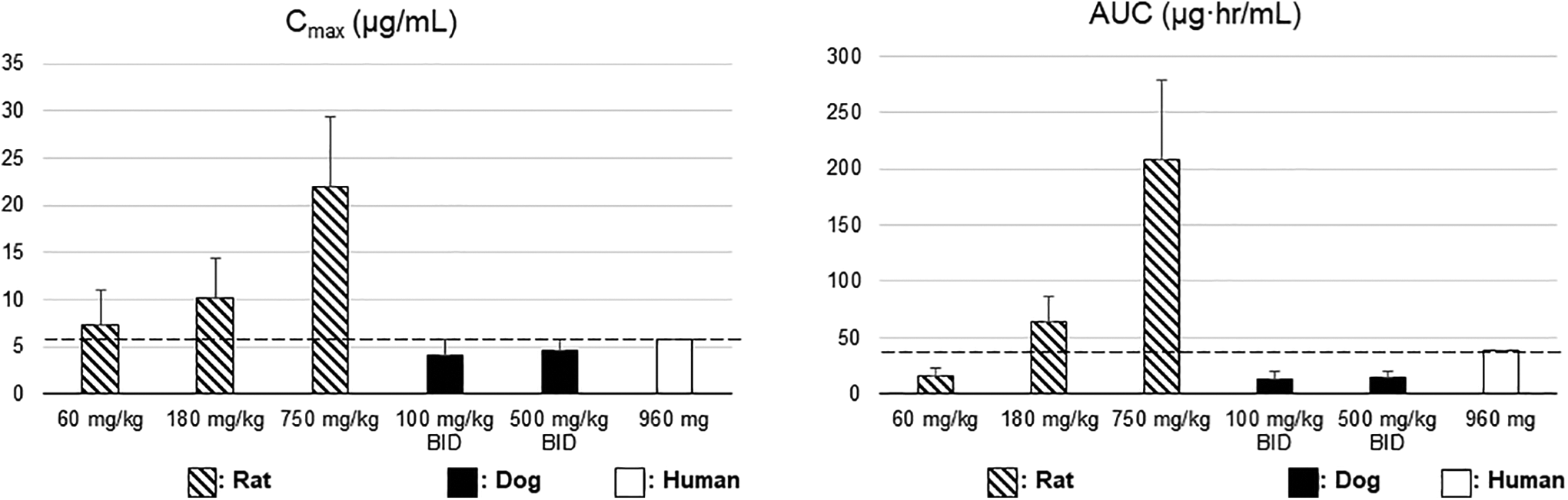
Comparison of sotorasib systemic exposures between the 3-month repeat-dose toxicology studies in the rat, dog, and human. Animal exposure: Cmax and AUC after the last dose (day 91) in the 3-month repeat-dose toxicology studies of rat/dog. Human exposure (vertical dotted lines): Cmax (5.78 µg/mL) and AUC0-24 h (38.2 µg·h/mL) on day 8 at the highest clinical dose (960 mg). AUC indicates area under the concentration–time curve from time zero to the time of the last quantifiable concentration; Cmax, maximum observed drug concentration during a dosing interval.
Sotorasib-related changes included increased liver and pituitary weight, decreased thyroid weight, abnormal content in the gall bladder, minimal to mild changes in hematology (decrease in RBC mass), and serum chemistry parameters (increase in total bilirubin, alkaline phosphatase, cholesterol, and triglycerides) (Table 2). Light microscopic changes were observed in the liver (hepatocellular hypertrophy), pituitary (hypertrophy of basophils of the pars distalis), and the thyroid gland (decreased colloid and hypertrophy of follicular epithelium); these microscopic changes were attributed to an adaptive or secondary response to hepatocellular enzyme induction.26,27
Rat Renal Mechanistic Study
Based on the site-specific renal tubular degeneration/necrosis that indicated a potential toxic metabolite etiology, a time course study was conducted. 21 Male SD rats received daily oral doses of sotorasib at 0, 60, or 750 mg/kg over a 7-day period. Sotorasib at 750 mg/kg induced renal toxicity characterized by tubular degeneration/necrosis of the S3 segment of the proximal tubule in the OSOM, consistent with the location and characteristics of tubular injury observed in the rat repeat-dose studies. Sotorasib-related light microscopic changes in the kidney correlated with increases in BUN, creatinine, kidney injury molecule 1, and clusterin. Metabolites detected in urine and kidney included M62 (lysine conjugate), M30 (cysteine-glycine conjugate), M10, M20, and M61 (oxidation of cysteine-glycine conjugate) and M21 (hydrogenation). Except for M62 and M21, the sotorasib metabolites in the kidney and/or urine were consistent with mercapturate pathway transformations. Compared to relative percentage of drug-related material in the kidney at 60 mg/kg (nonrenal toxic), M10 was increased and M62 was decreased at 750 mg/kg. The MALDI analysis revealed that metabolites M10 (cysteine conjugate) and M20 (acetylcysteine conjugate) were the most prominent metabolites in the kidney at 750 mg/kg and both were spatially restricted to the OSOM, particularly at early time points (2 and 4 hours postdose). Based on the results from this study as well as the metabolic scheme of sotorasib, the renal toxicity was attributed to the formation of a putative toxic reactive metabolite following metabolism of sotorasib by the mercapturate pathway. 21
Embryo-Fetal Development Toxicology Studies
Sotorasib was not teratogenic in the rat or rabbit embryo-fetal development toxicology studies (Supplementary Tables 6 and 7). In the rat, there were no effects on embryo-fetal development up to the highest dose (540 mg/kg) evaluated (3.9 times higher than the exposure at the human dose of 960 mg based on AUC). In the rabbit, at 100 mg/kg, 1 of 20 animals was euthanized due to body weight loss and severely reduced food intake on GD 21. Although all remaining animals survived to scheduled euthanasia on GD 29, sotorasib-related decreases in maternal food consumption and body weight gain occurred during the dosing period (Figure 3). Lower fetal body weights and a reduction in the number of ossified metacarpals were observed at the dose level associated with maternal decreased body weight gain and food consumption (100 mg/kg, 2.2 times higher than the exposure at the human dose of 960 mg based on AUC). There were no sotorasib-related effects on any ovarian or uterine parameters (corpora lutea, implantation sites, live or dead fetuses, early or late resorptions, and fetal sex) (Supplementary Table 8). There were no sotorasib-related fetal external, visceral, or skeletal malformations or variations. Lower fetal body weights (not statistically significant) and a reduction in the number of ossified metacarpals in fetuses (statistically significant) were observed at 100 mg/kg; however, the degree of these changes was minimal, and reduced ossification was not observed in the other sites examined; therefore, the changes were considered due to nonspecific maternal effects rather than direct developmental insult.28-32
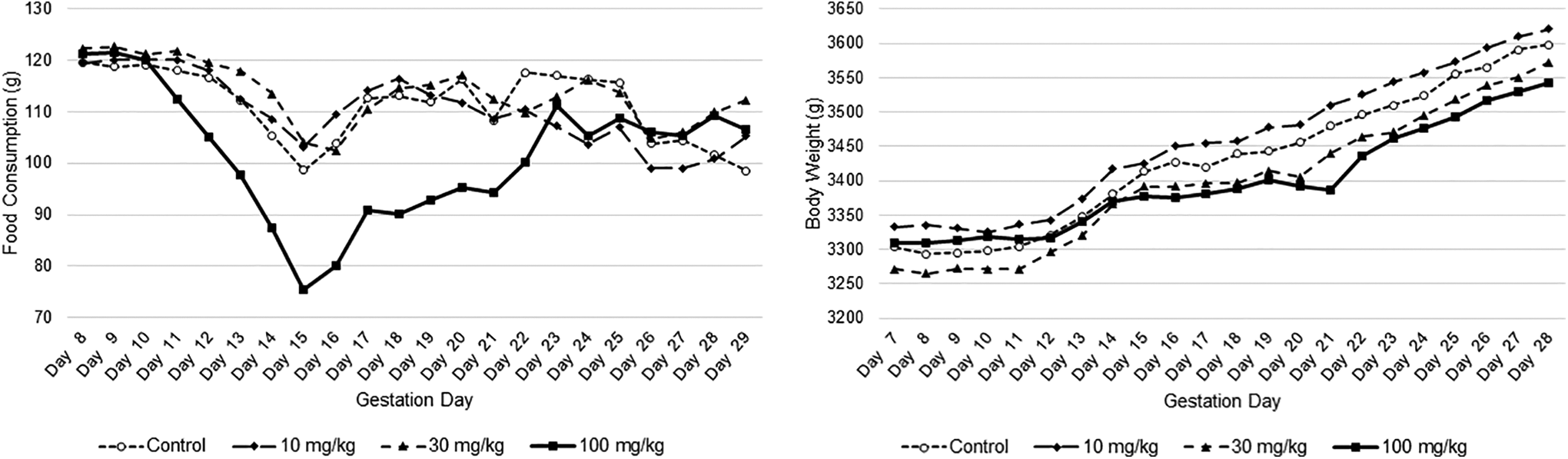
Group mean food consumption and body weight in the rabbit embryo-fetal development toxicology study.
Metabolite Safety Assessment
Among the 3 circulating metabolites (M24, M10, and M18) assessed in screening safety assessments (Supplementary Table 9), M18 has the same double bond covalent warhead as sotorasib while M24 and M10 do not (Supplementary Figure 1). Consistently, only M18 maintains primary pharmacology effects; however, the effect is markedly reduced when compared to sotorasib. Secondary pharmacology screenings did not indicate any clinically relevant off-target pharmacological activities. In vitro hERG assays did not indicate any clinically relevant interactions. Mutagenic potential for these metabolites was assessed in exploratory in vitro bacterial mutagenicity Ames assay for M24 or in silico mutagenicity predictions for M18 and M10. None of these assessments identified mutagenicity risk.
Discussion
Lack of On- and Off-Target Toxicities
A comprehensive safety assessment including primary, secondary, and safety pharmacology as well as toxicology studies was conducted to characterize the nonclinical safety profile of sotorasib and rat, dog, and human circulating metabolites (M10, M18, and M24). Sotorasib is highly selective for KRASG12C over the WT KRAS. The KRAS p.G12C mutation has only been found in tumor tissue, not normal tissue.10-13 Consistent with a tumor-specific target distribution, there were no primary pharmacology-related on-target effects identified in pivotal nonclinical GLP toxicology studies in normal nontumor bearing animals (rat and dog) as well as absence of direct embryo-fetal development effects (rat and rabbit). The absence of target-related toxicity for sotorasib is unique compared to other novel small molecule oncology drugs recently approved and/or developed.33-36 Although many novel small molecule drugs directed against cancer-specific targets, such as selective kinase inhibitors, were initially expected to result in improved efficacy with minimal adverse effects in the clinic, this promise has not been fully realized. Many of these novel targets are not unique to cancer cells, and most have critical roles in cell proliferation and survival; therefore, some on target toxicity associated with the intended primary pharmacology is expected in normal tissues/cells. Even mutant selective kinase inhibitors such as BRAF Val600Glu kinase inhibitors exhibit on target toxicity in normal tissues.37,38 Common targets of toxicity include the gastrointestinal tract, hematopoietic system, and reproductive organs; these toxicities can be dose limiting. In addition, many of the targets of novel oncology drugs have a role in developmental processes; therefore, adverse effects on embryo-fetal development, including teratogenicity, can be anticipated for most small molecule inhibitors of pathways involved in neoplastic proliferation. 33 In contrast to these novel selective kinase inhibitors, the target of sotorasib is only present in cancers with the KRASG12C mutation. Therefore, the safety profile of sotorasib is anticipated to be superior to other selective kinase inhibitors.
Lack of off-target toxicity is also an advantage of sotorasib compared to other small molecule anticancer therapeutics. Toxicities associated with closely related off targets can be observed with small molecule kinase inhibitors.36,39-43 A common target of selective kinase inhibitors is the conserved ATP binding domain of the kinase superfamily which inherently limits selectivity. Limited selectivity then complicates the distinction between on- versus off-target mechanism, identification of causal kinase(s), and the determination whether polypharmacology plays a role in the observed efficacy or toxicity. Many adverse effects including gastrointestinal, hematopoietic/bone marrow, dermatologic, and cardiac toxicities are considered to be associated with both on- and off-target effects of various kinase inhibitors.36,39-43 Sotorasib binds to a unique pocket represented by histidine 95 instead of the GTP binding pocket, 44 which might account for its high specificity and differentiated safety.
Toxicities associated with kinase inhibitors are frequently observed and can have a severe impact on patients. For example, serious cardiovascular toxicity has been observed with new small molecule kinase inhibitors that can impact clinical development.36,41 Many of the marketed kinase inhibitors have label warnings for cardiac toxicity.45-47 The standard battery of nonclinical in vitro and in vivo cardiovascular safety pharmacology studies has either failed to detect kinase inhibitor cardiac toxicity hazards or failed to provide sufficiently robust risk predictions to adequately inform governance decisions on clinical progression. 41 Thus, additional predictive nonclinical investigation including kinome and “omics” wide approaches has been proposed to better predict cardiac or other off-target toxicities and/or elucidate underlying mechanism.48,49 Sotorasib, therefore, was assessed not only in a standard battery of in vitro and in vivo cardiac safety pharmacology studies but also in extensive off-target screening including numerous various receptors, ion channels, transporters, and enzymes including various kinases. Sotorasib showed no clinically relevant off-target effects. Cysteine proteome analysis also indicated that sotorasib engaged only the Cys12-containing peptide from KRASG12C. 1 As discussed below, key findings observed in the repeat-dose toxicology studies were attributed to metabolism of sotorasib but not on- or off-target toxicity of unchanged sotorasib. Thus, sotorasib is not associated with any clinically relevant off-target effects.
Systemic Exposure Comparison Between Animal Species and Humans
Despite an excellent nonclinical safety profile, the exposure multiples based on unchanged sotorasib concentration in plasma in toxicology animal species versus humans tended to be low; exposure in the dog at 1,000 mg/kg was lower than that observed in the clinic (Figure 2). The low exposure multiple may be due, in part, to a higher metabolism rate in animal species as described hereafter. Based on mass balance studies in the rat, dog, and human, total radioactivity levels reflecting all sotorasib-related material exposures, including both unchanged sotorasib and its metabolites, appear to be higher in the rat and dog compared to the human (data not shown). The dose level used in the single dose rat mass balance study was 60 mg/kg, which was the lowest dose used in the rat 3-month repeat-dose study. The Cmax of unchanged sotorasib in the rat at 60 mg/kg was only slightly higher (1.3-fold) than that in humans; however, the Cmax of total radioactivity in the rat at 60 mg/kg was 3.3-fold higher than that in humans. The highest dose in the 3-month rat study (750 mg/kg) would suggest a much greater exposure to total sotorasib-related material as compared to the human clinical exposure. The dose level used in the dog single-dose mass balance study was 500 mg/kg. Although the Cmax of unchanged sotorasib in the dog at 500 mg/kg was even lower than that in humans, the Cmax of total radioactivity in the dog at 500 mg/kg was 5.6-fold higher than that in humans (data not shown). In the 3-month repeat-dose study in the dog, the highest dose (1,000 mg/kg divided BID) would be predicted to result in much greater exposure to total sotorasib-related material in the dog compared to human clinical exposure. The highest dose level in the 3-month repeat-dose toxicology studies was a maximum tolerated dose for the rat and a maximum feasible dose for the dog.
Key toxicological changes observed in the rat (renal toxicity attributed to putative nephrotoxic metabolite formed in the rat renal tissue) and dog (adaptive changes in the liver, pituitary, and thyroid secondary to hepatocellular enzyme induction) were attributed to metabolism of sotorasib. These results indicate that the rat and dog toxicology studies were assessed under greater exposures to total sotorasib-related materials compared to human. Such greater exposures to total sotorasib-related material in the animals were also considered to form greater amount of culprit metabolites involved in key toxicological changes in the animals. Therefore, the low exposure multiples based on only unchanged sotorasib plasma concentration do not indicate a low or a lack of safety margin. Instead, the higher exposures for the sum of sotorasib and its metabolites in rats and dogs as compared to the exposures observed in clinic suggest that the nonclinical toxicology evaluation sufficiently assessed potential safety liabilities for the clinic.
Rat Renal Toxicity
The kidney was identified as a target organ of toxicity in the rat toxicology studies. Renal toxicity in the rat was attributed to the formation of a putative toxic reactive metabolite following metabolism of sotorasib by the mercapturate pathway. 21 Based on the absence of renal toxicity in the dog and the lack of similar signals of renal toxicity in human clinical trials to date,6,7 sotorasib-related renal toxicity is considered rat specific. Our 7-day mechanistic study in the rat demonstrated much greater dispositions of M10 cysteine conjugate and other mercapturate pathway-related metabolites in the kidney and urine at the nephrotoxic dose compared to the nontoxic dose and based on MALDI, colocalization of M10 (as well as M20) in the tubular injury site (OSOM). The metabolite M10 cysteine conjugate is considered a source metabolite of the putative nephrotoxic reactive metabolite through β-lyase-mediated bioactivation. 21 The rat has been reported to be highly susceptible to β-lyase-mediated nephrotoxicity characterized by renal tubular epithelial cell degeneration/necrosis because of greater specific activities of cysteine S conjugate β-lyases in the rat renal tissue50-55 and allometric scaling. 56 Greater and more rapid formation of M10, a source metabolite of the putative nephrotoxic metabolite, in systemic circulation in the rat versus human is also proposed to contribute to greater susceptibility to sotorasib-induced nephrotoxicity in the rat. 21 Clinical trials with sotorasib have included monitoring of renal function, and to date, there has been no signal suggestive of renal toxicity similar to that observed in the rat toxicology studies.6,7
Changes in the Liver, Pituitary, and Thyroid in the Dog
In the 3-month repeat-dose toxicology study of dogs, there were sotorasib-related changes that were consistent with hepatic enzyme induction involving UGT and secondary hypothyroidism.26,27 The primary changes included centrilobular hepatocellular hypertrophy with increased liver weight, hypertrophy of thyroid follicular epithelium, decreased colloid in thyroid follicles with decreased thyroid weight, and hypertrophy of pituitary basophils of the pars distalis with increased pituitary weight. Two animals at the high dose (1,000 mg/kg/d) had morphologic features of atrophic thyroid glands, which were considered a result of exacerbation of a spontaneous age-related change.57,58 These microscopic changes in the liver, pituitary, and thyroid were also accompanied by changes in clinical pathology parameters (increased cholesterol, triglycerides, total bilirubin, and alkaline phosphatase) and a macroscopic observation of biliary sludge in the gallbladder. 59 The hepatocellular hypertrophy was not accompanied by any degenerative changes or changes in alanine aminotransferase or aspartate aminotransferase.
Hepatic microsomal enzymes are important in the maintenance of thyroid hormone homeostasis. One of the major metabolic pathways in the elimination of thyroxine (T4) and triiodothyronine (T3) is glucuronidation by UGTs. 60 Thus, increased glucuronidation enhances elimination of thyroid hormones, affecting the hypothalamic pituitary thyroid axis. Thyroid hormone production is tightly regulated by a feedback loop involving T3 and T4 levels, the hypothalamus, and the pituitary gland. Decreased T3 and T4 result in increased thyrotropin-releasing hormone by hypothalamic neurosecretory cells, which leads to increased production of thyroid-stimulating hormone from the pituitary. Prolonged stimulation of the pituitary with thyrotropin-releasing hormone can manifest morphologically as pituitary basophil (thyrotroph cell) hypertrophy correlating with increased pituitary weight.61,62 Increased thyroid-stimulating hormone stimulation of the thyroid gland leads to hypertrophy of follicular epithelium, with a concomitant decrease in colloid, which is consumed by the thyroid epithelium during thyroid hormone production.61,62
An in vitro enzyme induction assay with sotorasib in cultured beagle dog hepatocytes revealed increased mRNA expression of UGTs (UGT1A6 and UGT2B31) as well as some CYP isozymes. A dog mass balance study with 14 C sotorasib indicated that primary metabolism was mediated by nonenzymatic glutathione conjugation, oxidative N-dealkylation, and glucuronidation (data not shown). Taken together, the changes in the liver, pituitary, and thyroid observed in the 3-month dog study were interpreted as an adaptive response to reduced thyroid hormone levels by induced hepatic UGTs. In general, dogs are more resistant to the effects on hypothalamic-pituitary-thyroid axis than rodents.26,63 However, no sotorasib-related microscopic changes in the liver, pituitary, or thyroid were observed in the rat up to 750 mg/kg in the 3-month study. Therefore, species-dependent differences in sotorasib metabolism (such as a higher degree of dependency on glucuronidation among sotorasib metabolism in the dog vs a higher degree of glutathione conjugation in the rat) could be a driver for the species-specific effects on the thyroid. In general, the human is more resistant to effects on the hypothalamic-pituitary-thyroid axis than rodents or dog because of the longer half-life of T4 carried primarily by T4 binding globulin enabling stable reserves of T4. Adaptive changes in the thyroid observed in animal toxicology studies with several drugs such as lersivirine have not been considered indicative of a human health risk.26,61,63,64 To date, there has been no signal identified in the sotorasib clinical studies suggestive of hypothyroidism or thyroid dysfunction.6,7 Thyroid dysfunction is monitorable with thyroid function testing as well as measurements of serum cholesterol and triglyceride levels in the clinic, 65 and clinical trials with sotorasib have been monitoring thyroid function with regular measurement of serum T3, free T4, and thyroid-stimulating hormone as well as any clinical signs or symptoms indicating thyroid dysfunction.
Effects on RBC Mass
A decrease in RBC mass (hemoglobin, RBC count, and hematocrit) was observed in both rat and dog repeat-dose toxicology studies. The magnitude of the changes was dependent upon both sotorasib exposure and administration duration, that is, minimal in the 28-day rat study, minimum to moderate in the 3-month rat study, and minimal to mild in the 28-day and 3-month dog studies. A minimal increase in reticulocytes was observed in limited dose groups in association with decreased RBC mass; however, reticulocytes were more commonly decreased or not appropriately increased when compared to concurrent control values. Therefore, there was generally considered to not be an adequate erythropoietic response to the decreased RBC mass, suggesting that the decreased RBC mass was likely due to RBC/reticulocyte destruction and potentially in part erythropoiesis inhibition. Light microscopic correlates were observed only in the 28-day rat study (eg, increased erythroid cellularity in the bone marrow, extramedullary hematopoiesis in the spleen and liver), but not in the other studies. There were no clinical observations associated with the decreased RBC mass; therefore, the effects were observed only in hematology parameters and limited light microscopic examinations, and they were reversible. Thus, up to the maximum tolerated dose and maximum feasible dose in the 3-month study in the rat and dog, respectively, the sotorasib-related decrease in RBC mass in the nonclinical safety studies is considered to be classified as nonseverely toxic effects. Hematopoietic toxicity is commonly observed in preclinical safety studies with many small molecule anticancer therapeutics, especially kinase inhibitors since they affect rapidly proliferating tissues such as bone marrow.34,40,66,67 These molecules cause broader range of hematopoietic toxicity including not only anemia but also neutropenia, thrombocytopenia, and other various cytopenias. In addition, the magnitude of toxicity is often high, leading to the warnings and precautions in the product labels. 68 In contrast, sotorasib-related decrease in RBC mass is reasonably manageable and nonseverely toxic. Clinical trials with sotorasib have been monitoring any effects on RBC mass parameters with regular measurement of hematology as well as any clinical signs or symptoms indicative of anemia. The frequency of grade 3 anemia in the phase 1 trial with sotorasib was relatively low (3.1%). 6 To date, no dose-limiting toxic effects have been observed with sotorasib, even with extended treatment. Further accumulation of clinical safety data with sotorasib can determine more thorough profile for the effects on RBC mass in humans.
Lack of Teratogenicity or Direct Effects on Embryo-Fetal Development
Sotorasib was not teratogenic in the rat or rabbit embryo-fetal development toxicology studies. In the rat, there were no effects on embryo-fetal development up to the high dose tested (3.9 times the human exposure). In the rabbit, lower fetal body weights and a reduction in the number of ossified metacarpals in fetuses were observed only at the dose level associated with decreased body weight gain and food consumption in dams during the dosing phase (2.2 times the human exposure at the human dose of 960 mg based on AUC). The cause of fetal changes was considered to be nonspecific maternal effects rather than direct developmental insult.28-32 This secondary effect of sotorasib on embryo-fetal development is very different from other small molecule anticancer therapeutics. As many of the signaling pathways or cell markers targeted by small molecule anticancer therapeutics also have a role in developmental processes, adverse effects on embryo-fetal development, including teratogenicity, are inevitable for most small molecule inhibitors of pathways involved in neoplastic proliferation.33,34 Barrow and Clemann reported that fetal toxicity was observed in all of the 18 marketed small molecule oncology drugs with embryo-fetal development toxicology studies that they examined and the fetal toxicity occurred even at nonmaternally toxic exposure in two-thirds of the drugs. 69 Malformation and fetal lethality were observed in 12 and 13 of the 18 drugs, respectively. While cases of pregnancy in patients with cancer treated with small molecule anticancer therapeutics are very sporadic, the effects of treatment with small molecule kinase inhibitors on fetuses including skeletal malformations, soft tissue abnormalities involving abnormal vessel and organ formation, and low birth weights have been also reported in humans. 34 Thus, lack of teratogenicity or direct effects on embryo-fetal development with sotorasib demonstrated an improved safety profile for embryo-fetal development as compared to other typical small molecule oncology drugs.
Metabolite Safety Assessment
In general, nonclinical safety assessment for metabolites is not warranted for anticancer pharmaceuticals based on ICH S9 70 and ICH S9 Q&A clarification. 71 However, since sotorasib is a first-in-class small molecule inhibitor, several screening assessments were performed for 3 circulating metabolites, M24, M10, and M18, identified in humans, rats, and/or dogs. Among the 3 circulating metabolites, M18 has the same double bond covalent warhead as sotorasib while M24 and M10 do not. Consistently, only M18 maintains primary pharmacology effects; however, the effect is markedly reduced when compared to sotorasib. None of the secondary pharmacology screenings for off-target effects, in vitro hERG assays, or mutagenicity assessment raised clinically relevant concerns.
Conclusion
A comprehensive nonclinical safety assessment was conducted to characterize sotorasib. Sotorasib was negative in a battery of genotoxicity assays and negative in an in vitro phototoxicity assay. Based on in vitro assays, sotorasib had no off-target effects against various receptors, enzymes (including numerous kinases), ion channels, or transporters. Consistent with the tumor-specific target distribution (ie, KRASG12C), there were no primary pharmacology-related on-target effects identified. Renal toxicity, characterized by tubular degeneration and necrosis, was a target organ of toxicity in the rat but not the dog and was attributed to the local formation of a putative toxic reactive metabolite. In the 3-month dog study, adaptive changes of hepatocellular hypertrophy due to drug metabolizing enzyme induction was observed in the liver and associated with secondary effects in the pituitary and thyroid gland. Sotorasib was not teratogenic and had no direct effect on embryo-fetal development in the rat or rabbit. Human, dog, and rat circulating metabolites, M24, M10, and M18, raised no clinically relevant safety concerns based on the general toxicology studies, primary/secondary pharmacology screening, an in vitro hERG assay, or mutagenicity assessment. Overall, the results of the sotorasib nonclinical safety program support a high benefit/risk ratio for the treatment of patients with KRAS p.G12C-mutated tumors.
Supplemental Material
Supplemental Material, sj-docx-1-ijt-10.1177_10915818211022965 - Nonclinical Safety Profile of Sotorasib, a KRASG12C-Specific Covalent Inhibitor for the Treatment of KRAS p.G12C-Mutated Cancer
Supplemental Material, sj-docx-1-ijt-10.1177_10915818211022965 for Nonclinical Safety Profile of Sotorasib, a KRASG12C-Specific Covalent Inhibitor for the Treatment of KRAS p.G12C-Mutated Cancer by Katsu Ishida, Jonathan A. Werner, Rhian Davies, Fan Fan, Barbara Thomas, Jan Wahlstrom, James Russell Lipford and Thomas Monticello in International Journal of Toxicology
Supplemental Material
Supplemental Material, sj-docx-2-ijt-10.1177_10915818211022965 - Nonclinical Safety Profile of Sotorasib, a KRASG12C-Specific Covalent Inhibitor for the Treatment of KRAS p.G12C-Mutated Cancer
Supplemental Material, sj-docx-2-ijt-10.1177_10915818211022965 for Nonclinical Safety Profile of Sotorasib, a KRASG12C-Specific Covalent Inhibitor for the Treatment of KRAS p.G12C-Mutated Cancer by Katsu Ishida, Jonathan A. Werner, Rhian Davies, Fan Fan, Barbara Thomas, Jan Wahlstrom, James Russell Lipford and Thomas Monticello in International Journal of Toxicology
Supplemental Material
Supplemental Material, sj-docx-3-ijt-10.1177_10915818211022965 - Nonclinical Safety Profile of Sotorasib, a KRASG12C-Specific Covalent Inhibitor for the Treatment of KRAS p.G12C-Mutated Cancer
Supplemental Material, sj-docx-3-ijt-10.1177_10915818211022965 for Nonclinical Safety Profile of Sotorasib, a KRASG12C-Specific Covalent Inhibitor for the Treatment of KRAS p.G12C-Mutated Cancer by Katsu Ishida, Jonathan A. Werner, Rhian Davies, Fan Fan, Barbara Thomas, Jan Wahlstrom, James Russell Lipford and Thomas Monticello in International Journal of Toxicology
Supplemental Material
Supplemental Material, sj-docx-4-ijt-10.1177_10915818211022965 - Nonclinical Safety Profile of Sotorasib, a KRASG12C-Specific Covalent Inhibitor for the Treatment of KRAS p.G12C-Mutated Cancer
Supplemental Material, sj-docx-4-ijt-10.1177_10915818211022965 for Nonclinical Safety Profile of Sotorasib, a KRASG12C-Specific Covalent Inhibitor for the Treatment of KRAS p.G12C-Mutated Cancer by Katsu Ishida, Jonathan A. Werner, Rhian Davies, Fan Fan, Barbara Thomas, Jan Wahlstrom, James Russell Lipford and Thomas Monticello in International Journal of Toxicology
Supplemental Material
Supplemental Material, sj-docx-5-ijt-10.1177_10915818211022965 - Nonclinical Safety Profile of Sotorasib, a KRASG12C-Specific Covalent Inhibitor for the Treatment of KRAS p.G12C-Mutated Cancer
Supplemental Material, sj-docx-5-ijt-10.1177_10915818211022965 for Nonclinical Safety Profile of Sotorasib, a KRASG12C-Specific Covalent Inhibitor for the Treatment of KRAS p.G12C-Mutated Cancer by Katsu Ishida, Jonathan A. Werner, Rhian Davies, Fan Fan, Barbara Thomas, Jan Wahlstrom, James Russell Lipford and Thomas Monticello in International Journal of Toxicology
Supplemental Material
Supplemental Material, sj-docx-6-ijt-10.1177_10915818211022965 - Nonclinical Safety Profile of Sotorasib, a KRASG12C-Specific Covalent Inhibitor for the Treatment of KRAS p.G12C-Mutated Cancer
Supplemental Material, sj-docx-6-ijt-10.1177_10915818211022965 for Nonclinical Safety Profile of Sotorasib, a KRASG12C-Specific Covalent Inhibitor for the Treatment of KRAS p.G12C-Mutated Cancer by Katsu Ishida, Jonathan A. Werner, Rhian Davies, Fan Fan, Barbara Thomas, Jan Wahlstrom, James Russell Lipford and Thomas Monticello in International Journal of Toxicology
Supplemental Material
Supplemental Material, sj-docx-7-ijt-10.1177_10915818211022965 - Nonclinical Safety Profile of Sotorasib, a KRASG12C-Specific Covalent Inhibitor for the Treatment of KRAS p.G12C-Mutated Cancer
Supplemental Material, sj-docx-7-ijt-10.1177_10915818211022965 for Nonclinical Safety Profile of Sotorasib, a KRASG12C-Specific Covalent Inhibitor for the Treatment of KRAS p.G12C-Mutated Cancer by Katsu Ishida, Jonathan A. Werner, Rhian Davies, Fan Fan, Barbara Thomas, Jan Wahlstrom, James Russell Lipford and Thomas Monticello in International Journal of Toxicology
Supplemental Material
Supplemental Material, sj-docx-8-ijt-10.1177_10915818211022965 - Nonclinical Safety Profile of Sotorasib, a KRASG12C-Specific Covalent Inhibitor for the Treatment of KRAS p.G12C-Mutated Cancer
Supplemental Material, sj-docx-8-ijt-10.1177_10915818211022965 for Nonclinical Safety Profile of Sotorasib, a KRASG12C-Specific Covalent Inhibitor for the Treatment of KRAS p.G12C-Mutated Cancer by Katsu Ishida, Jonathan A. Werner, Rhian Davies, Fan Fan, Barbara Thomas, Jan Wahlstrom, James Russell Lipford and Thomas Monticello in International Journal of Toxicology
Supplemental Material
Supplemental Material, sj-docx-9-ijt-10.1177_10915818211022965 - Nonclinical Safety Profile of Sotorasib, a KRASG12C-Specific Covalent Inhibitor for the Treatment of KRAS p.G12C-Mutated Cancer
Supplemental Material, sj-docx-9-ijt-10.1177_10915818211022965 for Nonclinical Safety Profile of Sotorasib, a KRASG12C-Specific Covalent Inhibitor for the Treatment of KRAS p.G12C-Mutated Cancer by Katsu Ishida, Jonathan A. Werner, Rhian Davies, Fan Fan, Barbara Thomas, Jan Wahlstrom, James Russell Lipford and Thomas Monticello in International Journal of Toxicology
Supplemental Material
Supplemental Material, sj-tif-1-ijt-10.1177_10915818211022965 - Nonclinical Safety Profile of Sotorasib, a KRASG12C-Specific Covalent Inhibitor for the Treatment of KRAS p.G12C-Mutated Cancer
Supplemental Material, sj-tif-1-ijt-10.1177_10915818211022965 for Nonclinical Safety Profile of Sotorasib, a KRASG12C-Specific Covalent Inhibitor for the Treatment of KRAS p.G12C-Mutated Cancer by Katsu Ishida, Jonathan A. Werner, Rhian Davies, Fan Fan, Barbara Thomas, Jan Wahlstrom, James Russell Lipford and Thomas Monticello in International Journal of Toxicology
Footnotes
Acknowledgments
The authors thank the following toxicology study personnel: John R. Ciallella, Scott Sanderson, Elise M. Lewis, Jeffrey A. Solomon, Darol E. Dodd, Marilyn Registre, Zhuzai Xiang, and Annie Hamel.
Author Contributions
Katsu Ishida contributed to conception and design, contributed to analysis and interpretation, drafted the manuscript, and critically revised the manuscript. Jonathan A. Werner contributed to conception and design; contributed to acquisition, analysis, and interpretation; and critically revised the manuscript. Rhian Davies contributed to conception and design; contributed to acquisition, analysis, and interpretation; and critically revised the manuscript. Fan Fan contributed to conception and design; contributed to acquisition, analysis, and interpretation; and critically revised the manuscript. Barbara Thomas contributed to conception and design, contributed to analysis and interpretation, and critically revised the manuscript. Jan Wahlstrom contributed to conception and design; contributed to acquisition, analysis, and interpretation; and critically revised the manuscript. J. Russell Lipford contributed to conception and design; contributed to acquisition, analysis, and interpretation; and critically revised the manuscript. Thomas M. Monticello contributed to conception and design, contributed to analysis and interpretation, and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The authors are employees of and own stock in Amgen Inc.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Amgen funded this research. This research received no specific grant from any funding agency in the public, commercial, private, or not for profit sectors.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.