
Abstract
Etelcalcetide is a novel
Introduction
Secondary hyperparathyroidism (HPT) is a disorder characterized by parathyroid gland hyperplasia and increased concentrations of circulating parathyroid hormone (PTH) with associated abnormalities of serum calcium, phosphorus, and fibroblast growth factor 23. 1 The disease occurs most commonly as a consequence of chronic kidney disease (CKD). The biochemical changes are associated with abnormalities in bone histology, increased fracture risk, vascular and soft tissue calcifications, calciphylaxis, a variety of symptoms (eg, muscle weakness, fatigue, lethargy, bone and joint pain), and increased mortality. 2
The principal regulator of PTH secretion is the calcium-sensing receptor (CaSR) expressed on chief cells in parathyroid tissue. Calcimimetics target the CaSR in the parathyroid gland and provide a means of regulating PTH secretion without increasing serum calcium and phosphorus levels. 3,4 Cinacalcet (Sensipar®/Mimpara®), an orally administered small molecule drug, is the only calcimimetic currently approved for the treatment of secondary HPT in patients with CKD undergoing hemodialysis. 5 Etelcalcetide (also known as AMG 416) is a calcimimetic that functions as an allosteric activator of the CaSR and is being developed as an intravenous (IV) calcimimetic for the treatment of secondary HPT in patients with CKD on hemodialysis. 6 -8 Etelcalcetide is administered to patients as an IV bolus dose 3 times per week at the end of hemodialysis. Unlike cinacalcet, etelcalcetide has a low potential for drug–drug interactions due to the lack of interaction with CYP450 enzymes. 9 As an IV calcimimetic administered in the dialysis unit setting, etelcalcetide reduces the burden of medication adherence for patients, which may potentially lead to improved disease management.
Etelcalcetide is a synthetic peptide comprised of 7
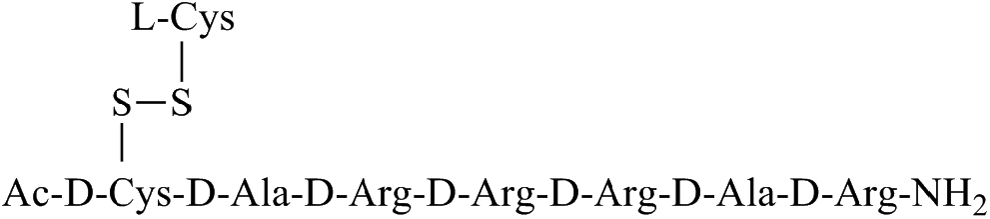
Structure of etelcalcetide. Ac denotes the acetyl group on N-terminus of the
Intravenous administration of etelcalcetide in rats and dogs resulted in dose-proportional increases in plasma exposure, with clearance being dependent on renal function.
9,10,12
Etelcalcetide is predominantly biotransformed into blood via the replacement of
Reported here are the safety pharmacology, pivotal chronic repeat-dose toxicology, genetic toxicology, carcinogenicity, and reproductive and developmental toxicology studies in support of global marketing authorization of etelcalcetide. The results of these studies support the use of etelcalcetide in the intended patient population.
Methods and Materials
Test and Control Article
Etelcalcetide (Ac-c[C]arrrar-NH2; 1,048 Da), or N-acetyl-
Nonclinical Test Systems
All animals were housed at Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) international-accredited facilities and all research protocols were approved by their respective institutional animal care and use committees, or similar animal welfare committees. Sprague Dawley (Crl: CD[SD]) rats were group-housed in polycarbonate housing on bedding or wire mesh or individually housed in nesting boxes for reproductive studies. Rats in the carcinogenicity study were individually housed. Animals had ad libitum access to water and pelleted feed or meal, except for an overnight food fast prior to blood collection at scheduled necropsy. Beagle dogs were housed in stainless steel cages on corncob bedding and had ad libitum access to food and water, except for an overnight food fast prior to blood collection at scheduled necropsy. Transgenic (Tg) rasH2 mice (CByB6F1-Tg[HRAS]2Jic [+/− hemizygous c-Ha-ras]) were individually housed in polycarbonate housing on Sani-chip hardwood bedding, with ad libitum access to food and water. Muta mice (CD2-lacZ80/HazfBR) were housed 3 per polycarbonate cage on softwood bedding, with ad libitum access to food and water. Time-mated New Zealand White (Hra:[NZW]SPF) rabbits were housed individually in stainless steel cages suspended above milled corncob bedding and had ad libitum access to water and pelleted feed during the dose administration period. All animals were maintained on a 12:12 hour light:dark cycle in rooms with appropriate temperature and humidity controls. Ages of animals are indicated below for their respective studies.
Nonclinical safety studies were performed in accordance with Good Laboratory Practices (United States Code of Federal Regulations, Title 21, Part 58: Good Laboratory Practice for Nonclinical Laboratory Studies). Etelcalcetide was pharmacologically active in all nonclinical species based on dose-dependent lowering of serum calcium and PTH as demonstrated in rat 12 and dog (noted below) as well as mouse and rabbit (data not shown). In nonclinical safety studies, etelcalcetide was administered by the IV route to mimic the therapeutic route of administration in patients, except in the carcinogenicity and Muta mouse studies where the subcutaneous (SC) route was used due to technical limitations of repeat IV dosing in these models. The formulated drug was well tolerated when administered by IV or SC route. High doses were considered maximally tolerated. Lower doses were selected in order to characterize the dose response and establish a no observed adverse effect level (NOAEL).
Etelcalcetide Biokinetic and Toxicokinetic Analysis
Analytical studies confirmed dose formulation concentrations, homogeneity, and stability using a validated reverse-phase liquid chromatography (LC) method with ultraviolet detection at 205 nm. For analysis of plasma drug levels, a sensitive LC method with tandem mass spectrometric detection (LC-MS/MS) was adapted and validated from previous methodology and used to measure etelcalcetide in acidified plasma. 9 All samples were prepared for LC-MS/MS analysis by a protein precipitation method, except for the mouse plasma method that utilized solid-phase extraction. Toxicokinetic (TK) parameters were derived from noncompartmental analysis of the individual plasma concentration–time data using Phoenix WinNonlin (Pharsight Corp, Sunnyvale, California).
Safety Pharmacology
The effects of etelcalcetide on the human ether-a-go-go (hERG) potassium channel current were evaluated in HEK-293 cell line stably transfected with hERG complementary DNA using the whole-cell patch clamp assay at concentrations up to 10 μg/mL (9.5 μmol/L).
An integrative safety pharmacology study to evaluate respiratory, neurological, and cardiovascular effects (electrocardiograms [ECGs], heart rate, mean arterial pressure) was conducted in male beagle dogs (n = 4, approximately 1 year of age) surgically implanted with a radiotelemetry transmitter (Data Sciences International, St Paul, Minnesota). Single IV bolus doses (0.5 mL/kg over 1 minute) were administered at 0.3 or 1.5 mg/kg (sequentially in the same dogs following drug washout), a high dose that exceeds the maximum tolerated dose in chronic repeat-dose studies (0.9 mg/kg every other day). Following a washout period, the same dogs were administered etelcalcetide 1.5 mg/kg again to further characterize the temporal relationship between plasma drug levels, serum calcium and plasma PTH, and the cardiovascular changes over a 48-hour period. Plasma PTH was measured using the canine intact PTH enzyme-linked immunosorbent assay kit (Immutopics International, San Clemente, California). All cardiovascular data were recorded using the Data Sciences computer system (Dataquest Acquisition software). Heart rate, mean arterial pressure, and body temperature were recorded from each animal for 30 seconds at 5-minute intervals. Electrocardiograms and arterial blood pressure waveforms were recorded from each animal for 30 seconds at 30-minute intervals. Electrocardiograms were also evaluated in dogs as part of the repeat-dose toxicity studies with a duration of 1 to 9 months at doses up to 0.9 mg/kg.
General Repeat-Dose Toxicology
Repeat-dose IV toxicity studies of up to 6 and 9 months in duration were conducted in rats and dogs, respectively. The safety profile established in a prior 28-day repeat-dose IV toxicity study in rats at the same dose levels was similar to that observed in the chronic study (data not shown). Etelcalcetide was administered to rats (approximately 7-8 weeks of age at the first dose) once daily by the IV route (2 mL/kg over ∼30 seconds) at doses of 0, 0.3, 1, or 3 mg/kg for up to 182 consecutive days to evaluate the toxicity (n = 10/sex/group) and TKs (n = 9/sex/group; test article groups only), followed by a 28-day recovery period (n = 5/sex/group in the vehicle control and high-dose groups) to evaluate reversibility. An interim necropsy on day 91 was also included, and the results were similar to the terminal necropsy on day 182 (data not shown). The high dose was based on poor tolerability at 5 mg/kg observed in a short-term (≤7 day) range-finding study.
Etelcalcetide was administered to dogs (approximately 6-7 months of age at the first dose) once every other day by the IV route (0.5 mL/kg over ∼30 seconds) at doses of 0, 0.2, 0.5, and 1.2/0.9 mg/kg over a 273-day period to evaluate the toxicity and TK (n = 3/sex/group), followed by a 28-day recovery period (n = 2/sex/group in the vehicle control and high-dose groups) to evaluate reversibility. The reduced dosing frequency in dogs was due to differences in tolerability related to a longer plasma half-life observed in dogs relative to rats. An interim necropsy on day 92 was also included, and the results were similar to the terminal necropsy on day 273 (data not shown). The high dose level was based on poor tolerability at 1.5 mg/kg in a prior 28-day repeat-dose IV toxicity study. The safety profile in this 28-day study was otherwise similar to that observed in the chronic study and so is not presented here. During the chronic toxicity study, the high dose was reduced from 1.2 mg/kg to 0.9 mg/kg on days 25 and 27 in males and females, respectively, due to poor tolerability (body weight decreases and clinical signs of lethargy, prostration, and reduced activity).
The following study parameters were evaluated in both rat and dog studies: mortality, clinical observations, body weight, food consumption, TK analysis, clinical chemistry, hematology, coagulation, ophthalmology, urinalysis, organ weights, macroscopic observations, and light microscopic evaluation of tissues.
Genotoxicity Assessment
In vitro, etelcalcetide was tested in a bacterial reverse mutation assay in Salmonella typhimurium (strains TA98, TA100, TA1535, and TA1537) and Escherichia coli (strain WP2 uvrA pKM101) using the plate incorporation method at concentrations up to 5,000 μg per plate in the presence and absence of a metabolic activation system derived from Aroclor-induced rat liver S9 fraction. The clastogenic potential of etelcalcetide was evaluated in an in vitro mammalian chromosome aberration assay using human peripheral blood lymphocytes in both the presence and absence of a metabolic activation system derived from an Aroclor-induced rat liver S9 fraction. Based on cytotoxicity range-finding studies, etelcalcetide was tested at concentrations up to 5,000 μg/mL for 4 and 20 hours in the absence of S9 and up to 4,000 μg/mL for 4 hours in the presence of S9. All cells were harvested 20 hours after treatment initiation. The mutagenic potential of etelcalcetide in mammalian cells was evaluated based on forward mutations at the hypoxanthine-guanine phosphoribosyl transferase locus in Chinese hamster ovary (CHO) cells at concentrations up to 5,000 μg/mL in the presence and absence of an Aroclor-induced rat liver S9 fraction.
In vivo, etelcalcetide was evaluated for genotoxic (clastogenic/aneugenic) potential in a bone marrow micronucleus study in male rats (6-8 weeks of age). In an initial 3-day dose range-finding study, an IV dose (5 mL/kg over 30 minutes) of 5 mg/kg once daily was determined to be the maximum tolerated dose based on mortality and clinical signs of toxicity (lethargy, piloerection) at doses of 7.5 mg/kg and higher. In the definitive micronucleus study, male rats (n = 5/group) were dosed IV (5 mL/kg) once a day with etelcalcetide at 0, 1.25, 2.5, and 5.0 mg/kg for 3 consecutive days and evaluated for micronucleated polychromatic erythrocytes in the bone marrow 24 hours after the last dose. Cyclophosphamide (40 mg/kg; n = 5 male rats) was administered IV 24 hours prior to bone marrow collection as a positive control. Etelcalcetide was tested for mutagenicity based on the induction of gene mutations in the lacZ transgene in liver and bone marrow from male Muta mice (CD2-lacZ80/HazfBR). Both tissues are expected to have high exposure based on tissue distribution data in the rat. 9 A previous study demonstrated that male mice showed slightly higher (<2×) exposure compared to females, and therefore, only male animals were used for both the range-finding and main experiment. In a range-finding study, Muta mice (n = 3/group) were dosed once daily by the SC route (10 mL/kg) with vehicle and etelcalcetide at dose levels of 8 mg/kg for 7 days, or 4 or 6 mg/kg for up to 28 days. Based on mortality and clinical signs of toxicity, 4 mg/kg was determined to be the maximum tolerated dose. Therefore in the main study, Muta mice (n = 6/group) were dosed once daily by the SC route at 1, 2, or 4 mg/kg for 28 days. A vehicle control and untreated control group were also included. Animals were euthanized 3 days after the last dose. Satellite animals were included for TK assessment. Following euthanasia, tissues were flash frozen in liquid nitrogen and stored at −80°C nominal. DNA was extracted from frozen liver and bone marrow of all animals, packaged into a bacteriophage lambda vector, and transfected into competent E coli C lacZ− galE − Kanr bacteria. Plating was performed for total titer of plaque-forming units and total lacZ− mutants. At least 200 000 plaque-forming units per tissue per animal, generated from at least 3 independent packaging reactions, were generated. Mutation frequency was calculated using the total number of viable bacteriophage (ie, phage that has been successfully packaged with the lacZ transgene and visible as plaques on titer plates), compared with the number of bacteriophage that contains mutated lacZ transgenes (ie, plaques on positive selection plates), and expressed as the number of mutants per 106 plaque-forming units. The mean mutant frequencies for each tissue in the vehicle group were within the 95% reference range of the laboratory’s historical control data. Correct functioning of the DNA packaging reaction was confirmed by data from concurrently evaluated positive control DNA samples from a previous study, thus confirming the validity of the study. Group differences in mutation frequency were evaluated by 1-way analysis of variance and 1-sided Dunnett test for pairwise comparisons of treated groups with the vehicle control, using the animal as the unit of analysis. A linear contrast test was also performed (excluding the untreated control) to evaluate the dose response.
Carcinogenicity Assessment
A 6-month carcinogenicity study in the transgenic rasH2 mouse, and a 2-year carcinogenicity study in the rat, was conducted to assess carcinogenic risk in support of marketing authorization. Study design and doses were as agreed upon by the US Food and Drug Administration Executive Carcinogenicity Assessment Committee. Dose selection for both studies was based on tolerability established in SC dose range-finding studies. Etelcalcetide was administered once daily by the SC route (10 mL/kg bolus) to Tg.rasH2 mice (5 weeks of age at the start of dosing) for 26 weeks (n = 25/sex/group) at doses of 0.3, 1, or 3 mg/kg to female mice and 0.375, 0.75, and 1.5 mg/kg to male mice. The difference in tolerability between sexes was likely related to the slightly higher Cmax and exposure (area under the concentration-time curve [AUC]) in males relative to females, which was also reflected by the greater magnitude of serum calcium suppression in males, as determined in a 28-day SC range-finding study (data not shown). A vehicle control and saline control group were also included (n = 25/sex/group). The TK profile of etelcalcetide was established in homozygous Tg.rasH2 mice on day 1 and during week 26 (n = 3/time point/sex/group). Urethane was included as a positive control group and administered by the intraperitoneal route (n = 10/sex) on days 1, 3, and 5 at a dose of 1,000 mg/kg. Positive control animals were euthanized on day 92 following the expected signs of toxicity due to splenic and lung tumors.
In a 2-year rat carcinogenicity study, etelcalcetide was administered once daily by the SC route (1 mL/kg bolus) to rats (6 weeks of age at the start of dosing; n = 65/sex/group) at doses of 0.2, 0.4, 0.8, or 1.6 mg/kg. A vehicle control and saline control group were also included (n = 65/sex/group). The addition of a fourth dose group at 1.6 mg/kg was due to uncertainty over tolerability based on the previous range-finding studies. The TK profile of etelcalcetide was established in rats on day 183 (n = 3/time point/sex/group).
The following study parameters were evaluated in both carcinogenicity studies: mortality, clinical observations, body weight, food consumption, TK, macroscopic observations, and light microscopic evaluation of tissues.
Reproductive and Developmental Toxicity Assessment
Potential effects on fertility were evaluated in the rat. Etelcalcetide was administered once daily by the IV route (2 mL/kg) to male and female rats at doses of 0, 0.75, 1.5, and 3 mg/kg (n = 25/sex/group). The high dose was the maximum tolerated dose based on previous range-finding studies. Males received 28 daily doses before cohabitation, during cohabitation, and continuing through the day before scheduled euthanasia following necropsy of the last female, for a total of 58 to 59 doses. Females received 14 daily doses before cohabitation and through gestational day (GD) 7 for a total of 22 to 36 doses. On GD 15, a laparohysterectomy was performed on females with evidence of mating or 10 days after the mating period if no evidence of mating was observed. The following parameters were evaluated: mortality, clinical observations, body weight, food consumption, serum calcium (8 hours postdose on the last day of dosing prior to cohabitation), mating index (percentage showing evidence of mating), fertility index (percentage of those siring a litter or confirmed pregnant/number used for mating), copulation or conception index (percentage of those siring a litter or confirmed pregnant/number confirmed mated), male reproductive organ weights, sperm parameters (testicular and epididymal sperm numbers and sperm production rate, motility, and morphology), estrous cycle length, and intrauterine survival (number of viable embryos, implantation sites, and corpora lutea).
Potential effects on embryo–fetal development were evaluated in the rat and rabbit. In pregnant rats (approximately 14 weeks of age at the start of dosing), etelcalcetide was administered once daily by the IV route (2 mL/kg over ∼60 seconds) at doses of 0, 0.8, 1.6, or 3.2 mg/kg (n = 25/group) from GD 6 to 17. A laparohysterectomy was performed on GD 20. In pregnant rabbits (approximately 6 months of age at the start of dosing), etelcalcetide was administered once daily by the IV route (2 mL/kg over ∼60 seconds) at doses of 0, 0.375, 0.75, or 1.5 mg/kg (n = 22/group) from GD 7 to 20. A laparohysterectomy was performed on GD 29. Doses in the rat and rabbit were based on maternal tolerability determined in dose range-finding studies. The following parameters were evaluated: clinical observations, body weight and food consumption, gravid uterine weight, and serum calcium (8 hours postdose on GD 7 and 17 for rat; 8 and 24 hours postdose on GD 7 and 20 for rabbit). At necropsy, the uteri, placentae, and ovaries were examined, and the numbers of live and dead fetuses, early and late resorptions, total implantations, and corpora lutea were recorded. Gravid uterine weights were recorded, and net body weights and net body weight changes were calculated. The fetuses were weighed, sexed, and examined for external, visceral, and skeletal malformations and developmental variations.
Potential effects on prenatal and postnatal development were evaluated in developing rat offspring (F1) consequent to exposure of dams (F0) from implantation through lactation and weaning. Etelcalcetide was administered to pregnant rats once daily by the IV route (2 mL/kg) at doses of 0, 0.75, 1.5, or 3.0 mg/kg (n = 25/group) from GD 7 through lactation day (LD) 20. Doses were based on tolerability in pregnant rats established in the dose range finding and definitive embryo–fetal development study, as described previously. The following parameters were evaluated in F0 rats: clinical observations, body weight, food consumption, duration of gestation and parturition, and maternal behavior. Plasma drug levels were evaluated in a separate cohort of pregnant F0 rats (n = 4/group) on GD 7 and 21. Fetal plasma drug levels (pooled by litter) were evaluated in the TK cohort following necropsy 1 to 2 hours postdose on GD 21. Prior to weaning, F1 rats were evaluated for viability, clinical observations, body weight, litter size, and sex ratio. On LD 21, F1 generation rats were weaned, and 1/sex/litter was selected for continuation on study for the assessment of any potential effects on growth, behavior, and reproductive capacity. Following weaning on LD 21, F1 rats were evaluated for viability, clinical observations, body weight, food consumption, sexual maturation (vaginal opening beginning on postpartum day 28 for females and preputial separation beginning on postpartum day 38 for males), and behavior (passive avoidance on postpartum day 24 [±1 day] for learning and memory, motor activity on postpartum day 60 [±2 days], M-shaped water maze on postpartum day 70 [±2 days] for overt coordination, swimming ability, memory, and learning). The test sessions for passive avoidance and water maze were separated by a 1-week interval, and the criterion was the same for both days of testing. F1 adults were cohabitated and evaluated for reproductive capacity starting on postpartum day 93. The ovaries and uteri of F1 females were examined for the number and distribution of corpora lutea, implantations, and/or viable and nonviable embryos.
Results
Safety Pharmacology
There was no appreciable change in hERG current up to the highest tested concentration (10 μg/mL; 9.5 μmol/L), indicating that etelcalcetide had no direct effect on hERG channel function.
There were no etelcalcetide-related effects on the respiratory or central nervous system (CNS) in the dog at doses of 0.3 or 1.5 mg/kg. At the high dose, animals showed clinical signs of tremoring, most notably at 24 hours after dosing coincident with maximal (22% to 36%) decreases in serum calcium and not correlative with plasma Cmax of etelcalcetide. The tremoring was consistent with the decrease in PTH (below the lower limit of quantitation) observed 6 through 18 hours postdose, which gradually returned to baseline by 48 hours postdose. Decreases in serum magnesium (23%-29%) and increases in serum phosphorus (up to 47%) were also observed and were most pronounced from 21 to 24 hours postdose. These serum ion concentrations had substantially or completely reversed to baseline values by 48 hours post dose.
All the ECGs in the dog safety pharmacology study were qualitatively within normal limits. Etelcalcetide at 1.5 mg/kg increased mean corrected QT interval (QTc) from 12% to 16% relative to controls, beginning from approximately 16 through 22 hours postdose, with intermittently higher mean values thereafter at 28 to 32 hours. Changes in QTc were temporally associated with maximum decreases in serum calcium levels and normalized following recovery of serum calcium levels (Figure 2). No change in QTc was observed immediately after dose administration when plasma drug levels were maximal. Similar increases in QTc (13% relative to vehicle controls at the end of the study) were also observed in male dogs in the repeat-dose chronic toxicity study at the high dose only (1.5 mg/kg). Although the increase was statistically significant, the values were within the range of historical control values in the laboratory.
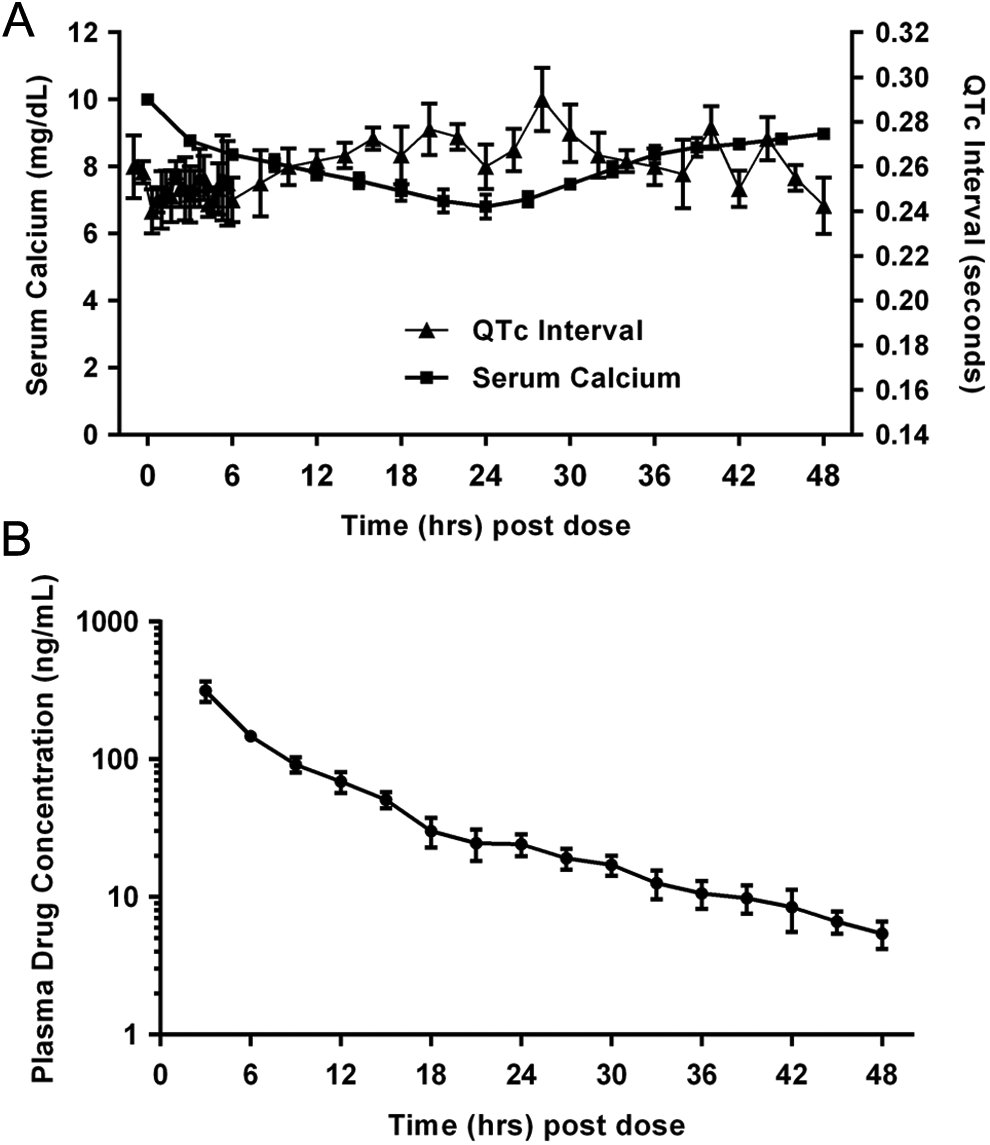
Serum calcium and corrected QT interval (QTc) following a single intravenous (IV) dose of etelcalcetide in the dog. Changes in serum calcium, QTc, and plasma drug concentrations were evaluated over a 48-hour period following a single dose of 1.5 mg/kg to male dogs (n = 4). A, Serum calcium (squares) was measured every 3 hours after dosing, up to 48 hours postdose. Corrected QT interval (triangles) was measured at 20-minute intervals for 30 seconds for the first 6 hours postdose and then at 2-hour intervals for 30 seconds from 6 to 48 hours postdose. B, Plasma drug concentrations were measured every 3 hours after dosing up to 48 hours postdose.
Small increases in heart rate and blood pressure were observed in the safety pharmacology study at 1.5 mg/kg, which was temporally associated with decreased serum calcium and not plasma drug concentrations. There was no etelcalcetide-related effect on heart rate at 0.9 mg/kg in the chronic dog toxicity study.
General Repeat-Dose Toxicology
Short-term (≤7 days) repeat-dose range-finding studies were conducted in the rat and dog to establish doses for longer term studies. Acute effects consisted of mortality (rats only), severe hypocalcemia, tremoring, decreased body weight, and food consumption at daily doses of 5 mg/kg in rats and ≥2 mg/kg in dogs. The hypocalcemia, tremoring, and changes in body weight and food consumption were reversible.
In the chronic repeat-dose toxicity study in rats, there was no etelcalcetide-related mortality after daily dosing for 6 months at 0.3, 1, or 3 mg/kg. Slight to mild tremors were observed in the 3 mg/kg group. Body weight and body weight gains were decreased in males in the 3 mg/kg group (29% lower than controls) and were associated with decreased food consumption. There was no effect on body weight or food consumption in females. There were no etelcalcetide-related clinical signs or effects on food consumption and body weight during the recovery periods.
There was a dose-dependent decrease in serum calcium (up to 22% at the high dose relative to controls on day 1) and increase in serum phosphorus (up to 19% at the high dose relative to controls on day 1) in all dose groups when measured 8 hours postdose on days 1, 45, 91, and 182. The changes in serum calcium were comparable among sexes and across time points and were considered adverse in the 3 mg/kg group due to associated tremors and body weight changes. Changes in serum calcium and phosphorus returned to control levels by the end of the recovery period. Other etelcalcetide-related effects on clinical chemistry and hematology parameters were small in magnitude, within the historical control range, had no correlated microscopic changes, and were all reversible. There was a decrease in urinary-specific gravity and an increase in urine volume and incidence of pale yellow or colorless urine at 3 mg/kg. As there were no microscopic changes in the kidneys, these changes were considered nonadverse.
Spleen weights (absolute and relative to brain) were decreased in males at doses ≥1 mg/kg. The change in spleen weight was correlated with microscopic changes in decreased splenic red pulp. Adrenal weights (absolute and relative to brain/body) were increased at 3 mg/kg in both sexes; however, there was no correlating microscopic change. No etelcalcetide-related organ weight changes were observed at the end of the recovery period. In the thymus, etelcalcetide-related increases in lymphoid depletion characterized by individual cell necrosis/apoptosis (minimal to moderate) were observed at ≥1 mg/kg. Focal or multifocal stomach erosions (minimal to mild) were observed at 3 mg/kg. The low incidence and severity of the stomach erosions and the absence of any reduction in red blood cell parameters suggest that these erosions are not adverse. There were no etelcalcetide-related microscopic changes observed at the end of the recovery period.
The sex-combined plasma TK parameters for pivotal toxicology studies are summarized in Table 1. The increases in Cmax and AUC were proportional to dose, and there was no difference in exposure between sexes, although females had slightly higher exposures (<2-fold) on day 1. Exposure values were slightly higher at the end of the study as compared to day 1 values, reflecting slight (<2-fold) accumulation in plasma.
Summary of Findings and Toxicokinetic Data for Pivotal Toxicology Studies of Etelcalcetide.
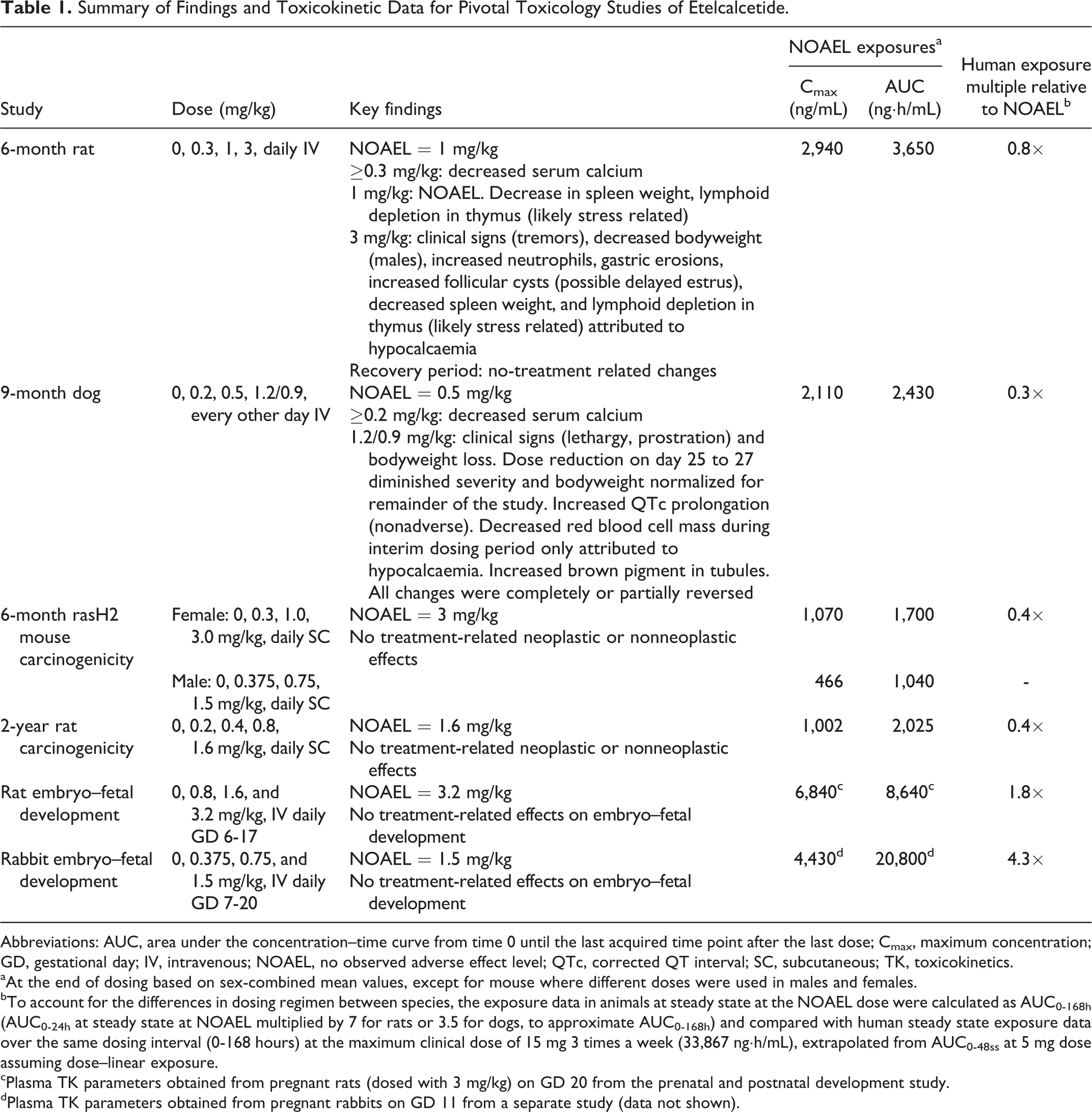
Abbreviations: AUC, area under the concentration–time curve from time 0 until the last acquired time point after the last dose; Cmax, maximum concentration; GD, gestational day; IV, intravenous; NOAEL, no observed adverse effect level; QTc, corrected QT interval; SC, subcutaneous; TK, toxicokinetics.
aAt the end of dosing based on sex-combined mean values, except for mouse where different doses were used in males and females.
bTo account for the differences in dosing regimen between species, the exposure data in animals at steady state at the NOAEL dose were calculated as AUC0-168h (AUC0-24h at steady state at NOAEL multiplied by 7 for rats or 3.5 for dogs, to approximate AUC0-168h) and compared with human steady state exposure data over the same dosing interval (0-168 hours) at the maximum clinical dose of 15 mg 3 times a week (33,867 ng·h/mL), extrapolated from AUC0-48ss at 5 mg dose assuming dose–linear exposure.
cPlasma TK parameters obtained from pregnant rats (dosed with 3 mg/kg) on GD 20 from the prenatal and postnatal development study.
dPlasma TK parameters obtained from pregnant rabbits on GD 11 from a separate study (data not shown).
There was no etelcalcetide-related mortality in dogs after 9 months of dosing every other day at 0.2, 0.5, or 1.2/0.9 mg/kg. Clinical signs of lethargy, prostration, and reduced activity were limited to the 1.2 mg/kg group prior to dose reduction on day 25/27. Etelcalcetide-related clinical signs were not observed at ≤0.5 mg/kg. Body weights and body weight gains were decreased in the 1.2 mg/kg group, and this effect was progressive through day 22 and associated with decreased food consumption. The dose-level reduction to 0.9 mg/kg diminished the severity of clinical signs and body weight changes. Body weights at 0.9 mg/kg eventually returned to the control group range by day 91 and remained similar to controls throughout the remainder of the study. There were no etelcalcetide-related clinical signs or effects on food consumption and body weight during the recovery periods.
There was a dose-dependent decrease in serum calcium (up to 26% in the high-dose group relative to controls on day 1) and increase in serum phosphorus (up to 20% in the high-dose group relative to controls on day 1) at all dose levels when evaluated 24 hours postdose on days 1, 45, 91, 183, and 273. Reductions in serum calcium and increases in serum phosphorus were notable 4 hours postdose and maximal at 24 hours postdose, and the changes were comparable in both males and females. The degree of reduction in serum calcium was considered adverse at the 1.2/0.9 mg/kg level due to its association with clinical signs and body weight changes. Comparable decreases in serum calcium were observed throughout the study, indicating the effect was not progressive. In contrast, the changes in serum phosphorus levels became greater as the study progressed and were increased approximately 57% relative to controls at the high dose. The changes in serum calcium and phosphorus were completely reversed at the end of the recovery periods. Other etelcalcetide-related effects on clinical chemistry parameters were small in magnitude, were within the historical control range, had no correlated microscopic changes, and were all reversible. There were no etelcalcetide-related changes in hematology, coagulation, or urinalysis.
There was no effect on organ weights. Microscopically, intracytoplasmic golden brown pigment was observed in the renal proximal tubules in all groups, including controls. This observation in etelcalcetide-treated animals was dose dependent in severity and so was considered etelcalcetide related. At the end of the 4-week recovery period, pigment was still present with reduced severity in the high-dose group, suggesting ongoing reversibility. As there were no associated degenerative microscopic findings in the kidney and no treatment-related changes in renal clinical pathology parameters (clinical chemistry or urinalysis), this finding was considered nonadverse.
The sex-combined plasma TK parameters for pivotal toxicology studies are summarized in Table 1. The increases in etelcalcetide Cmax and AUC were proportional to dose, and there was no difference in exposure between sexes and no evidence of accumulation.
Genotoxicity Assessment
Etelcalcetide was mutagenic in bacteria in 2 of the 5 tester strains (TA100 and TA1535). The increases in revertant colony count were greatest in TA1535 (up to 4- to 16-fold in the presence or absence of rat liver S9) and only slightly above background in TA100 (2- to 3.5-fold) in the absence of rat liver S9 only. Representative results are presented in Table 2. However, etelcalcetide was negative in an in vitro chromosomal aberration assay in human peripheral blood lymphocytes at concentrations up to 5,000 µg/mL in the presence or absence of rat liver S9 and negative in an in vivo bone marrow micronucleus study in rats up to the maximum tolerated dose. To further evaluate the relevance of the bacterial mutagenicity, etelcalcetide was evaluated for mutagenicity in additional in vitro and in vivo mammalian systems. Etelcalcetide was negative in an in vitro mammalian cell mutagenicity assay (hypoxanthine-guanine phosphoribosyl transferase assay in CHO cells) at concentrations up to 5,000 µg/mL in the presence and absence of rat liver S9. In vivo, etelcalcetide was evaluated for mutations in liver and bone marrow in the Muta mouse. Following 28 days of daily SC dosing at the maximum tolerated dose of 4 mg/kg, there were no statistically significant differences in mean mutant frequency in the liver between groups treated with etelcalcetide and the concurrent vehicle control. In bone marrow, there were also no statistically significant differences between groups treated with etelcalcetide and the concurrent vehicle control, but a significant (P < 0.05) dose response was observed. All bone marrow mutation frequencies for all animals treated with etelcalcetide were within the bone marrow historical control range and were comparable with the concurrent vehicle control range (Figure 3). Therefore, the significant dose response was considered to be of no biological relevance. Etelcalcetide was concluded to be not mutagenic in this study. Plasma exposures on days 28 demonstrated dose-proportional increases in Cmax and AUC (1,550 ng/mL and 1,980 ng·h/mL at 4 mg/kg), and exposures after the last dose were comparable to exposures on day 1.
Summary of Mutagenicity (Ames) Results for Etelcalcetide and Structurally Related Peptides in the Absence of Metabolic Activation.
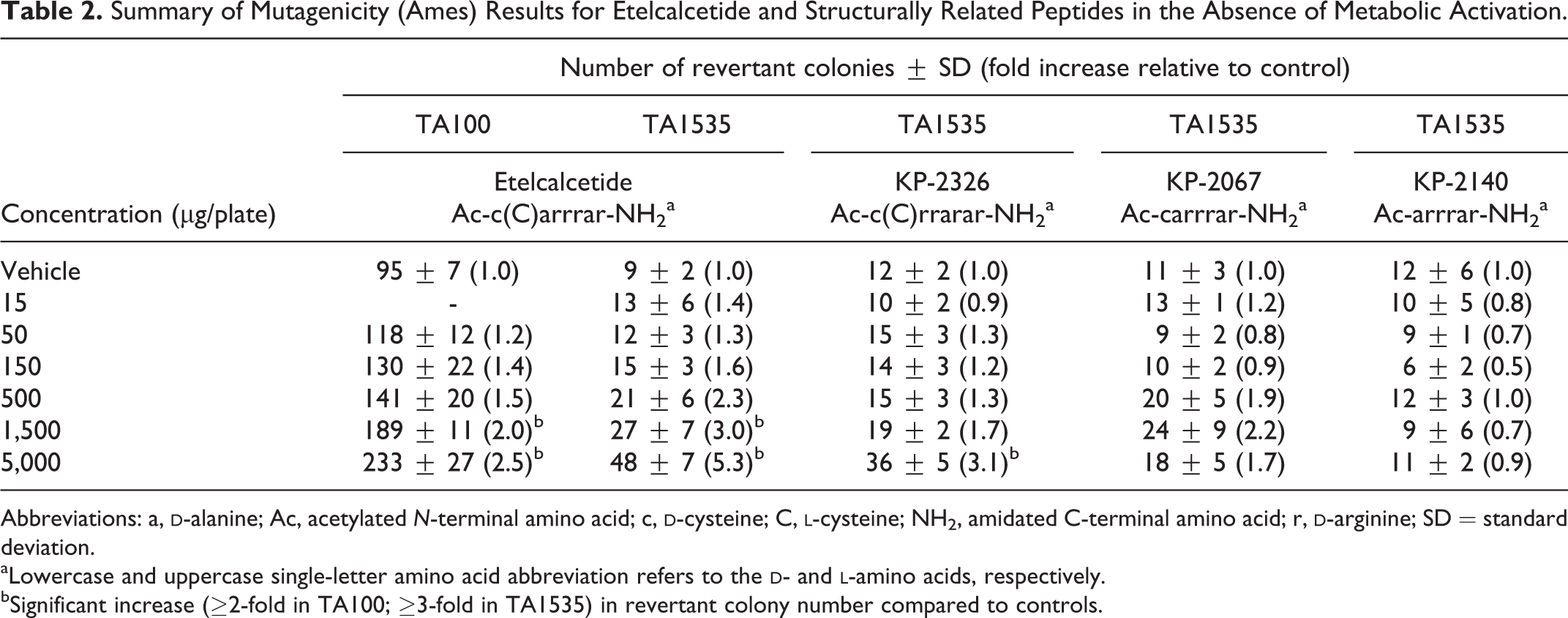
Abbreviations: a,
aLowercase and uppercase single-letter amino acid abbreviation refers to the
bSignificant increase (≥2-fold in TA100; ≥3-fold in TA1535) in revertant colony number compared to controls.
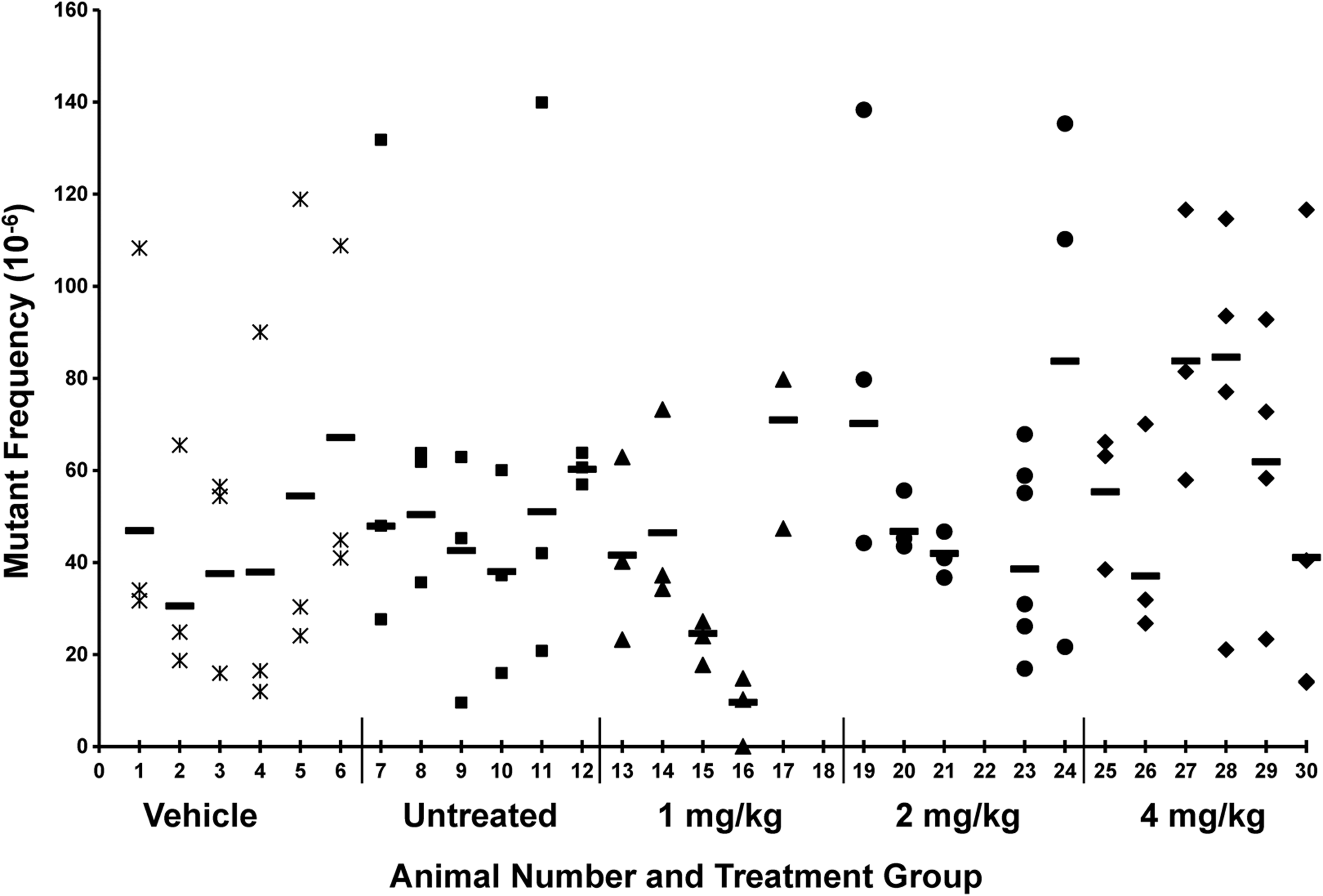
Mutation frequencies in bone marrow in the 28-day subcutaneous (SC) mutagenicity study in Muta mice. Symbols represent individual (n ≥ 3) packaging reactions for each animal. Horizontal bar (—) represents the mean mutation frequency for each animal. Data are not available for animals 18 (1 mg/kg) and 22 (2 mg/kg) due to DNA extraction failure. Observed range for historical control mutation frequencies is 10 × 10−6 to 186 × 10−6.
Results of follow-up Ames studies with etelcalcetide of higher purity suggested that the mutagenic activity was unlikely to be due to the presence of mutagenic impurities. A modified treat and plate assay
13
demonstrated that the revertant colonies were unlikely to be due to the release of free amino acids and compensation by histidine auxotrophy (data not shown). To further understand a potential mechanism of mutagenicity in Salmonella, etelcalcetide and structurally related peptides were evaluated for mutagenic activity to identify the structural components of the molecule important for activity (Table 2). The peptides were tested using the plate incorporation method in TA1535 in the absence of rat liver S9 metabolic activation, since reproducible mutagenic activity was previously established for etelcalcetide under these conditions. Etelcalcetide and the related peptide KP-2326 (rearranging alanine and arginine on the peptide backbone) both induced a significant increase (>3-fold) in revertant colonies (Table 2). Reduction in the disulfide bond within etelcalcetide (KP-2067) diminished but did not completely eliminate mutagenic activity. Although the slight increase in revertant colonies with KP-2067 did not meet the significance criteria of 3-fold increase relative to controls, the nonmonotonic dose response was reproducible in a confirmatory study (data not shown). Removal of the
Carcinogenicity Assessment
In the 6-month carcinogenicity study in rasH2 mice, there were no etelcalcetide-related effects on tumor incidence or nonneoplastic microscopic observations. There was a significant increase in the incidence of pulmonary tumors and hemangiosarcomas in the male and female positive control groups relative to the vehicle control groups, thus supporting the validity of the study. Evaluation of plasma drug concentrations in week 26 indicated that increases in Cmax and AUC were proportional to dose, similar among sexes and with no accumulation (Table 1).
In the 2-year carcinogenicity study in rats, animals were terminated early due to decreasing survival. Females in the 0.8 and 1.6 mg/kg group were terminated on week 89 due to reduced survival (n ≤ 15). The remaining groups/sexes were terminated at week 92 due to reduced survival (n ≤ 20) in male and female rats in the vehicle group. This was to ensure a sufficient number of animals per group to facilitate analysis and was done in accordance with regulatory agency input. The reduced survival was not statistically different from the saline groups and therefore not considered treatment related. There were no etelcalcetide-related effects on tumor incidence or nonneoplastic microscopic observations. Evaluation of plasma drug concentrations on day 183 indicated that increases in Cmax and AUC were proportional to dose, similar among sexes and comparable to levels measured on day 91 in the previous SC range-finding study, indicating that steady state levels had been achieved by day 183 (Table 1).
Fertility Assessment
In the rat fertility study, clinical signs (tremors, red and/or yellow material on various body surfaces, and/or salivation) were observed in both sexes at 3 mg/kg. Decreases in body weight, body weight gain, and food consumption were observed in males at 3 mg/kg throughout the dosing period, such that mean body weights were reduced 18% relative to controls at the end of the treatment period. In females, there was a decrease in body weight (6%) and food consumption during the premating and mating period of dosing but not during the gestation treatment period (GD 0-7). There were no effects on mean body weights, body weight gains, or food consumption at ≤1.5 mg/kg in males and females. Consistent with effects in nonpregnant rats, there was a dose-dependent decrease in serum calcium at all dose levels (up to 19% lower in the high-dose group relative to controls) when measured 8 hours postdose (the last day of the premating treatment period). There were no effects on mating and fertility parameters, male reproductive organ weights or sperm parameters (testicular and epididymal sperm numbers and sperm production rate, motility, and morphology), estrous cyclicity, or embryonic development at any dose level.
Embryo–Fetal Development Assessment
In a previous rat dose range-finding embryo–fetal development study, intrauterine survival and external fetal morphology were unaffected at dose levels up to 4.5 mg/kg, a level that exceeds the maximum tolerated dose in the chronic study. At this dose, lower mean fetal body weights were observed in association with maternal toxicity (mortality, body weight loss, clinical signs of tremoring). In the definitive rat embryo–fetal development study, the high dose was set at 3.2 mg/kg due to mortality at higher doses. Clinical signs (stained fur and vaginal discharge) were observed at all dose levels, with the changes being of higher incidence in the 3.2 mg/kg dose group. Decreases in maternal body weight gain and corresponding decreases in food consumption were observed at 3.2 mg/kg, such that mean body weights were reduced 6% relative to controls at the end of the treatment period. There were no treatment-related changes in maternal body weight and food consumption at ≤1.6 mg/kg and no changes in gravid uterine weights at any dose level. A dose-dependent decrease in serum calcium (up to 20% lower in the high-dose group relative to controls) at all dose levels was observed when measured 8 hours postdose on GD 6 and 17. Intrauterine growth and survival (number of viable and nonviable fetuses, early and late resorptions, and total number of implantation sites) were unaffected by etelcalcetide at all dose levels, and no etelcalcetide-related external, visceral, and skeletal malformations or developmental variations were observed. Plasma drug concentrations of etelcalcetide were evaluated in pregnant rats in a separate study on GD 7 and 21 as described below.
In a previous rabbit dose range-finding embryo–fetal development study, intrauterine survival and external fetal morphology were unaffected at nontolerated dose levels of 2.25 and 3 mg/kg. At these doses, lower mean fetal body weights were observed in association with maternal toxicity (mortality, body weight loss, clinical signs of tremoring). In the definitive rabbit embryo–fetal development study, the high dose was set at 1.5 mg/kg due to mortality at higher doses. A dose-dependent increase in the incidence of decreased defecation and other excreta-related changes (soft stool and small feces) were observed at all dose levels, with the changes being of higher incidence in the 1.5 mg/kg dose group. A decrease in maternal body weight and body weight gain (6% lower relative to controls) was observed in the 1.5 mg/kg dose group from GD 11 to 21, with corresponding decreases in food consumption. A dose-dependent decrease in serum calcium (up to 32% lower relative to controls) was observed at all dose levels when measured 8 hours postdose on GD 7 and 20. There were no etelcalcetide-related effects on any ovarian or uterine parameters (corpora lutea, implantation sites, litter size, embryo-fetal survival, fetal weights, and fetal sex) at any dose level, and no etelcalcetide-related external, visceral, or skeletal malformations or developmental variations were observed. Plasma drug concentrations of etelcalcetide were evaluated in pregnant rabbits on GD 11 in a separate study (data not shown), where increases in Cmax and AUC were dose proportional and did not increase over time (Table 1).
Prenatal and Postnatal Development Assessment
In the rat prenatal and postnatal development study, 1 F0 female rat in the 3 mg/kg group was euthanized on LD 17 due to clinical signs (tremors, salivation, and dehydration) and body weight loss, consistent with treatment-related hypocalcemia. Clinical signs in F0 rats were observed during gestation (tremors, salivation) and lactation (tremors, salivation, dehydration, hunched posture, stained fur) in the 3 mg/kg dose group. Slight excess salivation and stained fur were also observed at 1.5 mg/kg and tremors at 0.75 and 1.5 mg/kg during lactation. Etelcalcetide-related decreases in maternal body weight gain (15% lower relative to controls) were observed at 3 mg/kg during gestation and to a lesser extent during lactation, with corresponding reductions in maternal food consumption. There were no effects on maternal body weights, body weight gains, or food consumption at ≤1.5 mg/kg.
There was a small etelcalcetide-related increase in the mean duration of gestation at 1.5 and 3 mg/kg compared to controls (22.7 and 22.8 days, respectively, vs 22.4 in controls). At 3 mg/kg, fewer live-born pups were delivered (329 live born pups vs 348 in controls), and the number of stillborn pups was increased compared to controls (6 vs 1 in controls). The percentage of pups surviving from days 1 to 4 postpartum (viability index) was slightly reduced at 3 mg/kg (95.1% compared to 98.6% in controls), which reflected an increase in pup mortality (found dead or presumed cannibalized). There was an increased incidence of mild dehydration (based on skin turgor) in F1 pups in the 3 mg/kg group. Etelcalcetide-related reductions in mean pup weight were observed at 1.5 mg/kg (6% to 8% relative to controls) and at 3 mg/kg (8% to 15% relative to controls) during the postpartum period. This likely reflects a decrease in maternal body weights that correlated with lower pup weights noted at birth in the 3 mg/kg dose group. There were etelcalcetide-related reductions in mean body weights on days 22 to 29 postpartum (91% and 93% of controls, respectively) at 3 mg/kg in the F1-generation males, however, the reductions were transient as they were comparable for the remainder of the evaluation period (up to day 113 postpartum). There were no etelcalcetide-related effects on sexual maturation, behavioral end points (learning and memory or activity), mating, fertility, or ovarian and uterine parameters in the F1-generation rats at all dose levels. Evaluation of plasma drug concentrations of etelcalcetide on GD 7 and 20 indicated that increases in Cmax and AUC were dose proportional. Exposures on GD 20 were slightly higher than those after the first dose on GD 7 (≤2-fold). The fetal plasma to maternal plasma concentration ratio on GD 21 ranged from 2.4% to 3.0% across the dose levels, indicating low placental transfer of etelcalcetide.
Discussion
All of the adverse effects observed in toxicology studies in both rat and dog were related, either directly or secondarily, to the pharmacologic activity of etelcalcetide and the expected sequelae associated with dose-related reductions in serum calcium that ensues from suppression of PTH secretion. These pharmacodynamic responses were nonprogressive with repeat dosing and were reversible. At the highest doses tested in the repeat-dose toxicity studies, the degree of hypocalcemia was considered adverse and associated with tremoring/convulsions and a characteristic stress response consisting of clinical signs of poor health, decreases in body weight and food consumption, and anatomic and clinical pathology changes. 14 Clinical signs directly or indirectly related to decreases in serum calcium observed in rats and dogs included tremors, convulsions, prostration, decreased activity, lethargy, dyspnea, dehydration in rats, and salivation in dogs. Tremors are likely a direct effect on neuromuscular contraction as a result of the effect that reduced calcium has on the threshold for depolarization. 15 Additional pathological indicators of stress included altered organ weights (decreased for thymus and spleen and increased for adrenal gland), lymphoid depletion in the thymus, and gastric erosions/ulcerations in rats. These adaptive changes are well-established responses observed in animals subjected to stressors 14 and were fully reversible.
There were no adverse anatomic or clinical pathology findings or other effects that suggested toxicity of etelcalcetide unrelated to its mechanism of action on the CaSR. The intracytoplasmic golden brown pigment (resembling lipofuscin) observed in the proximal tubule of dogs was not associated with degenerative microscopic findings in the kidney or changes in renal clinical pathology parameters, and so this finding was considered nonadverse. The identity of the pigment and mechanism of increase is uncertain but may reflect an increase in etelcalcetide and/or the serum albumin conjugate formed with etelcalcetide 9 due to efficient tubular reabsorption of peptides and/or albumin. This is consistent with a relatively high amount of [14C]etelcalcetide-related radioactivity in the kidneys observed in a rat quantitative whole body autoradiography experiment. 9 The pigment observed in control dogs may reflect lipofuscin, a common background finding in the dog. 16,17 There was increased cytoplasmic eosinophilia of the parathyroid gland observed in rats, which was considered reflective of increased chief cell activity related to pharmacology. 17 These observations were not associated with any adverse changes in the parathyroid gland, including tumorigenicity following up to 92 weeks of treatment in the rat. In addition, there is no evidence that etelcalcetide is immunogenic in animals based on the absence of any unexpected change in plasma exposure or pharmacodynamic parameters (PTH and/or calcium) or evidence of immune-mediated reactions (immune complex disease, vasculitis, anaphylaxis, etc) in the repeat-dose toxicity studies.
The NOAEL in the chronic toxicity studies were 1 mg/kg/d in the rat and 0.5 mg/kg every other day in the dog, corresponding to a safety margin of 0.8× and 0.3×, respectively, relative to the maximum clinical dose based on plasma exposures (Table 1). Higher doses were associated with adverse hypocalcemia. Although healthy animals used in the toxicology studies are pharmacologically relevant, they are likely more sensitive to the hypocalcemic effects of etelcalcetide than are patients with CKD on dialysis. This is due to the greater effect of PTH on calcium reabsorption in intact healthy kidneys relative to patients with secondary HPT with little or no kidney function and who also have elevated PTH. Therefore, the safety margins likely overestimate the risk of hypocalcemia in patients with secondary HPT.
The safety pharmacology study in the dog indicated that etelcalcetide had no respiratory or CNS effects. The tremoring observed at high doses is considered secondary to decreases in serum calcium, which likely involves peripheral effects on muscle contraction. 15 Convulsions were observed in the repeat-dose toxicity studies in rat and dog at dose levels associated with tremoring and marked reductions in serum calcium. The convulsions observed may be reflective of seizures, as hypocalcaemia is known to reduce the threshold for seizures. 18 Consistent with this, CaSR gain-of-function mutations in humans are associated with hypocalcemia and neonatal seizures. 19 Tremoring and convulsions were reversed upon recovery of serum calcium changes following cessation of dosing. Direct effects on CNS function also seem unlikely since etelcalcetide is poorly distributed into the brain of the rat, 9 as expected for a positively charged peptide (Figure 1). The moderate prolongation of the QTc observed in acute and chronic studies in dog is an anticipated sequelae of hypocalcemia 20,21 owing to the role of calcium in cardiac depolarization. Further supporting an indirect mechanism for QTc prolongation, etelcalcetide did not affect the hERG ion channel current at concentrations up to the highest test concentration of 10 μg/mL, which is at least 300-fold greater than plasma drug concentrations where QTc prolongation was observed in the dog (24 hours postdose at 1.5 mg/kg). Furthermore, no QT prolongation was observed immediately following IV dosing when plasma drug levels are maximal. These data indicate that etelcalcetide does not have direct effects on cardiac repolarization.
The mechanism of mutagenicity in Salmonella is currently unknown. The weight of evidence supports etelcalcetide as not posing a genotoxic risk to humans since it was not genotoxic in mammalian cells in numerous in vitro and in vivo assays, including a mutation assay in liver and bone marrow in the Muta mouse. Etelcalcetide was also negative in rat and mouse carcinogenicity assays, which are sensitive to genotoxic compounds. Structure-activity relationship studies in Salmonella TA1535 demonstrated that mutagenic activity was completely eliminated when the
Some published in vitro data have linked enhanced expression and/or activation of the CaSR to a role in cell proliferation. 30 -34 However, the literature also indicates that CaSR activation is associated with a protective tumor suppressor role in certain tissues, namely colon and breast. 35 -41 There is no in vivo evidence that activation of the receptor is associated with an increased risk of cancer in any tissues, and there have been no reports of increased cancer rates in humans harboring gain-of-function mutations in the CaSR. 42,43 In addition, patients with secondary HPT have been shown to have an increased risk of cancer such that correction of the underlying disease may be expected to minimize risk factors. 44,45 Collectively, the weight of evidence argues that pharmacological activation of CaSR with etelcalcetide is not associated with an increased risk of carcinogenicity in humans.
There were no effects on mating and fertility parameters and no effects on reproductive organ weights or histopathology of reproductive tissues in chronic repeat dose toxicity studies up to maximum tolerated doses. Reproductive dysfunction does not appear to be associated with activation of the CaSR. The reported expression of the CaSR in rat testis and sperm, rat uterus, and human oocytes and cumulus cells suggest a possible role in fertility, however, the precise role of the receptor in these tissues is unclear. 46 -49 Reproductive abnormalities have not been observed in a mouse model bearing a gain-of-function mutation in the CaSR, and they are reported to breed normally. 50 Successful pregnancies have also been reported in mothers with autosomal-dominant hypocalcemia (ADH), a familial disorder caused by gain-of-function mutations in the CaSR. 51 -53 These data, together with the nonclinical safety data with etelcalcetide, indicate that activation of the CaSR is unlikely to affect reproductive function and fertility in humans. Etelcalcetide also presents a low risk for fetal harm, as there were no effects on intrauterine survival and external fetal morphology in both rats and rabbits, even at nontolerated dose levels, and no effects on postnatal behavior and reproduction. The decrease in fetal weight and pup viability were considered secondary to maternal toxicity due to hypocalcemia. Developmental abnormalities have also not been detected in a mouse model bearing a gain-of-function mutation in the CaSR 50 or in humans with ADH. 42,43
The reported distribution of CaSR expression and some in vitro studies suggest that the CaSR may have additional roles beyond regulation of PTH and calcium homeostasis, 54 including a role in cardiac physiology, 55 -59 immune cell function, 60 -62 cataract formation, 63 pulmonary function, 64 and neurodegeneration. 65 The nonclinical safety profile of etelcalcetide does not indicate that activation of the CaSR has appreciable effects beyond its role in regulating PTH and mineral homeostasis. Genetic and pharmacological models also indicate that the physiological relevance of CaSR activation beyond maintenance of calcium homeostasis appears minimal. Numerous (>80) activating, or gain-of-function, mutations of the CaSR have been documented in humans and are associated with ADH, a relatively benign condition characterized by hypoparathyroidism and hypocalcemia/hypomagnesemia, hypercalciuria, and hyperphosphatemia. 42,43 The activating mutations have been described to increase the sensitivity of the receptor to calcium stimulation (left shift the response curve) or increase cell surface expression. Autosomal-dominant hypocalcemia is typically asymptomatic, although neonatal seizures and carpopedal spasms have been reported, which is likely a sequelae of hypocalcemia. Nephrocalcinosis, renal insufficiency, and ectopic calcification can appear at a young age. These phenotypes are rare and are thought to be related to hypercalciuria owing to the role of the CaSR in renal calcium excretion. Bone mineral density in adolescents is reportedly normal. 66 An activating mutation within the mouse CaSR was found to mimic the ADH phenotype in humans. This mouse model appears to have more widespread ectopic calcification than humans, 50 however, this has not been observed in nonclinical safety studies with etelcalcetide. As with humans with ADH, these mice have otherwise been reported to be grossly normal. Other adverse consequences have not been associated with activating mutations in humans or mice. The genetics therefore suggest that activation of the CaSR is likely to play a minor physiological role in other tissues outside those involved in PTH and calcium homeostasis. This is consistent with pharmacological activation of the CaSR as supported by the nonclinical safety profile of etelcalcetide.
In summary, the safety pharmacology, repeat-dose toxicity, genetic toxicology, carcinogenicity, and reproductive and development toxicity studies demonstrate an acceptable safety profile for etelcalcetide. All adverse effects were attributed to the expected pharmacology of PTH and calcium lowering, and there were no adverse clinical or anatomic pathology findings or other effects that suggested toxicity unrelated to its pharmacological mechanism of action. These nonclinical data indicate no safety signal of concern for humans beyond that associated with hypocalcemia and QTc prolongation.
Footnotes
Author Contributions
Mark R. Fielden, Charles Dean, Kurt Black, Satin G. Sawant, Raju Subramanian, James E. Tomlinson, Sarah Walter, Cameron Zimmermann, and Ian Pyrah contributed to the conception, design, analysis, and interpretation of experiments and drafted and critically revised the article. Mark W. Griggs, Marie E. McKeon, Elise M. Lewis, and Carol Beevers contributed to the design, acquisition of data, analysis, and interpretation and drafted and critically revised the article. All authors approved the final draft.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: When the data were generated, Mark R. Fielden, Charles Dean, Kurt Black, Satin G. Sawant, Raju Subramanian, James E. Tomlinson, Sarah Walter, Cameron Zimmermann, and Ian Pyrah were employed by Amgen, Inc, which is developing etelcalcetide for use in humans. The authors Mark W. Griggs, Marie E. McKeon, Elise M. Lewis, and Carol Beevers were employees of Contract Research Organizations paid to conduct studies.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Amgen funded this research. This research received no specific grant from any funding agency in the public, commercial, private, or not-for-profit sectors.