
Abstract
Nonclinical toxicology studies are conducted to characterize the potential toxicities and establish a safe starting dose for new drugs in clinical studies, but the question remains as to how predictable/translatable the nonclinical safety findings are to humans. In many cases, there is good concordance between nonclinical species and patients. However, there are cases for which there is a lack of predictivity or translatability that led to early termination of clinical studies due to unanticipated toxicities or early termination of programs before making it to the clinic due to unacceptable nonclinical toxicities assumed to be translatable. A few case examples of safety findings that are translatable versus safety findings that are not translatable and why they are not translateable were presented as a symposium at the 38th Annual Meeting of the American College of Toxicology in Palm Springs, California, and are discussed in this article.
Introduction
Nonclinical toxicology studies in drug development consist of a comprehensive set of in vitro and in vivo studies for assessing safety of new small molecules and biologics before going into clinical trials. The in vivo studies are the most critical part of this assessment as they are designed to mimic the clinical study design (ie, route of exposure, dosing schedule, etc) in rodent and nonrodent species and extensively evaluate the potential toxic effects of new drugs. A multitude of parameters are assessed, including body weight, food consumption, clinical observations, clinical pathology, histopathology, and additional endpoints as needed (ie, safety pharmacology, genotoxicity, etc). The goal is to characterize the potential toxicities to determine whether they are monitorable and manageable and establish a safe starting dose for new drugs in clinical studies.
The science of toxicology is based on 3 general concepts: (1) The toxicity of a substance is an inherent property, (2) toxicity is directly related to the extent of exposure to a substance, and (3) toxicity observed in living organisms can predict effects in other living organisms. 1 The first concept (sometimes referred to as Paré law) is probably the most important task of a toxicologist: hazard identification. The second concept—commonly attributed to Paracelsus—is the one most often associated with the science of toxicology: “The dose makes the poison”. This is the basis of what we call risk assessment. It is the third concept whereby toxicity observed after exposing animals to poisons predicts effects in humans; that is, the central justification for experimental toxicology. Most often, this justification is supported by demonstration of concordance: When an adverse effect is seen in animals exposed to a substance—in drug development, this is commonly referred to as the test article (TA) or test substance (TS)—the same effect is seen in humans exposed to the same TA or TS. Toxicologists in the biopharmaceutical/pharmaceutical industry have spent considerable resources assessing this essential fact. The question is often framed as: Did nonclinical studies predict toxicity (or lack thereof) in clinical studies (and actual use)? This is an endless debate, and extensive analyses of concordance have demonstrated that toxicology studies are extremely valuable.
There is another aspect to this issue that is important in the development of drugs: It is anticipated that a TA or TS will have benefit for people with diseases. Thus, we have the concept of benefit verses risk. We generate a therapeutic index (TI), which is a ratio that compares the dose of drug that causes toxicity to the dose of drug that produces efficacy. A different risk/benefit (or TI) should be considered for S9 nonclinical evaluation of anticancer pharmaceuticals 2 versus non-S9 indications. The S9 guideline provides information on the type and timing of nonclinical toxicology studies for the development of both small molecule and biotechnology-derived pharmaceuticals intended to treat cancer in patients with late-stage or advanced-stage disease. More risk is acceptable in oncology than in nononcology applications because oncology patients are usually dying from the disease and would benefit from any therapeutic intervention that would extend their lives. In addition, patients with cancer are usually hospitalized so they can be safely monitored and managed. For these reasons, the highest nonseverely toxic dose (HNSTD) is identified in toxicology studies for S9 indications. Usually, nononcology indications require a much cleaner toxicology profile with good safety margins and are first dosed in healthy volunteers. Therefore, the no observed adverse effect level (NOAEL) is determined in toxicology studies for non-S9 indications. Some nononcology indications may be life-threatening and accordingly more risk may be acceptable, especially if there aren’t any current effective therapies available. Age of patient is also important, as minimal safety risk is preferred in pediatric patient populations.
What follows is a discussion of 3 drug development case examples of nonclinical to clinical predictivity/translatability of safety findings: bone marrow toxicity and peripheral neuropathy (PN) with microtubule inhibitor containing antibody–drug conjugates (ADCs), idiosyncratic drug-induced liver injury with several different classes of small molecule drugs, and drug-induced liver injury and rhabdomyolysis with the statins. This was presented as a symposium at the American College of Toxicology’s 38th Annual meeting held on November 7, 2017, in Palm Springs, California. The purpose of this symposium was to share case studies from different drug development programs on safety findings that are translatable versus safety findings that are not translatable and why they are not translatable, in order to better inform decision-making in the future for nonclinical safety assessments during drug development.
Case examples: Nonclinical to Clinical Translatability of Bone Marrow Toxicity and Peripheral Neuropathy with Microtubule Inhibitor Containing Antibody Drug Conjugates. Nicola J Stagg, Safety Assessment, Genentech
Antibody–drug conjugates (ADCs) are a therapeutic option for treating different cancers. ADCs consist of an antibody that targets a specific antigen on tumor cells attached to a linker and cytotoxic drug. The linker allows for release of the cytotoxic drug in the tumor and can be cleaveable (ie, payload release by proteolysis, pH, redox potential, etc) or noncleaveable (ie, payload release following degradation of antibody). Highly potent cytotoxic molecules are used in ADCs with different targets (ie, microtubules, DNA, etc). The microtubule inhibitors (MTIs) are the most common cytotoxic drugs used in ADCs and include maytansine (ie, DM1 and DM4) and auristatins (monomethyl auristatin E [MMAE] and monomethyl auristatin F). DNA damaging payloads are also commonly used in ADCs and include calicheamicin and pyrrolobenzodiazepines. Conventional linker–drug conjugation to antibodies occurs through lysine conjugation or conjugation to cysteines derived from reduction of interchain disulfide bonds. There is also site-specific conjugation consisting of engineered cysteines, unnatural amino acids, or enzymatic conjugation through glucotransferases and transglutaminases, 3 but clinical experience with this is limited. There are 5 approved ADCs on the market that include Kadcyla (ado-trastuzumab emtansine, T-DM1) for the treatment of breast cancer, Adcetris (bretuximab vedotin [BV]) for Hodgkin lymphoma and anaplastic large cell lymphoma, Mylotarg (emtuzumab ozogamicin) for acute myeloid leukemia, Besponsa (inotuzumab ozogamicin) for B-cell acute lymphoblastic leukemia, and Polivy (polatuzumab vendotin) for diffuse large B-cell lymphoma. There are also several others in clinical development for a multitude of different types of cancer. The focus of this translatability assessment will be on MTI ADCs, specifically T-DM1, BV, and several other valine citrulline (vc) MMAE ADCs in clinical development.
Trastuzumab emtansine contains a trastuzumab antibody, noncleavable linker, conjugation through lysine residues, and the DM1 payload. The nonclinical safety of T-DM1 was assessed in single-dose and repeat-dose toxicology studies in cynomolgus monkeys (binding species) and Sprague-Dawley rats (nonbinding species). 4 Findings included hepatic, bone marrow/hematologic (primarily decreased platelets), lymphoid organ, neuronal toxicities, and increased numbers of cells of epithelial and phagocytic origin in metaphase arrest. 4 These findings were attributed to the payload, DM1, and independent of the Her2 target. The safety of T-DM1 was assessed in 884 patients with HER2-positive metastatic breast cancer across 6 studies at a dose of 3.6 mg/kg every 3 weeks. 5 Thrombocytopenia and increased liver enzymes were the most common toxicities in addition to constitutional effects (ie, fatigue, nausea) and a low incidence of PN.
Bretuximab vedotin and the other vcMMAE ADCs use different antibodies, but the same cleavable linker, conjugation through reduced interchain disulfide bonds and the MMAE payload. The nonclinical safety of BV and other vcMMAE ADCs was assessed in repeat-dose toxicology studies in cynomolgus monkeys (binding species) and Sprague-Dawley rats (nonbinding species). The main toxicology findings observed with BV and vcMMAE ADCs in monkeys (up to doses of 6 mg/kg every 3 weeks for ∼4-5 cycles) and rats (up to doses of 12 mg/kg every week for 4 cycles) was bone marrow toxicity. 6 -9 Monkeys had dose-limiting neutropenia and rats had widespread bone marrow toxicity as well as liver enzyme changes and testicular atrophy. Patients treated with BV and other vcMMAE ADCs had dose-limiting neutropenia and later onset PN. 5 The toxicities were mainly mediated by the payload, MMAE, and independent of the targets. Patients with severe neutropenia were administered hematopoietic growth factors such as granulocyte colony-stimulating factor or granulocyte/macrophage colony-stimulating factor to regenerate neutrophils to enable higher doses and continuous treatment.
Two of the main clinical toxicities observed with conventional MTI ADCs are bone marrow toxicity and PN. Bone marrow toxicity (ie, neutropenia, thrombocytopenia, and anemia) is expected since MTIs tend to target rapidly proliferating cells and is easy to evaluate for translatability (ie, quantitative changes in hematology parameters in toxicology species and patients). Although peripheral nerves are not highly proliferating like bone marrow cells, they are susceptible to MTIs due to the long projections of axons and the critical role of the microtubule network in the nerve cell for axonal transport. 10 Peripheral neuropathy is harder to evaluate for translatability. Peripheral neuropathy is assessed in patients based on clinician-administered grading scales (ie, National Cancer Institute Common Terminology Criteria for Adverse Events [NCI-CTCAE]). 11 The NCI-CTCAE defines the grades as follows: grade 0: no symptoms; grade 1: asymptomatic, loss of deep tendon reflexes or paresthesia (including tingling), but not interfering with function; grade 2: sensory alteration or paresthesia (including tingling), interfering with activities of daily living (ADL); grade 3: sensory alteration or paresthesia interfering with ADL; and grade 4: disabling. Evidence of PN in nonclinical toxicology studies is based on histopathology changes to the sciatic nerve (ie, degeneration) and neurobehavioral signs.
Nonclinical to clinical translatability of bone marrow toxicity was observed with both T-DM1 and BV and other vcMMAE ADCs. Thrombocytopenia (decreased platelets in blood) was a common adverse event and the dose-limiting toxicity (DLT) in patients treated with T-DM1, and it was also observed in nonclinical species (monkeys and rat) administered T-DM1. 4,5 Although nonclinical to clinical translatability of bone marrow toxicity with T-DM1 (ie, decreased platelets that had a cyclic pattern of decline and recovery) was observed, it was more severe in patients than in nonclinical species and some patients exhibited a slow downward shift. Mechanistically, it was attributed to DM1-induced impairment of megakaryocyte differentiation. 12 Neutropenia (decreased neutrophils in blood) was often the most frequent and/or severe toxicity reported in patients and cynomolgus monkeys, and a commonly reported DLT in patients. 13 Similar severity and cyclic pattern of decline and recovery of neutrophils was observed between cynomolgus monkeys and patients. Mechanistically, decreased neutrophils is attributed to nonspecific uptake of the ADC in the bone marrow and release of MMAE that induces cytotoxicity of neutrophils. However, MTIs can also affect neutrophil function since an intact MT network is critical for migration/chemotaxis of neutrophils to the site of infection. 14,15
Translatablility of PN was not consistently observed with MTI ADCs. Trastuzumab emtansine had a very low incidence of PN in patients (mostly grade 1); 22% had PN of any grade with few grade 3 events (21 [2.4%] patients) and only 1 grade 4 event was reported. 5 Peripheral neuropathy with T-DM1 was primarily sensory and didn’t result in dose delays, reductions, or discontinuations. There was evidence of axonal degeneration of the sciatic nerve and spinal cord in monkeys administered T-DM1 at 10 mg/kg (6/14 animals) and 30 mg/kg (14/14 animals) every 3 weeks for 4 doses that was not reversible after 6 weeks of recovery. 4 There was a lack of translatability of PN with BV and the other vcMMAE ADCs. In a phase II study with BV, 42% of patients with Hodgkin lymphoma at 1.8 mg/kg every 3 weeks had PN (any grade), and 8% had a grade 3 or higher. 16 In several clinical trials with vcMMAE ADCs, PN was a frequent adverse event leading to treatment discontinuation and dose reduction. 13,16 -19 However, there was no evidence of sciatic nerve degeneration or neurobehavioral changes with BV or any of the other vcMMAE ADCs in monkeys or rats up to the maximum tolerated dose even with dosing up to every 3 weeks for 5 doses. 6 -9,20
The mechanism of action (MOA) for MTI ADC-mediated PN is believed to be attributed to distribution and nonspecific uptake of the ADC in the peripheral nerves, release of the cytotoxic MMAE payload, and microtubule inhibition leading to axonal degeneration. Four hypotheses were evaluated for the lack of translatability of PN with vcMMAE ADCs: (1) species differences in exposure, (2) insensitivity of animal models, (3) species differences in target biology and other vcMMAE ADC properties in peripheral nerves, and (4) increased susceptibility of patient population 20 (Figure 1). The results of this hypothesis-based approach identified no differences in pharmacokinetics between nonclinical species and late-stage oncology patients and evidence of nonspecific uptake of vcMMAE ADCs in the peripheral nerves of rats even with a single dose that supports the MOA. However, it also identified key challenges with predicting PN with MTI ADCs due to the delayed onset of this toxicity in patients, challenges in measuring PN nonclinically versus clinically and differences in patient sensitivity compared to healthy animals. 20 The duration of dosing and exposure of vcMMAE ADCs in nonclinical species may not be long enough (ie, up to 15 weeks vs onset of grade ≥2 in patients from 20-32 weeks). Patients report symptoms in the fingers and toes, whereas the sciatic nerve (which is a distal peripheral nerve) is routinely collected in nonclinical toxicology studies and evaluated by histopathology for nerve degeneration. Furthermore, oncology patients receiving ADCs have typically been late stage and usually heavily pretreated with prior chemotherapy, comorbidities, and are older, all of which are risk factors for PN (either they already have PN or have an increased susceptibility of developing it).
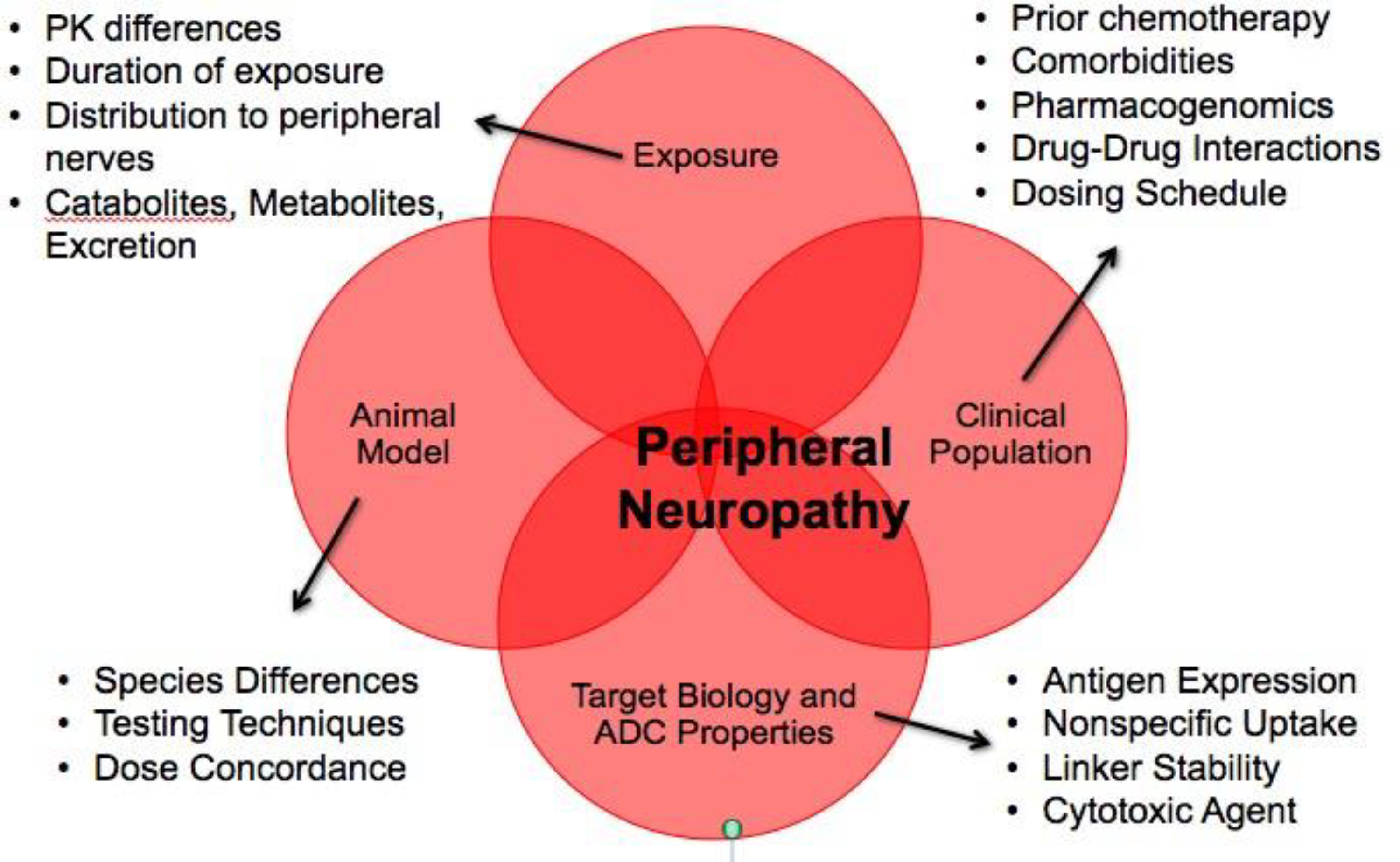
Four hypotheses for the lack of translatability of peripheral neuropathy in nonclinical toxicology studies with valine citrulline monomethyl auristatin E antibody–drug conjugates.
Although it is challenging to model MTI ADC-induced PN in nonclinical toxicology species, we identified opportunities to improve the predictivity of PN in our animal models. These included increasing duration of exposure and adding an expanded neurohistopathology assessment of peripheral nerves (ie, including foot pads for intraepidermal nerve fiber [IENF] analysis). We designed a toxicology study with a vcMMAE ADC to be able to reproduce the PN observed clinically. We selected rats (Sprague-Dawley) based on published literature of peripheral nerve degeneration in rats with both paclitaxel and vincristine.
21,22
The dose selection was 4 and 8 mg/kg weekly for a duration of up to 12 weeks based on some internal repeat-dose tolerability data of vcMMAEs in rats and exposure-response modeling (using patient and animal data). We evaluated standard toxicology end points (body weight and clinical observations) and toxicokinetic end points. We also included peripheral nerve-specific end points (ie, toxicity of peripheral nerve via hematoxylin and eosin of sciatic nerve, dorsal root ganglion, spinal cord, tibial nerve and sural nerve, and protein gene product 9.5 [PGP9.5] IENF of foot pad sections; exposure in peripheral nerve via matrix-assisted laser desorption/ionization mass spectrometry [MS] imaging and liquid chromatography [LC]-MS; other mechanistic end points including RNAseq). Preliminary data demonstrated evidence of nerve degeneration (minimal) in rats treated with 8 mg/kg of vcMMAE ADC for 12 weekly doses and an accumulation of MMAE in peripheral nerves. Other end points are still in progress. By modifying the design of toxicology studies, we were able to improve the predictivity of PN, which also has the potential to be used to provide possible mitigation strategies in the clinic for existing MTI ADCs and/or to screen next-generation MTI ADCs for reduced PN. 2. Predicting the Potential of Drug Candidates to Cause Idiosyncratic Liver Injury in Human Patients: Is There Hope? Robert Roth, Pharmacology and Toxicology, Institute for Integrative Toxicology, Michigan State University
Drug-induced liver injury is often classified as “intrinsic” or “idiosyncratic.” 23 Intrinsic toxicity from drugs usually occurs in overdose situations and results in distinctive liver lesions that are clearly dose related and occur after a predictable latent period. Injury occurs in all individuals at toxic doses (although the dose that is toxic can vary among individuals), and the injury is usually reproducible in experimental animals. Preclinical toxicity testing strategies that have been used for decades are usually successful in predicting intrinsic hepatotoxicity during drug development. Acetaminophen is a classic example of a drug that causes intrinsic hepatotoxicity. In contrast, idiosyncratic, drug-induced liver injury (IDILI) occurs at therapeutic treatment regimens, not necessarily in overdose situations, and typically occurs in a small, susceptible fraction of people. Liver lesions demonstrate variable pathologies, and the relationship between duration of drug exposure and the onset of toxicity is variable. In addition, the relationship of drug dose to liver injury is unclear. In contrast to intrinsic toxicities, idiosyncratic reactions are not reproducible in typical preclinical animal tests. Accordingly, nonclinical safety findings do not usually translate well to the prediction of IDILI in human patients. The antibiotic, trovafloxacin, and the nonsteroidal anti-inflammatory drug, diclofenac, are 2 examples of the many drugs that are associated with IDILI in human patients.
Effective assays to predict toxicity liability early in drug development could result in better lead compounds and safer drugs. Simple in vitro assays with high specificity (low false positives) and that are based on known mechanism(s) of IDILI pathogenesis would be particularly attractive. Unfortunately, although it is clear that these reactions depend on both the properties of the drug and the susceptibility of the patient, much remains unproven regarding the mechanisms by which IDILI arises. Several hypotheses have arisen to explain IDILI pathogenesis, and attempts at developing in vitro assays to predict the IDILI potential drug candidates have been made. These assays have employed various cell types and end points measured (Table 1). The end points include physical–chemical properties of drugs, factors related to toxicokinetics (eg, transporter inhibition; maximum serum concentration, C max), reactive metabolites, gene expression, cytotoxicity (eg, enzyme release, adenosine triphosphate decline), cytokine production, and hepatocellular function. 24 -28 Many of these published attempts to classify drugs according to IDILI potential are associated with issues such as a complex, costly system, and the absence of a phenotypically relevant end point that could render them less than ideal in a preclinical testing situation. Nevertheless, some have demonstrated good performance (sensitivity and specificity) and represent palpable progress toward useful predictive assays.
Cell Types and End Points Used in Attempts In Vitro to Classify Drugs According to IDILI Liability.
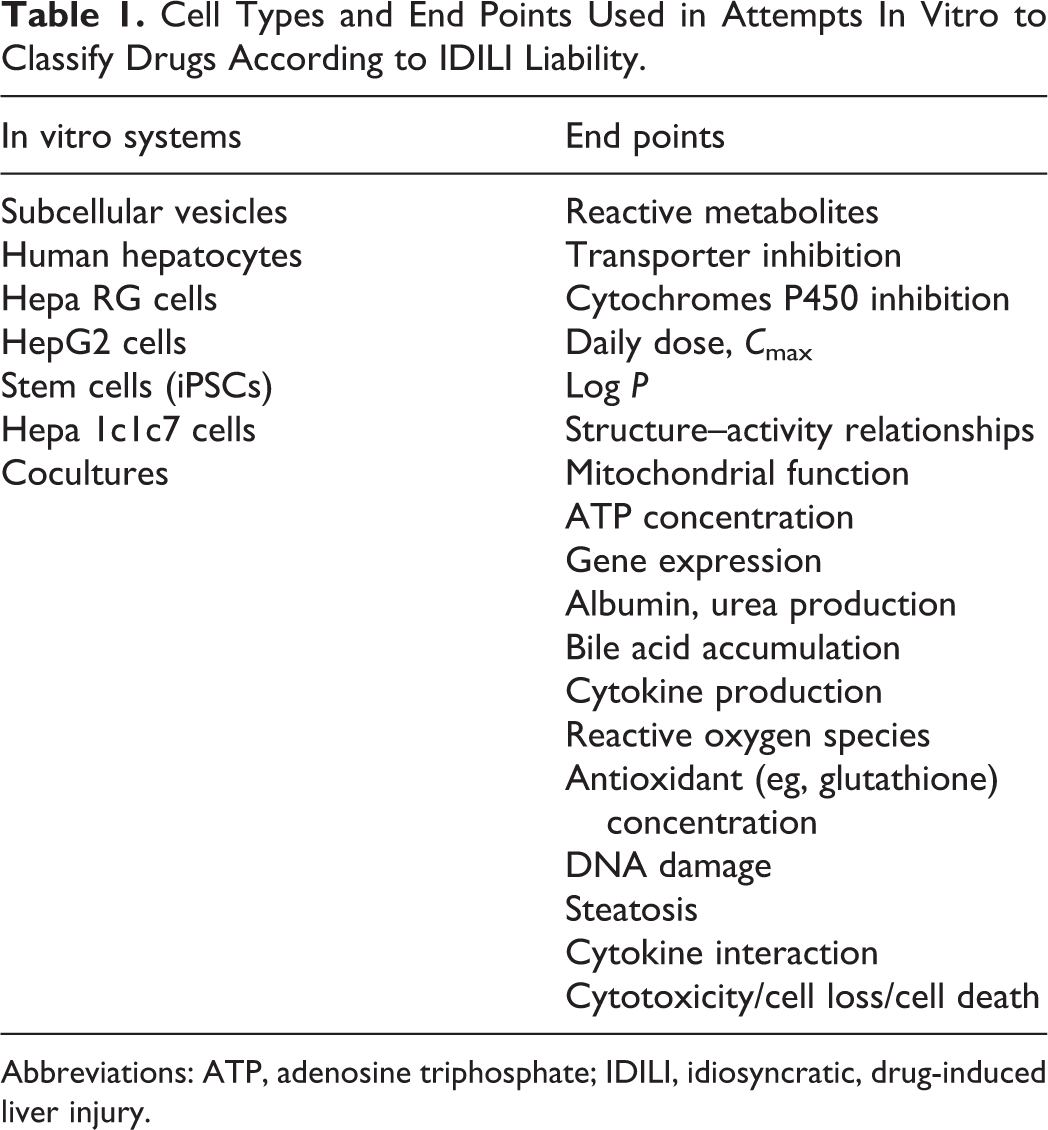
Abbreviations: ATP, adenosine triphosphate; IDILI, idiosyncratic, drug-induced liver injury.
An ideal assay for preclinical prediction of IDILI liability would use a cell type(s) that is easily obtained and maintained in culture and that yields consistent results. The assay would also be inexpensive, require minimal amounts of test compounds, and employ a single, easily measured end point that is directly relevant to the IDILI phenotype (eg, hepatocellular death). Moreover, the underlying basis for the assay should be consistent with the IDILI mode of action.
As mentioned above, there remains disagreement about the mode of action of IDILI, and consequently many theories have been proposed. The most popular of these is the adaptive immunity hypothesis. 29 According to this theory, an offending drug is bioactivated to a reactive metabolite that binds to protein. This adducted protein is recognized as a hapten, which is processed by antigen-presenting cells, leading to the clonal expansion and activation of leukocytes of the adaptive immune system (eg, T cells). In the presence of additional signals, activation of these cells results in the production of inflammatory cytokines and other mediators that have the potential to damage hepatocytes. A related theory, the inflammatory stress hypothesis, suggests that cells of the innate immune system activated during an inflammatory response can interact with drugs, resulting in a hepatotoxic episode. 30,31 Based on this hypothesis, rodent models have been developed in which cotreatment with an inflammatory stimulus (eg, lipopolysaccharide) and an IDILI-associated drug results in pronounced liver injury. 32 Exploration of these models has resulted in the identification of tumor necrosis factor (TNF)-α as a central mediator in the pathogenesis. 31 Importantly, activation of either the adaptive or the innate immune system results in production of TNF and other cytokines so that the mode of hepatocellular killing is likely similar irrespective of the initiating event in these 2 hypotheses (Figure 2).
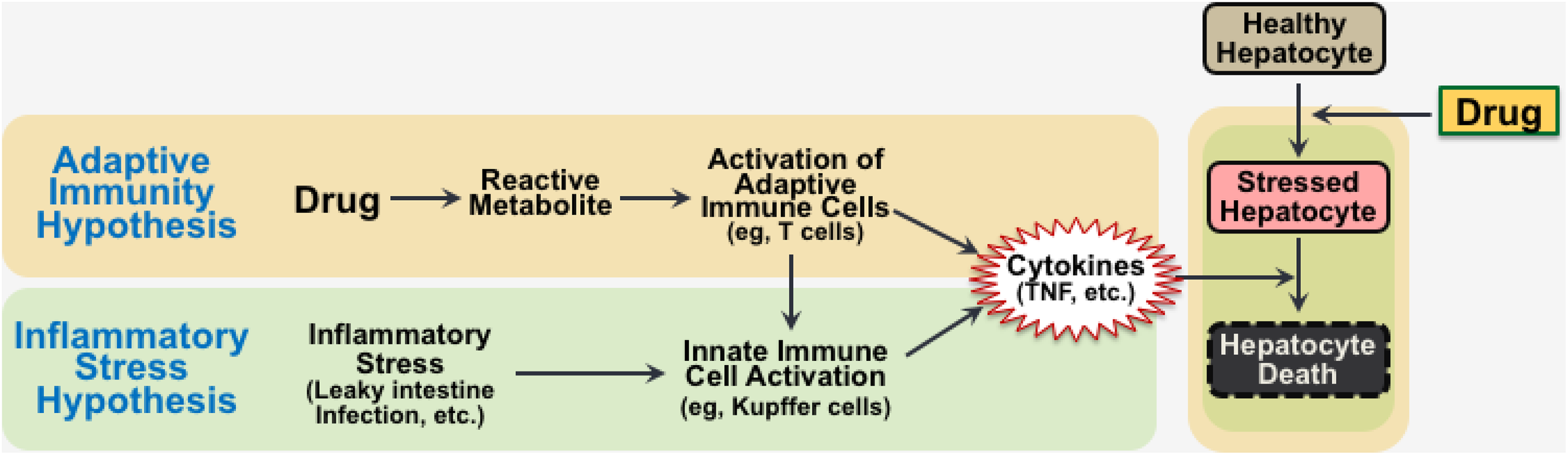
Hypotheses for the pathogenesis of idiosyncratic, drug-induced liver injury.
Tumor necrosis factor has dichotomous effects on hepatocytes: It is capable of activating both cell survival and cell death pathways. Which of these predominates depends on the state of the cell. In healthy hepatocytes, the activation of cell survival pathways (eg, nuclear factor κB) dominates, so that hepatocytes do not normally die from TNF exposure. In contrast, in hepatocytes that are stressed, cell death pathways predominate, leading to hepatocellular killing by TNF (Figure 1). 31,33,34 This has led to the idea that IDILI-associated drugs might impose a nonlethal stress on hepatocytes, rendering them susceptible to killing by TNF. This hypothesis has received considerable support from recent in vitro studies (for review, see 31 ).
Based on this hypothesis, we undertook a proof-of-concept study to determine whether drug–TNF interaction in vitro could classify drugs according to their IDILI liability. 35 We exposed the human hepatocyte cell line, HepG2, to various drugs in the presence and absence of TNF and elucidated detailed concentration–response curves, with cytotoxicity (lactate dehydrogenase release) as the sole end point. Our initial study employed 14 drugs that have been associated with human IDILI and 10 that have had little or no IDILI association. Various curve characteristics (maximal responses, EC50 s, etc), singly or in combination, were used to construct several logistic models, which were evaluated for their abilities to classify drugs according to their IDILI liability. The performance of some of these classification models was modest, but others demonstrated high sensitivity and specificity with this drug set. The best-performing model employed covariates derived from maximal responses and EC50 s in the presence and absence of TNF and included C max (obtained from published studies) as an additional covariate. With the drug set we employed, this classification model yielded a sensitivity of 0.93, specificity of 1.0, and an area under the receiver operating characteristic curve of 0.99, suggesting a remarkable ability to classify the drugs.
These results are consistent with the hypothesis that cytotoxic interaction of drugs with immune cytokines like TNF causes death of hepatocytes in IDILI. Current efforts focus on further evaluation of the model using additional drugs and on improvements in the modeling that will lead to enhanced utility in a preclinical testing scenario, for example, by developing related models that do not require delineation of a complete concentration–response curve and that incorporate additional covariates. The hope is that such an assay will inform go–no go decisions regarding candidate drug selection early in drug development. Based on our results and those of others, better preclinical prediction of IDILI potential seems in reach in the near future.
3. Lack of Concordance between Nonclinical and Clinical Findings with Statin Class of Compounds Kenneth Hastings, Hastings Toxicology Consulting LLC
Cardiovascular disease (CVD) is one of the most prevalent causes of morbidity and mortality in the world. According to one estimate, 17.3 million people died of CVD worldwide in 2013. 36 There are numerous similar estimates of the death toll due to this disease, and there are sufficient data to indicate that the problem is persistent and that prevalence (both in relative and total numbers) is increasing. 37 There are many forms of CVD (eg, coronary artery disease, peripheral artery disease) and many associated causes of death (eg, myocardial infarction, stroke). In addition, there are many comorbidities, such as hypertension and diabetes, and many known risk factors, such as obesity and tobacco consumption.
There does appear to be a common underlying pathology: atherosclerosis. 38 The common characteristic of atherosclerosis is the formation of atheroma: vascular plaques composed of foam cells (leukocytes), smooth muscle cells, and lipids. Atheroma formation is a chronic inflammatory process with calcification of the vascular endothelium, loss of blood vessel elasticity, arterial enlargement, and formation of unstable, thrombogenic plaques. 39 This process can result in hypertension, coronary artery occlusion, thromboembolism, ischemia, and infarction. This is the series of events that result in death due to heart attacks or stroke.
A common feature of atherosclerosis is elevated blood lipid levels, especially cholesterol. 40 Referred to as dyslipidemia, this phenomenon has long been considered the key causative risk factor for development of atherosclerosis, a theory developed based both on animal models and epidemiological observations such as familial susceptibility to dyslipidemia associated with significantly increased risk of death due to heart attacks and stroke. The obvious conclusion is that prevention of atherosclerosis would involve, in some way, reduction in blood lipid levels. Two methods for accomplishing this have long been established: modification of diet and pharmacotherapy.
One of the first class of drugs demonstrated to effectively lower blood lipid levels was the fibrates: These (probably) work by increasing endogenous lipase activity. 41 In a very large World Health Organization-sponsored study (208,000 man/years of observation), clofibrate was demonstrated to be very effective in lowering blood lipid (primarily cholesterol) levels. Unfortunately, clofibrate therapy was actually demonstrated to increase mortality compared to untreated control patients—an outcome that has never been adequately explained. 42 Although clofibrate (as well as other fibrates) can reduce blood lipid levels, reduced blood lipid levels has not been shown to decrease the risk of CVD.
In 1960, Konrad Block and Feodor Lynen received a Nobel Prize for demonstrating the biochemical steps in the de novo synthesis of cholesterol. 43 An important observation was that the key enzyme for this process is 3-hydroxy-3-methyl-glutaryl coenzyme A reductase (HMG-CoA reductase). 44 A drugable target had been discovered. As happens so often in drug development, by accident Akira Endo and his colleagues found that a mycotoxic fungus, Penicillium citrinum, produced a secondary metabolite that could inhibit HMG-CoA reductase—a discovery repeated independently by a group in the United Kingdom in 1976. 45,46 Unfortunately, the metabolite (mevastatin) failed in clinical trials, for reasons never disclosed. 47 However, a related compound was discovered by scientists at Merck & Co: lovastatin—which was the first statin approved for marketing in the United States (in 1987). Six other statins have been approved since and all remain on the market, including atorvastatin, which is perhaps the most widely used drug in the class.
All of the statins were demonstrated in clinical trials to significantly lower blood lipid levels and are indicated for a variety of dyslipidemias, but most importantly they have been shown to reduce the risk of CVD and associated morbidity and mortality. 48 Simply put, they have proven to be among the most efficacious drugs ever marketed for a serious and prevalent public health problem. Given the toxicities that were commonly observed in nonclinical toxicology studies, this could be considered even more remarkable—the issues were numerous and apparently serious.
Rosuvastatin—the most recently approved statin—can serve as an excellent example of the toxicities observed in nonclinical toxicity studies. Nonclinical studies were conducted in mice, rats, dogs, and nonhuman primates (cynomolgus monkeys).
49
Findings were typical of approved statins. Targets of toxicity in repeat-dose toxicology studies included: Liver (rats, mice, dogs) Gallbladder (dog, mouse) Rodent forestomach Cornea, lens, and retina (dogs) Kidney Muscle
Some of these adverse effects were observed at exposure levels close to those that were achieved at therapeutic doses and could have been clinically relevant. But the effect that tended to be of the greatest concern—not just with rosuvastatin but with the entire statin class—was hepatotoxicity, and it is this effect that is most informative of the concordance issue.
Adverse liver effects were seen in rodents and dogs and included hepatocyte hypertrophy, necrosis, and elevated liver transaminases. Although hepatotoxicity was seen at significant multiples of clinical exposure in mice and dogs (×11 and ×35, respectively, based on comparison of systemic area under the plasma drug concentration–time curve at a clinical dose of 20 mg/d), the dose multiple at which adverse liver effects were observed in rats was only ×5 clinical exposure. Hepatotoxicity was observed in nonclinical toxicity studies (including those conducted in rabbits and guinea pigs) with all of the statins, was considered to be an obvious class effect, and was the source of considerable concern. In fact, the observation of liver toxicity in animals (mainly rats) was important in interpretation of findings in clinical trials—that is, occasional elevations in alanine aminotransferase (ALT) levels. Although observed at higher rates than seen in placebo or diet management control participants, elevations in ALT levels in TA-exposed participants rarely exceeded ×3 upper limit of normal. However, the rates of mild elevations in ALT levels combined with findings suggesting that statins were potentially potent liver toxins in rats led to the assumption that they had the potential to cause serious liver toxicity in patients.
Consider the 3 concepts of toxicology discussed earlier. Without question, a hazard was identified: Statins could cause liver injury. As to risk, only in the rat was there evidence of clinical relevance. It is the third concept that should be carefully considered: Is there reason to consider the rat an inappropriate model for statin safety in clinical use? It is here that comparative biology becomes important. Several questions needed to be answered. First, was there reason to believe that rats were uniquely sensitive to statin hepatotoxicity? A great deal of work has gone into investigating this issue, but the conclusion seems to be that rats indeed are uniquely sensitive. The second question was, Is there any reason to assume that humans might be like rats—uniquely sensitive? It is interesting to note that hepatotoxicity was not a significant finding in nonhuman primates—at least for rosuvastatin. But this finding seems to be consistent with observations with other statins. A third question is, Does liver disease increase the probability of severe hepatotoxicity in patients exposed to statins? Actually, not only does the answer—at least in general—seem to be no, there are even examples where patients with chronic inflammatory liver diseases (such as chronic hepatitis C) actually benefited from statin therapy. 50
Global assessment of existing data resulted in Food and Drug Administration concluding that warnings about hepatotoxicity included in the labels of marketed statins were no longer needed—and in general medical practice, this issue seems to have been resolved. There are occasional reports of severe liver toxicity associated with statin therapy, but these appear to be idiosyncratic—low incidence, high severity reactions that have proven to be essentially unpredictable using standard nonclinical toxicology studies. 51 Nevertheless, there is a persistent belief that statins are liver toxicants—an idea that continues to haunt the scientific literature. It is also worth noting that liver toxicity is not a serious concern with marketed statins—as is unfortunately the case with many types of drugs, if serious safety issues are observed in clinical trials, development stops, and results may never be reported in the open literature. But it is important to consider an important lesson learned: A hazard was identified in nonclinical studies, attention was paid to this potential adverse effect in clinical trials, and when enough data were available from both pre- and postmarketing experience to indicate that the hazard did not translate into clinical risk, appropriate measures were taken by regulatory authorities to communicate these findings.
As to the issue of concordance between toxicity findings in nonclinical studies with other adverse clinical effects, there are examples of both. Evidence of muscle toxicity was observed in nonclinical studies, perhaps predictive of a serious adverse effect known to be a risk of statin therapy: rhabdomyolysis. Although the mechanism(s) of this toxicity are still not completely understood, animal studies have provided some clues. Ocular toxicity, observed in dogs at clinically relevant exposure levels, may have been predictive of adverse eye effects in humans, especially patients with preexisting cataracts. Tumor findings in rodent bioassays have not been concordant with postmarketing experience: No epidemiological studies have indicated that statins are human carcinogens. Two types of effects are likely related to the pharmacology of the statins: fetal toxicity (seen in nonclinical studies and the basis for labeled contraindication in pregnancy) and an emergent clinical effect: signs of neurotoxicity in elderly patients. In both cases, inhibition of de novo cholesterol synthesis could be the basis of the findings. The developing fetus requires cholesterol for development and it is also important for maintenance of neural structure. Another emergent issue with statin use has been increased risk of developing type 2 diabetes: Occasional signs of pancreatic toxicity were observed in nonclinical toxicity studies with the statins, but were not associated with diabetic effects.
What has emerged as an important pharmacological property of the statins is that they have potent anti-inflammatory activity. 52 Originally described in transplant medicine, this property of the statins could, at least in part, accounts for the impressive efficacy of these drugs in preventing CVD. 53 An essential component of CVD is chronic vascular inflammation. This anti-inflammatory property has been associated with the observation that elderly patients on statin therapy are less likely to die from pneumonia secondary to influenza infections. Studies in animals have demonstrated the possible mechanism for statin anti-inflammation: Inhibition of interferon-γ induced MHC II expression on vascular endothelia. Other mechanisms have been proposed based on animal studies.
The statins represent a fascinating example of real-world toxicology: hazard identification, risk assessment, and evaluation of cross-species concordance. All were components in bringing to market a class of drugs that have proven to be valuable for public health.
Discussion
Our goal in conducting toxicology studies in drug development is to establish a safe starting dose and to characterize the potential toxicities that may be observed in the clinical trials (ie, healthy volunteers or patients). It is imporant to understand the potential drug-induced toxicities in order to determine whether they are monitorable, manageable, and reversible before progressing them into the clinic. The acceptability of the toxicology profile depends on a variety of factors, including patient population, indication (ie, oncology vs nononcology), whether it’s life threatening, if there are current effective therapies for this indication, and so on. In this article, we discuss 3 drug development case examples, where we have retrospectively analyzed how well the nonclinical toxicology studies predicted safety findings oberved in the clinic.
The first case example is MTI ADCs. Microtubule inhibitor ADCs are for the treatment of cancer (S9 anticancer indication, ie, TI is determined based on the nonclinical toxicology HNSTD and not the NOAEL), which means more safety risk is acceptable. The main toxicity observed in the nonclinical toxicology studies and in patients is bone marrow toxicity, and there is fairly good concordance between animals and humans with T-DM1 and BV. This was predicted based on the MOA of the MTIs and was easily tested for in nonclinical toxicology studies and monitored for in patients because it is acute and can be assessed by quantitative changes to hematology parameters in blood. Whereas there was not consistent concordance with PN due to discordance in dosing duration, the lack of a quantitative measure that is used nonclinically and clinically (ie, histopathology of peripheral nerves vs patient reporting) and the increased susceptibility of the patient population to developing PN. The magnitude of severity of PN observed with many vcMMAE ADCs led to their discontinuation. By leveraging a hypothesis-based approach for the lack of translatability of PN, we were able to design a study with extended duration and more sensitive end points to improve the predictivity in animals. This could be used in future for assessing next-generation MTI ADCs with reduced PN.
The second example is IDILI. Idiosyncratic adverse drug reactions remain a major problem because the potential for toxicity is often not recognized until drugs have been on the market for some time and consumed by large numbers of patients. Better preclinical prediction of idiosyncratic reactions could inform decisions as to which drug candidate(s) to move forward in the drug development process. The liver remains a frequent target for idiosyncratic reactions. Development of animal models of IDILI as well as in vitro explorations have increased our understanding of potential mechanisms, but much remains unknown about what underlies IDILI responses. There have been numerous attempts to develop in vitro systems to predict IDILI potential of drug candidates based on physical–chemical properties of compounds and plausible molecular mechanisms of injury. These have met with various degrees of success, and there remains much room for improvement. It is unlikely that a single assay will be able to predict accurately all drugs or drug candidates that have IDILI potential, inasmuch as the mechanism by which IDILI likely arises is not the same for all drugs. Indeed, a battery of in vitro assays, each of which addressing a different mechanism of toxicity, might be needed. Other issues arise in our ability to assess assay performance. Such assessment typically involves comparison of sets of drugs known to cause or not to cause IDILI in humans. A problem arises in that different criteria have been used to assess whether or not a drug causes IDILI in people, and categorization of a drug as associated or not with human IDILI can change with time as more data accumulate postmarketing. Thus, the gold standard needed to assess assay performance carries considerable uncertainty, which consequently imposes uncertainty in assessing assay performance. This has the potential to limit confidence in any predictive assay. That said, increased understanding of mechanisms underlying IDILI should lead to assays with toxicity-relevant end points in which confidence exists and that are useful in preclinical prediction of compounds with IDILI liability.
The third case example is DILI and rhabdomyolysis with the statins. Liver toxicity was observed in nonclinical studies, but has not proven to be a serious adverse effect in most patients. Muscle toxicity was observed in nonclinical studies, and myalgia is a common cause of statin intolerance. Rhabdomyolysis—a sudden catastrophic destruction of skeletal muscle tissue—is the most serious adverse effect associated with statins, but the mechanism is unknown and the potential relationship with nonclinical observation of muscle toxicity remains speculative. Statin intolerance—usually characterized as myalgia but also associated with vague neurological effects—may be a warning sign for rhabdomyolysis, but this remains speculative.
In conclusion, nonclinical toxicology studies are a very important component of drug development. In the 3 case examples discussed, some of the toxicities showed good concordance between nonclinical species and patients (ie, bone marrow toxicity and rhabdomyolysis). However, there were also a few examples demonstrating a lack of translatability/predictivity (ie, PN, IDILI, and DILI). Additional work in some cases revealed why and/or alternative approaches. Investigations of MTI ADC-induced PN uncovered opportunities to modify nonclinical toxicology study designs and include end points to improve predictivity in animals. Work on IDILI has shown that, although some animal models exist, they are not conducive to routine toxicity testing, but a battery of mechanistically relevant in vitro assays might improve predictivity. Lastly, the serious liver toxicity that was predicted with statins based on rat data did not translate clinically and it was determined that rat was just uniquely sensitive. We conclude that animal studies are valuable and, in many cases, can predict toxicities in human. As evidenced by the additional investigative work, many factors can contribute to lack of translation and understanding these is important to improve translation.
Footnotes
Authors' Note
The views expressed are those of the authors. No official support or endorsements by the US Food and Drug Administration is provided.
Author Contribution
All authors contributed to conception and design; contributed to acquisition, analysis, and interpretation and drafted manuscript. All authors critically revised the manuscript and gave final approval. All authors agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.