
Abstract
Many antisense oligonucleotides (ASOs) from several classes of molecules are currently in drug development. Despite over 20 years of pharmaceutical research, few ASOs have been marketed due to problems with clinical efficacy or preclinical toxicologic challenges. However, a number of recent developments have renewed interest in this class including the registration of mipomersen, the advent of successful screening strategies to eliminate more toxic molecules, and new understanding of the risks of off-target nucleotide binding and mitigation of potential off-target effects. Recent advances in backbone chemistries, conjugation to other moieties, and new delivery systems have allowed better tissue penetration, enhanced intracellular targeting, and less frequent dosing, resulting in fewer toxicities. While these new developments provide invigorated interest in these platforms, a few lingering challenges and preclinical/clinical toxicity issues remain to be completely resolved, including: (1) proinflammatory effects (vasculitis/inflammatory infiltrates); (2) nephrotoxicity and hepatotoxicity unrelated to lysosomal accumulation; and (3) thrombocytopenia. Recent investigative work by several laboratories have helped elucidate mechanisms for these issues, allowing a better understanding of the clinical relevance and implications of particular toxicities. It is important for toxicologists, pathologists, and regulatory reviewers to be familiar with new developments in the ASO field and their implications, as a greater number of new types of antisense molecules undergo preclinical toxicity testing.
Keywords
Historical Background
The field of antisense oligonucleotide (ASO) therapeutics has been around since the early 1990s. In those early days, there were constant supply chain delays, synthesis methods limited available quantities of drug substance, and analytical methods were poorly developed. Since then, the delivery and science of ASO drug development has reached new heights of understanding and clinical/regulatory acceptance. Indeed, only a little over a decade ago, questions and controversy arose from publications that suggested ASOs were incapable of sufficient mRNA knock down to affect clinical disease and/or that many ASO experimental results were nonspecific effects (Stein 1997; Dias and Stein 2002; Vickers et al. 2007). However, early failures were largely due to the fact that initial ASO molecules were either of low affinity or did not effectively enter cellular compartments because of low membrane permeability. The original ASOs consisted of unmodified deoxyoligonucleotides. As a group, they suffered from poor solubility and rapid degradation by exonucleases, which led to the development and clinical introduction of the so-called first generation ASOs with substitutions of the phosphate backbone. Phosphorothioate (PS) ASOs were the most common of this type and were of higher solubility with improved membrane penetration as compared to the unmodified oligonucleotides (Fiset and Gounni 2001). PS-modified DNA differs from natural DNA in that one of the nonbridging oxygen atoms in the phosphodiester linkage is substituted with sulfur. These molecules were known to induce sequence-independent, but length-dependent binding to various cellular proteins, especially heparin-binding molecules, such as laminin and fibronectin (Stein 1997). Additionally, due to their relatively rapid degradation in vivo, first-generation ASOs tended to have poor clinical efficacy but benign safety profiles. When combined with nonspecific binding and proinflammatory activity, equivocal clinical results led to much of the controversy over mechanism of effect. Preclinical toxicologic changes, often limited to high doses, primarily resulted in accumulation of basophilic granules within specific tissues and cells of macrophage lineage, resulting in degenerative effects in affected organs at very high dose levels. Few of these first-generation ASOs reached late-stage clinical trials, and only fomivirsen (Vitravene®) was approved and marketed, for the intraocular treatment of cytomegalovirus retinitis (Dias and Stein 2002).
Second-generation ASOs were subsequently developed that included additional modifications of the backbone sugar moieties, which allowed much greater nuclease resistance and increased binding affinities (the latter often related to reduced flexibility of nucleoside rings) as compared to the earlier PSs or phosphodiesters (Dean and Bennett 2003). The intention of the pharmacology was to provide inhibition of mRNA through stimulation of RNAse H mechanisms while providing enhanced stability and improved cellular penetration. RNase H is a ubiquitous enzyme that hydrolyzes the RNA strand of an RNA/DNA duplex. Oligonucleotide-assisted RNase H-dependent activity can efficiently reduce targeted RNA expression, approaching 80 to 95% downregulation of protein and mRNA expression (Dias and Stein 2002). Further backbone modifications (e.g., methyl or methoxyethyl substitutions at the 2′ position forming 2′-OMe or 2′-MOE ASOs) have led to even greater stability and longer tissue half-lives, and many of these molecules are still in active clinical trials. Most recently, this has included the successful FDA regulatory approval and U.S. marketing of mipomersen 1 for familial hypercholesterolemia (Food and Drug Administration [FDA] 2012). The patterns of tissue distribution for second-generation PS ASOs are very similar to those of the first-generation, with kidney and liver containing the highest concentrations, but with greater intracellular drug concentration (Henry et al. 2008). The complexity of design continues to grow, with the recent introduction of conjugated molecules to second-generation ASOs. Raw materials are now entirely synthetic. Compound synthesis has been scaled up over a thousand fold, and there is a general understanding of process impurities (Stanton et al. 2012).
In more recent iterations, the so-called generation 2.5 and third generation ASOs include a wide variety of molecules such as locked nucleic acids (LNAs), N-methyl substituted bicyclic nucleic acids (BNAs), peptide nucleic acids (PNAs), and morpholinos, and several of these (especially LNAs) are currently in clinical trials by a number of biotechnology firms. While their safety and efficacy attributes differ slightly from those of the PS ASOs, each class of agent has stereotypic toxicity profiles and these nuances are important for the toxicologic pathologist to be aware of when evaluating an ASO toxicity study. LNAs operate primarily through RNAse H (Wahlestedt et al. 2000), but PNAs and morpholinos function by a simple steric block mechanism (Summerton 2004). PNAs were intended to be used predominately where very high binding affinities were required, but efforts to utilize PNAs therapeutically have been hampered by poor cellular penetration and inadequate in vivo pharmacokinetic properties (Larsen, Bentin, and Nielsen 1999) and they have lost favor as a treatment modality. Morpholinos are used predominately for applications requiring very high target specificity in systems such as in developing embryos. Morpholinos bind RNA with a higher affinity than DNA binds RNA and much higher affinity than PS ASOs for RNA (Summerton 2004). Because of their unusual structure, morpholinos do not interact to any meaningful extent with cellular proteins and tend to have fewer systemic toxicities as compared to other ASOs (Summerton 2004). LNA platforms are currently utilized by several different companies pursuing ASO therapeutics due to their potentially lower immunostimulatory potential (than PS ASOs), relatively high potency, short sequence, and favorable binding affinities. However, as noted later in this article, they appear to have a higher potential for hepatotoxicity and some other toxic effects (Stanton et al. 2012; Swayze et al. 2007).
In addition to the ASOs which depend on RNAse H for much of their pharmacologic activity or the morpholinos and PNAs which work through steric blocking, there is another class of nucleic acid therapies which use an entirely different mechanism for pathophysiologic effects based on the principle that double-stranded RNA can be used to silence specific genes. Short interfering RNAs (siRNAs) utilize inactivation of target mRNA through the RNA-induced silencing complex (RISC) and work in the cytosol rather than the nucleus. One strand of the siRNA duplex combines with cellular proteins to form the RISC, which then acts to block translation of partially complementary RNA sequences and degrades highly complementary RNA target sequences. RISC unwinds the 2 strands of RNA molecules, allowing the antisense strand to bind to the targeted RNA molecule, and endonuclease activity hydrolyzes the target RNA at the site where the antisense strand is bound (Zamore et al. 2000; Hutvagner and Zamore 2002). siRNA toxicologic profiles differ from those of the more traditional ASOs and a safety concern unique to therapeutic siRNAs is the unintended suppression of nontarget mRNAs and proteins via RNA interference (RNAi); however, while the mechanisms differ, both ASOs and siRNAs can induce hybridization-dependent off-target effects.
In addition to RNAse H, steric blocking and RISC inhibition of mRNA, a wide range of mechanisms of target modulation are now being pursued through novel technologies that all exploit Watson and Crick base-pairing, including exon skipping strategies, splice switching, mRNA sequestration, synthetic mRNA utilization, and miRNA inhibition. The toxicity profiles for many of these different platforms may vary slightly from the formulaic ASO “class toxicities” pathologists and toxicologists have recognized over the last decade. Importantly, there are some potential lingering challenges with ASO preclinical toxicities and potential implications for patient safety that have not been fully addressed, and these issues will be discussed from the perspective of the toxicologic pathologist. Great progress has been made in mitigating many of the previous problems associated with ASO drug development. Although there remain other issues pertinent to the chemists and pharmacologists such as optimal organ/cellular delivery, formulation, and obtaining peak efficacy, these challenges for the oligonucleotide industry are beyond the scope of this review. Designing toxicologic studies with this class of agents also involves many unique challenges, but are likewise beyond the scope of this article, and those interested are referred to other reviews on those aspects of ASO development (Dias and Stein 2002; Henry et al. 2008; Levin and Henry 2008).
Common Microscopic Toxicologic Changes among ASO Classes
The common histopathologic features associated with ASO administration and well-recognized class-wide toxicities have been previously and extensively reviewed (Henry et al. 2008; Levin and Henry 2008; Marquis and Grindel 2000; Monteith and Levin 1999; Monteith et al. 1999). However, there are a few misconceptions that make it worth revisiting for the toxicologic pathologist evaluating ASO preclinical studies. There are 2 broad categories of potential toxicities for ASOs: hybridization-dependent toxicities due to on- or off-target pharmacology and hybridization-independent toxicities due to nonantisense effects of the ASO. Pharmacologically based side effects result from hybridization-dependent ASO binding to the desired target RNA or, alternatively, to off-target RNA due to complete or partial complementarity. Target-based toxicities are specific to the individual ASO and will not be discussed in this review. Techniques for mitigating hybridization-dependent off-target effects (OTEs) will be discussed later in this article. Hybridization-independent toxicities represent effects that are not due to Watson/Crick base-pairing between an ASO and RNA sequences and have (wrongly, or at least confusingly) also sometimes been referred to as OTEs. This category of toxicities may exhibit some sequence dependency despite the fact it does not involve basepairing, and the mechanism is related to the specific chemical class of ASO involved. Hybridization-independent (nonpharmacologic) toxicities fall into 3 general subcategories due to either ASO accumulation effects, proinflammatory mechanisms (including immune complexes), or aptameric binding (as a consequence of ASO interactions with extracellular, cell-surface, and/or intracellular proteins). Both immunomodulation and aptameric protein interactions have persisted, imposing challenges for ASO development, but accumulation-related effects are the most commonly encountered changes in preclinical toxicity studies. While these 3 types of hybridization-independent toxicities do impact unintended targets, in this article, the term “OTEs” is considered limited to those hybridization-dependent changes resulting from specific but unintended mRNA knockdown other than drug target.
Cytoplasmic granule accumulation is commonly noted within particular epithelial cells with many of these molecules (Marquis and Grindel 2000; Henry et al. 2008). When noted within kidney or liver epithelium, cytoplasmic granules have been labeled “basophilic granules,” and based on ultrastructural analysis, are considered to reflect accumulation of drug-related material (Monteith et al. 1999; Monteith and Levin 1999; Levin and Henry 2008; Figure 1). Less commonly, basophilic granules have been noted in other tissue types, including adrenal gland, pituitary gland, choroid plexus, synovium, and very rarely in other epithelial cells. In these locations, they generally do not result in cellular degenerative effects.
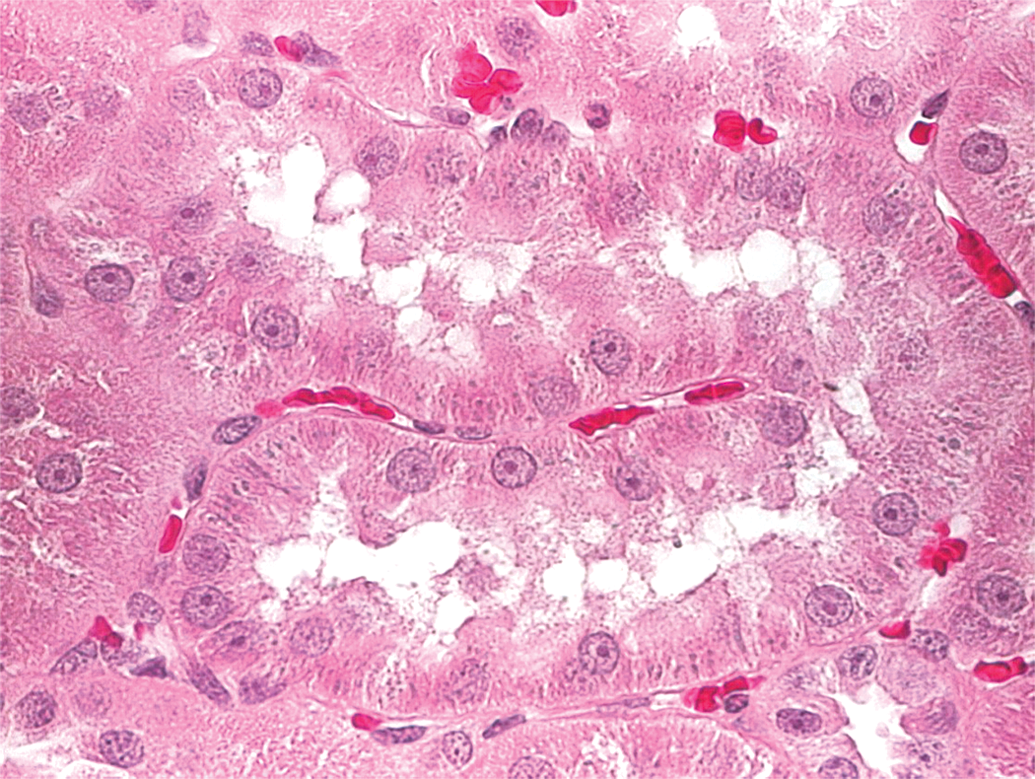
Rat kidney. Basophilic granules are noted as dark grey spots within tubular epithelium.
Their staining qualities can vary from basophilic to amphophilic and even eosinophilic (especially in monkeys), but “basophilic granules” is still the preferred histologic term (Frazier et al. 2012). Repeated dosing results in accumulation in these tissue compartments, which reaches a steady state, and the degree of toxicity then depends on concentration and the inherent potency of the accumulated ASO. At high doses, the prominence of the granules in epithelial cells correlates with an increased incidence or severity of degeneration in the kidney or liver. This is likely as a result of lysosomal breakdown, or in the liver also as a result of cytokine release by activated kupffer cells (Henry et al. 2002; Henry et al. 2008). In chronic studies, ASO accumulation in the kidney has also been associated with an increase in chronic progressive nephropathy in rats, as occurred with mipomersen (FDA 2012).
In addition to the commonly observed basophilic granules, a second cytoplasmic change can also be observed preclinically in tissue macrophages and should not be confused with basophilic granules. Most often, the granular cytoplasm of macrophages in animals given ASOs exhibit staining features slightly different than granules seen in the epithelial cells lining the renal tubules or in hepatocytes and are thought to contain secretory substances such as cytokines (Engelhardt et al. 2014). Affected macrophages have a distended, weakly stained and finely vacuolated or granular cytoplasm, and many pathologists have diagnosed them as increased granular macrophages or increased vacuolated macrophages. Moreover, unpublished internal studies using 2′-MOE ASOs failed to demonstrate oligonucleotide accumulation in cytoplasmic granules in macrophages, whereas they were noted universally in the basophilic granules of renal and hepatic epithelium. While it is true that macrophages do accumulate ASO material within phagosomes (Henry et al. 2008), it appears that the granular and/or vacuolated appearance of macrophage cytoplasm noted in many ASO studies is instead related to cellular activation and cytokine production by the proinflammatory properties of many of these compounds (Engelhardt et al. 2014). This concept is supported by the observation that granular/vacuolated macrophages may still be present at the end of recovery (off-dose) phases of toxicity studies, when serum exposures of an ASO are undetectable.
Hybridization-independent Proinflammatory Effects (Challenge #1)
Immunostimulatory effects have been associated with a number of siRNAs and many ASOs, particularly those of the PS class, in several different preclinical species (Agrawal and Kandimalla 2004; Henry et al. 1999; Monteith and Levin 1999; Ravindran, Jeng, and Liang 2010; Krieg 2002). The potency of the proinflammatory effects is dependent on several factors including basepair sequence and base modifications, but backbone chemistry is a major contributor (Senn, Burel, and Henry 2005). For instance, PSs have been demonstrated to have innate immunostimulatory activity (Hartmann et al. 1996). The shorter basepair length of most LNA gapmers (based on their higher binding efficiency) seems to reduce their proinflammatory potential as compared to other backbones. The basis for the sequence specificity in rodents are sequence motifs that are recognized by pathogen-associated molecular pattern (PAMP) receptors, such as the nucleic acid–sensing TLR receptors (Agrawal and Kandimalla 2004; Senn, Burel, and Henry 2005; Richardt-Pargmann and Vollmer 2009; Burel et al. 2012; Burel et al. 2013). These attributes have been exploited for immunomodulatory drug indications by industry (Krieg 1998, 1999; Henry et al. 1999). As a result of their activation of TLR, particularly TLR9, and other innate immune receptors, cytokines are released in a TH-1 like pattern resulting in the characteristic histologic findings in rodents of lymphoid hyperplasia and multiorgan lymphohistiocytic cell infiltrates (Henry et al. 2008; Henry et al. 1997; Choi, Chung, and Jung 2010; Krieg 1998). In monkeys, ASO-mediated inflammation is largely a result of activation of the alternative pathway of complement (Farman and Kornbrust 2003; Henry et al. 2002; Levin and Henry 2008). This again has been most often associated with PS ASOs, and like rodents, microscopic effects in monkeys such as lymphoid hyperplasia are noted in lymph nodes, spleen, and lymphohistiocytic infiltrates are found with increased severity in multiple organs. However, the preclinical proinflammatory lesions of greatest concern are related to glomerulonephritis (as described subsequently) and vasculitis associated with some types of ASO administration in monkeys. While these changes are reversible, due to the long tissue half-life of many of these drugs, as well as the self perpetuating nature of inflammatory lesions, recovery from ASO-mediated proinflammatory effects in preclinical species may take weeks to months.
ASO-associated vasculitis differs substantially from vasculopathies associated with administration of small molecules and tends to be more endothelial-centric, whereas small molecule vascular lesions are generally centered on the tunica media (Engelhardt et al. 2014). ASO-mediated vascular lesions often have pronounced adventitial inflammatory infiltrates, and they lack the vasopressor or vasodilatory activity noted with small molecule vasculotoxicants. A Society of Toxicologic Pathology (STP) working group has recently completed an article detailing ASO-induced vascular injury and describing the pathophysiologic mechanism for these classes of drugs, as well as providing guidance on risk assessment strategies for toxicologic pathologists when this type of injury is encountered (Engelhardt et al. 2014).
Both ASO-mediated vasculitis and glomerulonephritis share a similar pathogenesis in monkeys, related to complement activation and initial injury to the endothelium (Frazier et al. 2014). ASOs have been shown to interact with Factor H and disruption of the Factor H interaction with C3 convertase is thought to result in dysregulation of complement activation and in failure of clearance of circulating immune complexes (Alexander, Clarkson, and Fulgham 1985; Henry et al. 2002; Levin and Henry 2008). While direct interactions with complement factors by ASOs are the primary means through which these changes occur, there may be other indirect pathways that also play a role in the pathophysiology of these changes. For instance, cross talk between complement pathways and TLR receptors has been demonstrated, which reinforces innate immunity and inflammation through synergistic interactions (Hajishengallis and Lambris 2010; Heikenwalder et al. 2002), so TLR stimulation by ASOs may also be contributory to these responses in monkeys.
Based on the proposed mechanism and available clinical information to date, vasculitis does not appear to be a significant problem with clinical ASO administration, as the monkey appears to be much more sensitive to ASO-mediated complement activation than humans (Henry et al. 2008; Levin and Henry 2008; Kwoh 2008; FDA 2012; Engelhardt et al. 2014). However, it must be emphasized that proinflammatory effects have still been noted clinically in patients, characterized by flu-like symptoms or especially as injection site reactions when an ASO has been given subcutaneously or intramuscularly. As a greater number of ASOs with proinflammatory and/or vasculotoxic potential reach phase II or III clinical trials, more data may be available to determine whether any clinical risk for vasculitis will be apparent or whether there is greater confidence that the vascular changes are peculiar to the nonhuman primate based on its sensitivity to systemic complement activation. Given the uncertain relationship of preclinical vasculitis to patients and the clinical occurrence of other local immune effects, identification and proper risk assessment of proinflammatory issues will remain a priority for ASO drug development. At present, with at-risk backbone chemistries, screening is often performed on potential drug candidates via subchronic (≥4 week) in vivo toxicity studies and simultaneous in vitro complement fixation assays by many or most pharmaceutical companies. While somewhat resource intensive, there are a few alternatives to this combination of studies for ranking the inflammatory potential of prospective candidates.
Hybridization-independent Toxicities in the Liver and Kidney Unrelated to Accumulation (Challenge #2)
It has been demonstrated repeatedly that relatively minor modifications to the basepair sequence or backbone of an ASO can have drastic consequences for affinity or toxicity (Stanton et al. 2012). In many cases, these toxicities have been associated with microscopic evidence of degeneration or necrosis in the liver in subacute rodent studies at doses/exposures well below those generally accepted to result in accumulation-related hepatic effects. While the mouse appears to be the most sensitive species, hepatic toxicity has been reported in the rat, cynomolgus monkey, and human (Burdick et al. 2014). Although multiple antisense platforms, including 2′-MOE PS ASOs and siRNAs, have exemplar molecules associated with these effects, unexpected hepatic toxicities have been most commonly associated with the LNA platform (Hagedorn et al. 2013; Burdick et al. 2014; Kakiuchi-Kiyota et al. 2014). In some cases, mice treated with LNAs have demonstrated significantly increased plasma aminotransferase (ALT), and aspartate aminotransferase (AST) levels as well as histopathological evidence of liver necrosis and apoptosis (Swayze et al. 2007). For LNA chemistry, these effects appear to be largely independent of immunostimulatory mechanisms. However, for other platforms and for explicitly immunostimulatory LNAs, immunomodulation could potentially exacerbate liver toxicity through Kupffer cell activation. In most cases involving liver toxicity, the histopathologic lesions are largely comprised of hepatocellular degeneration with single-cell necrosis, without a primary inflammatory response (Burdick et al. 2014). In some of the more severe cases, it can involve overt necrosis of larger areas of hepatic acini. Some of the differences between related molecules may be related to increased hepatocellular uptake. However, in many cases of hepatotoxicity, interaction with intracellular hepatic proteins has been suspected as the cause of these differences between molecules. PS ASOs are known to cause nonspecific binding to proteins due to their polyanionic nature, and this phenomenon has largely been associated with heparin binding like growth factors and extracellular receptors. In contrast, recent work has suggested, or at least caused speculation, that much of the LNA hepatotoxicity may be due to aptameric binding to intracellular proteins (Kakiuchi-Kiyota et al. 2014; Hagedorn et al. 2013; Burdick et al. 2014).
As noted previously, antisense-induced hepatotoxicity can be elicited by small changes in the basepair sequence, but these effects do not appear to be truly hybridization-dependent (not based on Watson/Crick pairing) and are instead related to protein interactions with certain classes of ASOs. Recent research has provided new and valuable insights into the pathogenesis of these lesions and the potential intracellular pathways that are being perturbed. Different LNA gapmers modulate distinct transcriptional pathways that can result in hepatotoxicity in mice (Kakiuchi-Kiyota et al. 2014). Hepatotoxic mechanisms involving DNA damage were described with 1 LNA, and disruption of clathrin-mediated endocytosis with another related molecule (Kakiuchi-Kiyota et al. 2014). In another study, structure-toxicity analysis using results from 2-week toxicity studies in mice given LNA-modified ASOs demonstrated specific hepatocellular effects by ASOs on specific cellular proteins (Burdick et al. 2014). These ASO–protein complexes were thought to inactivate critical cellular processes or potentially activate hepatocellular antiviral responses resulting in apoptosis. The authors postulated that toxicity of LNAs might be mediated through p53 pathways secondary to LNA-induced cellular modifications. Structure–activity relationships in the basepair sequence are beginning to be explored in detail, and examples of potentially hepatotoxic basepair sequences have been identified. The aforementioned study found that LNA hepatotoxicity was strongly associated with TGC and TCC sequence motifs in 3-8-3 gapmers (Burdick et al. 2014). ASOs containing these sequence motifs tended to exhibit higher binding to mouse liver proteins. In other studies, the CTGT motif was found to increase the likelihood of hepatotoxicity with 2′-MOE PS ASOs (A. Levin: personal communication, DIA oligonucleotide conference, 2013). Very recently, a bioinformatic analytic program called a “random forests classifier set” was developed, which predicted with 80% accuracy, the low- or high hepatotoxic potential of LNAs based on basepair sequence and backbone modifications (Hagedorn et al. 2013). These authors effectively demonstrated how to redesign a potentially hepatotoxic ASO to mitigate hepatic effects. However, it appears that basepair sequence alone may not be the sole factor responsible for the hepatic effects. When other companies have compared sequences of some of their own historic candidates to those published, several molecules with “offending sequences” actually had less hepatotoxic signals in in vivo studies than those without the hepatoxic targeted sequences. This institutional difference may reflect the fact that different lengths and types of gapmers were being analyzed, with different melting temperatures (Temperature Measurement System) and different 3D conformations. It is expected, however, that with time and refinement of current bioanalytic programs, we will eventually identify those specific sequences within closely related ASO classes with the greatest hepatotoxic risk. A few hepatotoxic molecules will inevitably escape identification by these prescreening criteria, and proceed into preclinical development. Historically, in vitro cytotoxicity screens have not been reliable predictors of hepatic toxicity in preclinical in vivo testing (Burdick et al. 2014). This may be a reflection of incomplete ASO transporter systems in many of these hepatocyte cultures. However, when used in combination with 14- or 28-day mouse toxicity studies, the use of more recent in vitro assays and in vivo screening studies together appear to be more effective in deselecting molecules with untoward hepatotoxic potential earlier in preclinical development (Burdick et al. 2014). It is therefore critical for toxicologic pathologists to be aware of, and identify these types of microscopic hepatic changes in candidate selection studies, even when lesions are relatively subtle.
Most renal toxicities with ASOs are generally considered to be due to accumulation of oligonucleotides within lysosomes of the proximal tubule, resulting in physiologic perturbation of tubular absorptive capacity and in some cases, increased tubular proteinurea (Henry et al. 2008). However, as in the case of the liver, there are some candidate molecules that have demonstrated profound preclinical renal toxicity at doses below those where accumulation-related degenerative effects would be expected, thus implying other, currently undefined, mechanisms of toxicity. Based on workshop discussions at the 2011 and 2013 DIA/FDA oligonucleotide conferences, anecdotal reports from several sponsors are similar, but there are no published examples of this type of oligonucleotide-related nephrotoxicity and no experiments concerning pathogenesis are as yet published in the literature. In these uncommon cases, histologic changes including single cell necrosis, tubule necrosis, and/or dilation and casts have been described. Elevations in blood urea nitrogen (BUN) and serum creatinine (CR) have been reported, as well as changes in urine protein:creatine ratios (uPC). Several different backbone chemistries have been implicated, especially where a particular molecule has unusually high renal clearance, but LNAs have appeared to be associated with these unexpected kidney toxicities more often than other platforms. Due to their somewhat less common occurrence, the mechanism for these types of kidney injury are much less studied than hepatotoxicity, and ASO-protein binding within proximal tubule cells has not been conclusively demonstrated. However, based on their more frequent presentation with LNAs, protein interactions (similar to hepatic protein binding) are highly suspected. To date, no basepair sequences related to nephrotoxicity have been identified, and no particular protein or signaling pathways have been implicated in the pathogenesis of lesions considered unrelated to lysosomal accumulation. Based on similar experience with hepatotoxicity, assessment of the renal toxicologic potential of candidate molecules in 14- or 28-day mouse toxicity studies may be the best screening tool available at this time to deselect molecules with significant renal liabilities unrelated to lysosomal accumulation.
Due to use of these in vitro and in vivo preclinical screening strategies, human clinical cases of ASO-mediated nephrotoxicity have been rare. A case of acute tubular injury was reported in a healthy woman participating in a first-in-human trial of an LNA (van Poelgeest et al. 2013). The volunteer was given SPC5001, a typical LNA type ASO targeting proprotein convertase subtilisin/kexin type 9 (PCSK9) for the indication of lipoprotein cholesterol reduction. Acute kidney injury was noted 5 days after the patient received her third weekly subcutaneous doses of the drug. Serum creatinine increased and coincided with the presence of white blood cells, granular casts, and hematuria on urine microscopy. Kidney biopsy showed multifocal tubular necrosis and evidence of ASO accumulation. Upon conservative treatment, the patient’s serum creatinine level gradually decreased and reached her baseline level 44 days after the last oligonucleotide was administered. The patient recovered fully, and kidney function was normal at every follow-up visit. Importantly, other participants in the trial treated with an equal dose of SPC5001 also showed signs of transient tubular dysfunction, implicating a causal drug-related toxicity (van Poelgeest et al. 2013). The lesion appeared to be clinically monitorable, as retrospective analysis demonstrated increases in α-GST, KIM-1, and β2-microglobulin levels after only the first dose. Target pharmacology was unlikely to be the mechanism since PCSK9 loss of function mutations have not been associated with reduced kidney function (Zhao et al. 2006). No adverse histologic or biochemical effects were noted in cynomolgus monkeys given 20 mg/kg subchronically of SPC5001.
Acute tubular necrosis was also associated with long-term 2′-MOE PS ASO anticancer treatment after 74 weekly doses of 10 mg/kg in a human patient with metastatic melanoma. A causal mechanism for kidney injury was not definitively determined (Herrington et al. 2011). In that case, the authors felt accumulation effects were confounded by other patient-specific factors (including renal comorbidities and concomitant administration of other drugs). Tissue drug concentrations 28 days after stopping the ASO exceeded the predicted clinical range by 38-fold. Renal changes were reversible and complete recovery of renal function was noted 6 months after drug discontinuation (Herrington et al. 2011).
In addition to tubular lesions, another type of accumulation-independent nephrotoxic reaction attributable to ASOs is glomerulonephritis. Glomerular changes have been found in a small percentage of subchronic or chronic preclinical (rodent and monkey) toxicity studies of ≥3 months duration, most commonly with PS ASOs. In monkeys, lesions were characterized by enlarged glomeruli, variably increased cellularity of the tufts, increased mesangium, and occasional infiltration of inflammatory cells (Frazier et al. 2014). Immune-mediated dense deposits along the subendothelial glomerular basement membrane were identified by electron microscopy in monkeys given a 2′-oMe PS ASO, and complement fragments were noted along vascular tufts with anti-C3c immunofluorescence (Frazier et al. 2014). The endothelial cell was considered the initial cellular site of glomerular injury, representing a potential target for complement-mediated injury. Tubuloreticular inclusions were noted within the endothelium in monkey glomeruli, which in humans have been associated with immune deposition disease (Frazier et al. 2014). Glomerular changes were described in mice given the same 2′-oMe PS ASO, and have also occasionally been noted in rats with some ASOs. In mice, glomerular changes were identified as early as 56 days after initiating weekly injections and were characterized by slightly increased mesangial matrix, increased glomerular cellularity, and occasional inflammatory cells or nuclear debris. The mechanism of the changes was considered related to immune pathogenesis and local inflammatory activity in the kidney (Frazier et al. 2014). With chronic treatment over 6 months, however, there was marked mesangial accumulation of immunoglobulin fragments mixed with amyloid, consistent with the murine syndromes of hyaline glomerulopathy and contributory amyloidosis. This resulted in mortalities and, in some mice, secondary papillary necrosis from vascular disruption of the medulla. These chronic obliterative glomerular lesions in mice were considered a result of upregulation of murine-specific pathways and not necessarily relevant to humans (Frazier et al. 2014). However, the potential for clinical glomerular effects from ASO-mediated immunomodulation and especially complement-mediated damage (as in monkey) remains a relevant clinical concern.
Potentially, ASO-associated adverse events related to glomerulonephritis appear to be uncommon in human patients, with a low reported incidence in clinical trials, but there have been a few examples including with mipomersen (FDA 2012) and drisapersen (K.E. Meyers: personal communication, DIA oligonucleotide conference, 2013). 2 Fortunately, in the few cases where clinical drug-related glomerulonephritis has been noted, relatively rapid recovery has occurred after drug withdrawal. While proteinuria is noted commonly in preclinical and clinical studies with many ASOs (Kwoh 2008), this has generally been attributed to tubular changes rather than glomerular disease, as proteinuria, and especially albuminuria, may be associated with tubular as well as glomerular injury (Frazier and Seely 2013). It is important to distinguish between more medically manageable tubular toxicity and more deleterious glomerular injury, when proteinuria is identified in a patient (or a preclinical study animal) administered an ASO. Large increases (e.g., >1.5 g/L) or the presence of large molecular weight proteins in urine may aid in identifying glomerular origin and signal a risk for clinical glomerulonephritis. Small or trace amounts of protein and a predominance of albumin would be consistent with tubular effects, and of considerably less clinical concern.
Thrombocytopenia (Challenge #3)
Thrombocytopenia has been occasionally noted following ASO treatment in preclinical test species but appears to be compound-specific rather than a common oligonucleotide class effect. Published experience suggests that reduced platelet counts have been observed with approximately 10% of 2′-MOE PS ASOs and resulted in typically mild decreases at high doses in monkeys and rats, and more rarely mice (Henry et al. 2008). With several second-generation PS ASOs, doses of more than 20 mg/kg/wk in monkeys resulted in platelet reductions. The condition did not generally progress nor result in adverse effects on hemostasis (Levin and Henry 2008; Marquis and Grindel 2000). These drug-related effects appeared to have characteristics of sequence specificity, yet were non-selective with respect to pharmacologic targets (Levin and Henry 2008). In these cases, the mild thrombocytopenic episodes were reversible over the course of weeks or months depending on half-life (Levin and Henry 2008). Based on posters and discussions among participants at the 2013 DIA/FDA oligonucleotide conferences and the 2014 TIDE conferences (e.g., Younis et al. 2013), a slightly different preclinical presentation has been noted in recent years, in which sporadic monkeys in toxicity studies have exhibited marked thrombocytopenia with platelet counts below 40,000/µL, and without an apparent dose response. In most of these cases, it has taken multiple doses for the effect to occur, and precipitous drops in platelets have been noted. The clinical presentation is one of multiple hemorrhages and lethargy. In these unpublished anecdotal reports, platelet levels have increased after drug withdrawal, but thrombocytopenia recurred with drug rechallenge. The time of recurrence was sometimes protracted, which would not be consistent with a type I antibody–mediated hypersensitivity reaction. Although thrombocytopenia has occurred with some of the same proinflammatory PS that involved idiosyncratic cases of ASO-mediated vasculitis, the incidence of thrombocytopenia did not necessarily correlate with the incidence of vasculitis or occur in the same individuals.
Thrombocytopenia has also been noted occasionally in clinical studies with ASOs (Kwoh 2008), but platelet reductions have largely been mild and reversible and have more closely resembled the situations with early cases of ASO-mediated monkey thrombocytopenia. In an anticancer trial, parenteral administration of a PS ASO in patients with tumors reportedly led to “transient thrombocytopenia occuring only during the first course of therapy. The platelet count normalizes and causes no bleeding episodes” (Cuddihy et al. 1999). Rare instances of more severe cases of clinical thrombocytopenia have been reported. In a phase I study of an ASO-targeting Protein kinase C for a cancer indication, dose-limiting toxicities of thrombocytopenia, and fatigue were noted at a dose of 3.0 mg/kg/day (Yuen et al. 1999). In another phase I trial with an anti-BCL2 ASO, thrombocytopenia was also dose limiting (Waters et al. 2000). As rare events, these appear to be idiosyncratic responses in humans participating in clinical trials and have resulted in drug withdrawals or patient discontinuation in isolated instances (Kwoh 2008). Based on the recent preclinical and clinical experiences, the mechanism for these ASO-mediated changes in monkeys and humans is therefore under active investigation.
While several different potential mechanisms for thrombocytopenic episodes have been proposed, it is quite possible that the mechanism could be different between species and in the mild versus severe events. One general theory is that there may be an indirect effect on adenosine diphosphate (ADP) activation by ASOs, but so far no published data are available to support this hypothesis. Recent preliminary data presented in poster form (Younis et al. 2013: 2013 DIA oligonucleotide conference) indicates there are potential differences in platelet activation between humans and monkey strains given 2′-MOE ASOs. Some small molecule drugs are known to antagonize the P2Y12 receptor, which is responsible for ADP activation (Cattaneo and Lecchi 2007). Another theory suggests that there may be inappropriate binding and/or activation of platelet factor-4 (PF4). PF4 is released after platelet activation and can bind to negatively charged molecules. This type of polyanionic binding occurs with heparin and is responsible for heparin induced thrombocypenia (HIT) in humans. A similar pathogenesis for an HIT-like phenomenon with ASOs is based on the fact that charge-dependent binding of RNA to PF4 and even binding of PF4 to oligonucleotide aptamers has been described (Jaax et al. 2013). In support of this hypothesis, ASOs have been shown to bind platelets with the same type of electrostatic binding as heparin (Hartman et al. 1996; Jaax et al. 2013), and PF4 forms large multimolecular complexes with heparin that are highly immunogenic and can get cleared from the bloodstream (Greinacher et al. 2006). While this line of inquiry is promising, questions remain. For instance, it was noted that only about 60% of the anti-PF4/heparin antibodies that induced platelet activation in the presence of heparin also caused platelet activation in the presence of nucleic acids (Jaax et al. 2013). There is little or no published data as of yet to support any particular theory, but it is possible that the mechanism may be elucidated in the coming years, given the level of interest in this topic by industry.
Hybridization-dependent OTEs
Many recent advances in the understanding of ASO toxicity have come in the area of OTEs. While OTEs do not pose the current developmental challenges to industry that are related to immunostimulation, thrombocytopenia or hepatotoxicity, they are a constant consideration in choice of molecule to progress. Sequence specificity refers to the capacity of an ASO to distinguish between its intended target RNA sequence and all other RNAs in the cell. OTEs can potentially occur due to inadvertent inhibition (binding or blocking) by an ASO to unintended target RNAs with very similar basepair sequence and one or two mismatches. Designing oligonucleotide therapeutics free of complementarity to unintended RNA targets is desirable but almost unrealistic and is analogous to the situation with small molecule therapies where they are designed to specifically target a single receptor but interact with other related receptors (Lindow et al. 2012). The specter of these types of toxicities has haunted the industry for a longtime, but cases of demonstrable OTEs have been increasing in number with the shorter sequences associated with LNAs, some siRNAs and miRNA targeting ASOs. Risks for OTEs vary by backbone chemistry and the number of consecutive perfect nucleotide matches required to elicit an OTE depends on mechanism. There have been concerns that siRNAs can silence multiple genes in addition to the one intended and produce unpredictable OTEs (Scacheri et al. 2004). siRNAs in RISC can potentially elicit miRNA-like effects on mRNAs with as few as 7 nucleotide matches to the seed region of the siRNA (Jackson et al. 2006). In this respect, siRNAs also have the disadvantage of competition with endogenous microRNAs for RISC components, resulting in further OTE complications (Vickers et al. 2007). In contrast, because steric blocking is highly dependent on target sequence position in the intended RNA target, effects on unintended off-target RNAs containing perfect matches are not expected with oligonucleotides using this mechanism (Obad et al. 2011).
There has been a legitimate concern that human OTEs may not show up as toxicologic effects in preclinical species due to sequence differences between species. The monkey is most analogous to man in terms of potential OTEs based on sequence homology, and the use of the nonhuman primate in early toxicity testing should provide greater confidence that OTEs will not show up unexpectedly in human trials. Bioinformatic-based homology searches against human genomic databases for a given ASO or siRNA will frequently find “hits” with 1 or 2 human mismatches, but searches including 3 or more mismatches can result in lists of homologous genes in the hundreds, particularly with shorter (e.g., LNA) sequence lengths. Based on analysis of hybridization affinities, basepair mismatches of 1 or 2 bp are much more physiologically relevant than multiple mismatches, but there are other reasons why OTEs do not appear more frequently preclinically or clinically. The first reason has been the successful implementation of rigorous selection of molecules with the fewest relevant mismatches. This prescreening of potential candidates has become routine in most institutions and has resulted in filtering among closely related molecules prior to ever introducing an ASO into preclinical in vivo studies. Even with this screening, OTEs should have been noted with greater abundance in preclinical or clinical studies than they have to date. Instead, the toxicities noted have been overwhelmingly associated with those expected by class. Preclinical toxicities with ASOs are not generally related to pharmacology (on- or off-target), but rather are instead commonly associated with intracellular accumulation or proinflammatory activity. OTEs would more likely result in 1 or more unexpected toxicities arising within a study or clinical trial, and this just has not happened with great frequency. The reason is that not all off-target binding actually leads to functional effects. Not all sites on mRNA are accessible to an ASO, nor are all off-target mRNAs in tissues that receive pharmacologic concentrations of an ASO as they are differentially and temporally expressed in different tissues. As has been noted, tissues such as kidney and liver appear to receive higher concentrations than many other tissues where an off-target mRNA may be expressed and off-target mRNAs may be limited in less accessible tissues like brain, gastrointestinal, or testes. Finally, due to threshold effects or synergistic pathways, not all off-target genes produce toxic effects when they are knocked out. For these reasons, toxic responses due to off-target pharmacology are actually much more common with small molecules than they are with ASOs. We (as toxicologists or pathologists) are not as successful at predicting these small molecule OTEs, as we have become with ASOs because we do not have similarly efficient bioanalytic screening readily available. Pragmatic genomic screening strategies, especially regarding stringency, are in place in most companies to delineate potential OTEs. The in silico algorithms used in these bioinformatic approaches are constructed based on data and experience from previous drug discovery knowledge of compounds with similar mechanism and chemistry (Lindow et al. 2012). These methods are not foolproof, but OTEs should in most cases still be anticipated or predicted preclinically. Even where knockdown of a human-specific mRNA is suspected, in vitro screening assays in human cell lines using quantitative PCR and looking for off-target gene knockdown may be employed early in development. However, from the pathologist’s perspective, one tends to have to agree with the Oligonucleotide Safety Committee’s recommendation concerning definitive testing for OTEs: “Although animal toxicology studies do not guarantee that all potential safety issues will be identified for humans, preclinical toxicity studies remain the best and most well characterized way to predict the presence of all toxicities, including those related to hybridization” (Lindow et al. 2012).
Recent Success Stories Provide More “Reasons to Believe”
Much of the promise of oligonucleotide therapies lies in recent successes in demonstrating efficacy. A long list of ASOs are currently under evaluation in phase II or III clinical trials for treating cancer, autoimmune disease, genetic and neuromuscular disorders, and life-threatening viral diseases (Jarver et al. 2012; Stanton et al. 2012). Recently, mipomersen (Kynamro®) was approved by the FDA for the treatment of familial hypercholesterolemia and was a first-in-class systemically administered antisense agent targeting apolipoprotein B-100 (apoB-100). Despite a preclinical toxicity profile that included the usual range of class-wide effects and proinflammatory activity, this 2′MOE had a clinical safety profile considered acceptable by the regulatory reviewers for the indicated population (FDA 2012). Discontinuations due to serious adverse events were higher in mipomersen patients than in those given placebo, and this fact coupled with some clinical hepatotoxicity led to refusal of marketing approval by the EMA (EMA 2012). However, the FDA considered these potential issues with the medication were countered by significant efficacy of the drug as demonstrated by significant decreases in LDL-C in patients (FDA 2012). Preclinical toxicity findings included lymphohistiocytic cell infiltrates and inflammatory changes in numerous organs; increased plasma cytokines and chemokines such as MCP-1 in mice; increased splenic weights and total serum IgG in monkeys in chronic studies with associated decreased C3 concentrations; local injection site reactions in multiple species; and vascular lesions in 2 of the 6 monkeys treated for 12 months. Finally, mean decreases of up to 30% in platelet count were observed in monkeys and rats. Encouragingly, similar findings (other than flu-like symptoms and injection site reactions related to immune stimulation) did not appear to be present in the clinical population and adverse events of concern were more focused on hepatotoxicity. Thus, much of the preclinical toxicity risks identified with mipomersen administration were not noted in clinical patients. While this does not preclude clinical relevance of these and other ASO-related preclinical toxicities with other therapies, it does provide potential pathways for future development of related agents. Since the class of RNA interfering therapies covers a range of indications, and therefore drug candidates are being reviewed across a variety of potential divisions among the FDA (CDER/CBER) and EMA, differences in experience and tolerance to class-wide toxicities are bound to arise within the various regulatory agencies and divisions. Like small molecule drugs, the particular indication for a specific drug is an important factor in the risk assessment, and risk:benefit must be carefully evaluated for patients where an ASO therapy is considered.
Successful Screening Strategies to Eliminate More Toxic Compounds
A common theme among the approaches by the pharmaceutical and ASO biotechnology community to address the various toxicologic challenges has been to implement early and comprehensive screening strategies. A wide variety of these have already been described in this review including a series of in vitro (e.g., complement fixation assays, bioinformatic genomic homology screens, and hepatocyte cytotoxicity assays ) and in vivo assays (e.g., 28-day or longer mouse hepatotoxicity/nephrotoxicity screens). These have become increasingly important in establishing the best choices for drug candidates to progress into clinical trials. Because of predictable pharmacokinetic properties, a single mid- to high dose of multiple candidates can be run effectively in the same study for comparison. By the time the toxicologist or pathologist encounters an ASO in a subacute or subchronic preclinical study, most ASOs have been vetted through a robust gauntlet of tiered assays to potentially identify unwanted effects due to severe nephrotoxicity, hepatotoxicity, or pronounced proinflammatory activity. Many or most molecules that are in phase II and III at the current time do not share many of the more severe toxicities described earlier in this article. However, for the toxicologic pathologist evaluating oligonucleotide candidate selection studies, these types of toxicities may be present in subtle form. It is the pathologist’s job to make an accurate comparison to other viable candidates or tool molecules, so an accurate rank order of candidates can be established. In this regard, the pathologist has played, and continues to play, a critical role in successful ASO screening strategies.
New Generation Novel ASOs and Delivery Systems
Recent advances in ASO backbone chemistries, including conjugation to other moieties and new delivery systems, have allowed better tissue penetration, enhanced intracellular targeting and less frequent dosing of ASOs (Fiset and Gounni 2001; Akinc et al. 2010). Targeting specific tissues other than liver or kidney has always posed a problem in ASO pharmacology. Due to preferential transporter uptake by proximal tubules, macrophages, and hepatocytes, tissue levels in other organs tend to be much lower. Novel approaches to intracellular delivery systems that would create higher oligonucleotide levels in the correct tissue (and proper intracellular compartment of the target) would permit lowering the overall circulating exposures. The use of 10-fold lower concentrations, as some of these platforms provide, may ultimately result in the potential for fewer systemic preclinical and clinical toxicities (Akinc et al. 2010; Wang et al. 2010). Biomolecular delivery systems, utilization of vectors and other delivery targeting moieties, liposomal nanoparticles, peptide linkers, or conjugation to N-acetylgalactosamine (GalNAc), are some of the innovative designs that have been implemented to enhance cellular/nuclear uptake of ASOs (Ozbaş-Turan, Akbuğa, and Sezer 2010; Choksi, Poonawalla, and Wilkerson 2010; Wang et al. 2010; Frank-Kamenetsky et al. 2008; Akinc et al. 2010). One of the major goals of these cellular targeting strategies is to decrease the incidence of one or more of the current challenging toxicities (immunomodulation, thrombocytopenia, and hepato/nephrotoxicity). Cationic delivery systems internalize ASOs via endocytosis. To avoid having the ASO in the wrong subcellular compartment, modifications to delivery systems through the use of basic peptides or carrier proteins have been made, which modulate plasma membrane permeability and provide rapid entrance into the cytoplasm without high lysosomal concentrations (Jarver et al. 2012; Henke et al. 2008). Such peptides are synthetically conjugated, used as noncovalent complexes, or used in combination with polymer, liposomal or exosome formulation techniques. In the case of naked siRNA, for all except kidney and perhaps skin applications, a delivery system of some sort is necessary, or at least very advantageous, to obtain significant and sufficient in vivo activity. For other types of ASOs, peptide linkers may also help directed cellular targeting and in some cases, transportation into the nucleus (Jarver et al. 2012; Henke et al. 2008).
There has been increasing interest of late in the use of N-acetylgalactosamine (GalNAc)-conjugation with both therapeutic siRNAs and with PS ASOs. Using these types of conjugated exogenous ligands, the ASO or siRNA multivalent molecule binds with high affinity to the asialoglycoprotein receptor (ASGPR) expressed on hepatocytes and gets rapidly incorporated into the cytoplasm of the hepatocyte (Akinc et al. 2010). ASGPR is a c-type lectin that is abundantly expressed in parenchymal cells in the liver (Geuze et al. 1983). It seems from early experiments from several companies that this technique provides a more desirable compartmentilization of the molecules, improves potency, and lowers the necessary efficacious dose (Prakash et al. 2014). These GalNac conjugates have so far been utilized specifically for liver localization, but other endogenous receptors may be targeted by other exogenous ligand conjugates for other organs. Based on data from subacute studies, the class-wide ASO toxicities related to accumulation are still present with conjugated molecules, but appear to be lessened in severity, as compared to unconjugated PS ASOs. Chronic studies are still pending with the GalNac conjugates, as with most of these other new delivery technologies, so any differences from classic ASO toxicity profiles (including those associated with the “challenging 3 toxicities”) remain to be determined with the latest generation of ASO molecules. For the immediate future, ASOs are likely to be most successful in targeting indications where drug delivery is most efficient (liver, kidney, or lung diseases), and in indications with well-defined risk:benefit ratios such as orphan diseases, life-threatening conditions such as cancer, or those where no other alternative therapies currently exist.
Conclusions
The advent of successful screening strategies to eliminate more toxic molecules, a new understanding of the risks of off-target nucleotide binding and mitigation of potential OTEs has improved the process of oligonucleotide development. The main toxicologic challenges facing oligonucleotide development programs are considered to be proinflammatory effects (vasculitis/inflammatory infiltrates), nephrotoxicity and hepatotoxicity unrelated to lysosomal accumulation, and thrombocytopenia. Recent investigative work by several laboratories has helped elucidate mechanisms for these issues, allowing a better understanding of the clinical relevance and implications of these particular toxicities. However, the field of oligonucleotide development is expanding rapidly, with many new platforms and methodologies for targeted delivery, and vigilance must be maintained in monitoring the potential toxicity of this compound class.
Footnotes
Acknowledgments
I appreciate the assistance of Mike Ringenberg, Joel Parry, Jim Ridings, Justin Vidal, and Jan Losos of GSK in the critical review of this article.
Author Contribution
K. S. Frazier contributed to conception and design, drafted article, gave final approval, and agrees to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.