
Abstract
The naturally produced, nonprotein amino acid β-N-methylamino-
Introduction
Exposure to β-N-methylamino-L-alanine (BMAA) has been proposed as a risk factor for the development of sporadic neurodegenerative diseases (reviewed in the study of Bradley and Mash 1 ). This link was suggested because of the relatively high concentrations of BMAA in the unique food sources consumed by the Chamorro people living on Guam, 2 an island population that was plagued by a high incidence of the neurodegenerative disease complex amyotrophic lateral sclerosis/Parkinsonism dementia complex (ALS/PDC). 3 The subsequent detection of BMAA in a number of trophic systems outside of Guam 4 –12 and in brain samples of deceased Alzheimer's disease and ALS patients from North America 13,14 has increased worldwide awareness and concern for potential human exposure to this neurotoxin. Although a highly debated topic, 15 –22 the link between exposure to BMAA and neurodegenerative disease development is supported by the findings of Scott and Downing 23,24 who showed that a single dose of BMAA administered at a susceptible age in an animal model (corresponding to second trimester human fetal exposure) results in the development of the full spectrum of symptoms and neuropathologies characteristic of ALS/PDC. The observed symptoms and neuropathologies cannot, however, be fully explained by the current proposed mechanisms of toxicity, suggesting that additional mechanisms of BMAA toxicity need to be identified in order to fully understand the role of BMAA in neurodegenerative disease development.
Compelling in vitro evidence suggests that the spontaneously formed β-N-carboxy BMAA adduct acts as an excitotoxin at both ionotropic and metabotropic glutamate receptors, supporting this as a mechanism by which BMAA contributes to neurodegenerative disease development (reviewed in the study of Chiu et al 25 ). However, only 9% of bioavailable BMAA is converted to the excitotoxic β-N-carboxy BMAA adduct, 26 and BMAA is therefore a relatively weak excitotoxin 18,21 requiring high exposure concentrations to induce toxicity. Misincorporation of BMAA into proteins in the place of L-serine was also suggested as a potential mechanism of toxicity. 27 However, numerous studies have shown that misincorporation of BMAA does not occur 28 –31 and that the widely reported association of BMAA with proteins is most likely the result of strong surface associations. 32
Liu et al 33 demonstrated that BMAA can induce oxidative stress by inhibiting cystine uptake through the cystine/glutamate antiporter system, with a subsequent depletion of the intracellular hydroxyl radical scavenger glutathione. De Munck et al 34 observed oxidative damage in the livers of rats 6 months post intraperitoneal administration of BMAA, in addition to an alteration in the activities of the stress response enzymes. Esterhuizen-Londt et al 35 also demonstrated an overall decrease in the activities of the oxidative stress response enzymes in the aquatic macrophyte Ceratophyllum demersum following 24 hours of BMAA exposure as well as in the zooplanktonic crustacean Daphnia magna. 36 Changes in the oxidative stress response enzyme profiles were also reported for 2 freshwater mussel species in response to BMAA exposure. 37 Despite these findings, and the well-described role of oxidative stress in neurodegenerative disease development (reviewed in 38 –46 ), the direct effect of BMAA on human oxidative stress response enzymes has received little attention. Van Onselen and Downing 32 showed the inhibition of various enzymes containing functional hydroxyl groups, offering a plausible mechanism for direct enzyme inhibition in the generation of excess reactive oxygen species (ROS). We therefore sought to investigate the effect of BMAA on human catalase, an enzyme directly involved in the detoxification of β-amyloid-linked cellular toxicity. 47
Materials and Methods
The direct effect of BMAA on catalase (EC 1.11.1.6) activity was assessed in a cell-free exposure using a commercial preparation of catalase from human erythrocytes (2.2 mg·mL−1 solution in 50 mmol/L Tris, pH 8.0, molecular weight ∼250 kDa, 71 350 units·mg protein−1; Sigma Aldrich, St Louis, MO, USA) and compared to the effect of the known catalase inhibitor 3-amino-1,2,4-triazole (3AT). Catalase, diluted in 10 mmol/L sodium phosphate buffer pH 7.0, was incubated with either BMAA (β-N-methylamino-L-alanine hydrochloride, Sigma Aldrich) or 3AT (Sigma Aldrich) at various concentrations ranging from an equimolar concentration of enzyme to BMAA/3AT to 100 times the concentration of BMAA/3AT to enzyme. A 1:1 molar ratio of BMAA to catalase concentration was selected as the starting concentration to investigate inhibition based on the hypothesized interaction of 1 molecule of BMAA with a single exposed hydroxyl group. 32 Controls without BMAA/3AT supplementation were also prepared as well as controls of BMAA or 3AT in the absence of catalase to assess whether hydrogen peroxide degradation could be facilitated by direct interactions between the substrate and the inhibitors. Following an incubation period of 1 hour with continuous agitation at 37°C, the enzyme activity was assessed as described by Sinha 48 by measuring the formation of chromic acetate from dichromate in the presence of hydrogen peroxide spectrophotometrically at 610 nm. In order to determine the type of inhibition, enzyme kinetics were assessed over a range of substrate (hydrogen peroxide) concentrations for catalase in the presence of either BMAA or 3AT at the lowest inhibitory concentration and compared to the untreated control kinetics.
The effects of BMAA and 3AT on catalase activity were also assessed in a human colorectal epithelial adenocarcinoma cell line, Caco-2 cells (Highveld Biological, Johannesburg, Republic of South Africa). A nonneural cell line was selected to investigate catalase inhibition so as to eliminate any cellular responses linked to the well-described excitotoxicity of BMAA in neural cells. We have previously shown that BMAA does not induce apoptosis or necrosis in Caco-2 cells, as measured by the MTT assay and phosphatidylserine translocation, after 48 hours of exposure.
29
The cells were routinely maintained in RPMI-1640, with 2.05 mmol/L
Results and Discussion
In both the cell-free (Figure 1) and the cell culture (Figure 2) exposures, BMAA inhibited catalase activity to the same extent as 3AT, the known catalase inhibitor, without directly affecting substrate concentration (control data not shown). Based on the kinetic data that were obtained (Figure 1B), BMAA inhibition appears to be noncompetitive in nature. A possible site for the noncompetitive interaction with the enzyme, based on findings by Van Onselen and Downing, 32 is the NADPH (nicotinamide adenine dinucleotide phosphate) binding site. Interactions between BMAA and proteins appear to occur at amino acid residues that contain hydroxyl groups in their side chain, that is, serine, tyrosine, and threonine, and the NADPH-binding site contains a functional serine residue (Ser201) that ensures proper docking of NADPH through hydrogen bond formation with the N6 amino group of the adenine ring. 50 Prevention of NADPH binding by BMAA would interfere with the electron relay chain from NADPH to the heme oxyferryl group that forms during the first step of the catalysis. 51 When electron transfer from NADPH is inhibited, electron transfer is from the tyrosine residue (Tyr358) that is ligated to the heme group, instead. However, only a single electron can be transferred from the tyrosine residue, resulting in incomplete oxidation and the release of an inactive form of catalase referred to as Compound II. 51,52 This could possibly explain the reduced catalase activity that was observed in the presence of BMAA, but structural data are required to confirm this hypothesized interaction.
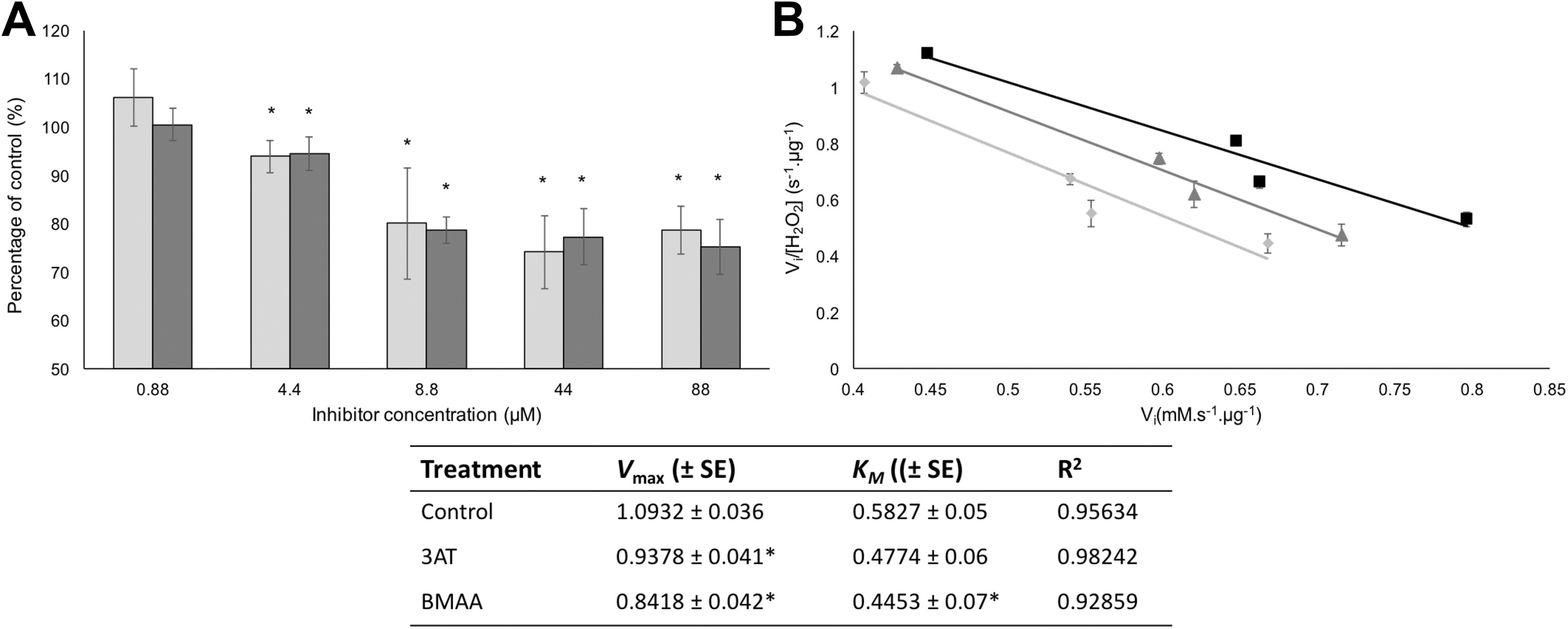
The effect of β-N-methylamino-L-alanine (BMAA) and 3-amino-1,2,4-triazole (3AT) on commercial catalase. Panel A depicts the activities, relative to untreated controls, of commercial catalase that was incubated with either BMAA (light gray bars) or 3AT (dark gray bars) at various concentrations depicted on the x-axis. An Eadie-Scatchard enzyme kinetic plot for catalase in the presence of either 4.4 µM BMAA or 3AT is depicted in panel B. The kinetics for the untreated controls are indicated by the black squares (□), the kinetics for catalase incubated with 3AT are indicated by the dark gray triangles (▴), and the kinetics for catalase that were incubated with BMAA are indicated by the light gray circles (•). The kinetic data derived from the Eadie-Scatchard plot with standard errors (±SE) are indicated in the table. The averages of 5 replicate samples (n = 5) were plotted and error bars denote the standard deviation from the mean. An asterisk (*) indicates significance from control samples (P < 0.05).
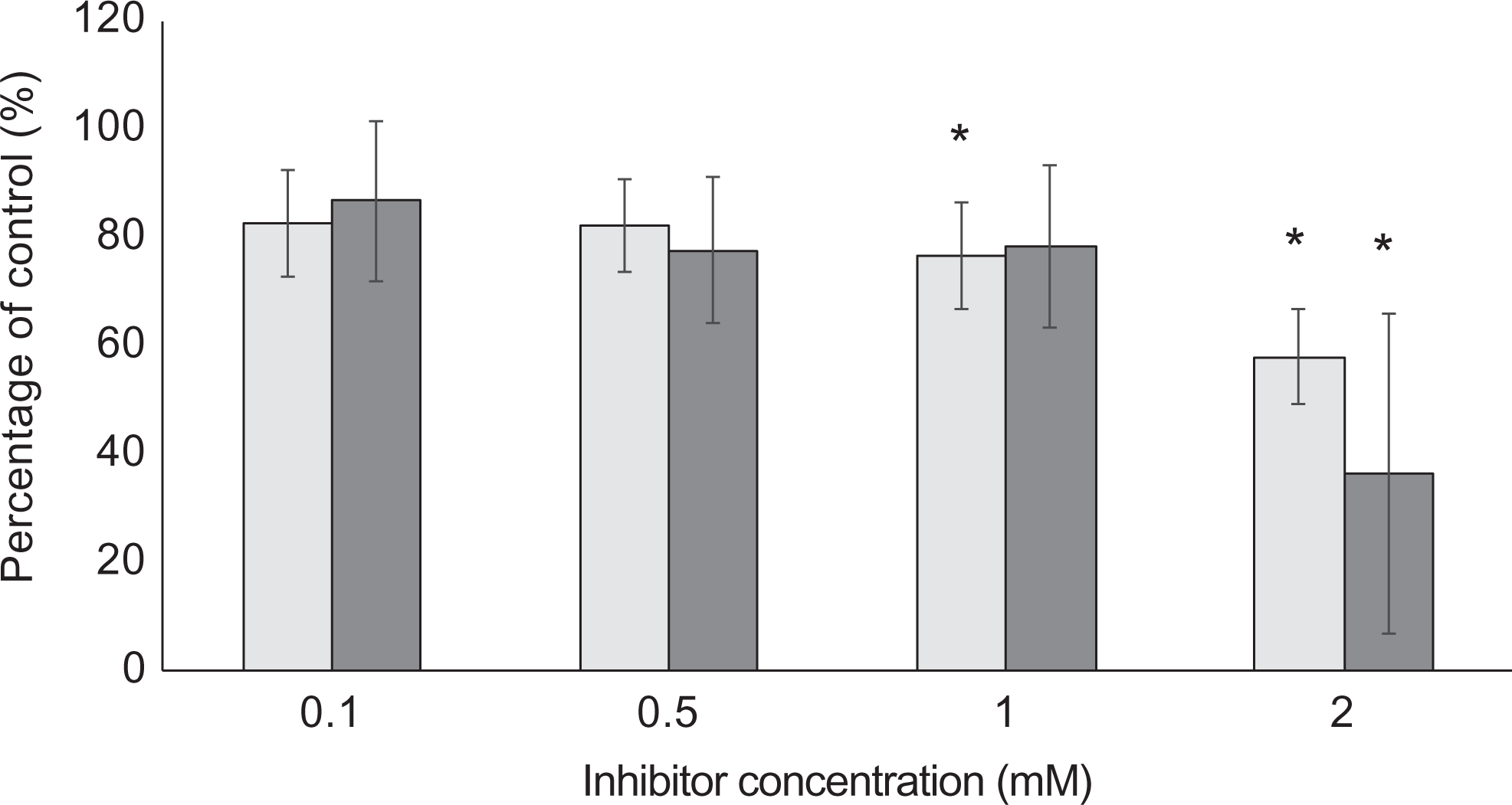
The effect of β-N-methylamino-L-alanine (BMAA) and 3-amino-1,2,4-triazole (3AT) on the catalase activity of Caco-2 cells. Following 6 hours of exposure to either BMAA (light gray bars) or 3AT (dark gray bars) at various concentrations indicated on the x-axis, the catalase activity of the cell lysates was determined and plotted against the untreated controls. The averages of 6 replicate samples (n = 6) were plotted, and the error bars indicate the standard deviation from the mean. An asterisk (*) indicates significance from control samples (P < 0.05).
The kinetic data that were obtained for 3AT inhibition (Figure 1B) indicate noncompetitive inhibition. However, structural analysis has revealed that catalase inhibition by 3AT is most likely due to binding of 3AT in the active site, 51 suggesting competitive inhibition. Interestingly, however, Margoliash et al 53 described 2 types of inhibitions of catalase by 3AT: a reversible inhibition that is obtained at relatively high concentrations (20 mM) of the inhibitor and an irreversible inhibition that occurs between 3AT and the catalase–hydrogen peroxide complex I. The structural analysis of catalase with 3AT by Putnam et al 51 only accounts for 1 type of inhibition. Since the irreversible inhibition requires the formation of the catalase-hydrogen peroxide complex I, 3AT cannot be bound in the active site for this type of inhibition, since it would interfere with complex I formation. In addition, the hydrogen peroxide substrate concentration that was used in our study far exceeded the concentrations of 3AT that were used, making competitive inhibition at the active site highly unlikely. Our kinetic data therefore support the findings of Margoliash et al 53 who showed that 3AT inhibits catalase by multiple mechanisms. Whether this might also be the case for BMAA is unknown.
A general mechanism of toxicity such as catalase inhibition implies that BMAA might not necessarily be a tissue-specific toxin. However, BMAA has only been linked to the development of neurodegenerative diseases. This may be a function of localization, which has been shown for BMAA to occur in the brains and spinal cords of fetuses following BMAA administration to pregnant dams on gestational day 14 and in the brains and spinal cords of 10-day old neonatal rats that received a single subcutaneous injection of BMAA. 54 –56 If this holds true for humans, a large hippocampus, at the same developmental age, is <1 mL in volume. 57 To achieve a BMAA concentration of 4.4 μmol/L in that area, 520 ng of BMAA needs to cross the blood–brain barrier and accumulate in the hippocampus. Based on the hippocampal accumulation data presented by Karlsson et al, 56 primate bioavailability data (estimated around 80%) 58 and placental transfer, 54 a pregnant woman needs only to consume 190 μg of BMAA once to achieve that concentration. That is equivalent to <30 grams (dry weight) of mussels collected in the Thau lagoon (French Mediterranean Sea) based on the HILIC-MS/MS (hydrophilic interaction chromatography-triple quadropole mass spectrometry) data presented by Réveillon et al. 59 Therefore, it can be argued that the concentration of BMAA used in the cell-free study is environmentally relevant. Furthermore, neuronal cells are naturally more susceptible to oxidative damage than other types of cells, mainly due to the high rate of oxygen metabolism with resultant ROS formation and the post mitotic nature of neuronal cells that accumulate oxidative damage with aging (reviewed by the study of Radak et al 60 and Manoharan et al 61 ). Although not very reactive by itself, hydrogen peroxide is relatively stable and can diffuse away from its site of formation to react with redox metals such as copper and iron, which are present in relatively high concentrations throughout the central nervous system (CNS), to form the very reactive hydroxyl radical that is central to oxidative damage of DNA, proteins, and lipids that are typically found in neurodegenerative disease patients (reviewed in Smith et al 62 ). The demonstrated BMAA-induced inhibition of catalase and the subsequent accumulation of hydrogen peroxide is therefore more likely to occur in the CNS and could therefore be a significant contributor to the neurotoxicity of BMAA.
Footnotes
Author Contributions
R.v.O. and T.G.D. designed the experiments; R.v.O. performed all the experiments and analyzed the data; R.v.O. and T.G.D. wrote the manuscript. R.v.O. contributed to conception and design, contributed to acquisition, analysis, and interpretation, and drafted manuscript. T.G.D. contributed to conception and design and critically revised manuscript. Both authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by the National Research Foundation of South Africa.