
Abstract
The peptide-based radioactive compound [68Ga]Ga-DOTA-Siglec-9 is a novel agent for imaging of inflammation with positron emission tomography. The drug target of [68Ga]Ga-DOTA-Siglec-9 is vascular adhesion protein 1. Previous studies have obtained promising results with [68Ga]Ga-DOTA-Siglec-9 in experimental animals. However, before taking this novel imaging agent into clinical trials, safety and toxicological studies need to be performed with the nonradioactive precursor compound DOTA-Siglec-9. This extended single-dose toxicity study was designed to provide information on the major toxic effects of DOTA-Siglec-9 and to indicate possible target organs after a single intravenous (iv) injection in rats. The study was performed using 60 adult Hsd: Sprague Dawley rats and included a control group and a treatment group to investigate the toxicity of DOTA-Siglec-9 solution at a final concentration of 0.2 mg/mL after a single iv injection of 582 µg/kg. The maximum dose tested was 1,000-fold the clinical dose on a mg/kg basis as indicated in European Medicines Agency International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use guideline M3(R2). The planned human clinical dose is approximately 0.582 µg of DOTA-Siglec-9 per kg of body mass. This study demonstrates that iv administration of DOTA-Siglec-9 at a dose of 582 µg/kg was well tolerated in rats and did not produce toxicologically significant adverse effects.
Introduction
Vascular adhesion protein 1 (VAP-1), currently known as a primary amine oxidase (AOC3, EC 1.4.3.6), is a leukocyte homing-associated molecule. 1 For several years, we have investigated VAP-1 as a potential target for sensitive whole-body positron emission tomography (PET) imaging of leukocyte trafficking. Under normal conditions, VAP-1 is stored in intracellular storage granules, but upon an inflammatory stimulus it is rapidly translocated to the endothelial cell surface, where it is readily accessible to intravenously administered PET ligands. Our leading VAP-1 targeted radioligand is gallium-68-labeled 1,4,7,10-tetraazacyclododecane-N′, N″, N‴, N‴′-tetraacetic acid-conjugated sialic acid-binding immunoglobulin-like lectin 9 motif containing peptide ([68Ga]Ga-DOTA-Siglec-9, Figure 1). 2 The basis for this is the finding that Siglec-9 is a leukocyte ligand of VAP-1, and [68Ga]Ga-DOTA-Siglec-9 is useful for detection of atherosclerotic, skin, synovial, and lung inflammation, as well as infection-induced inflammation in mouse, rat, rabbit, and pig models. 3 –9 [68Ga]Ga-DOTA-Siglec-9 is prepared from the precursor compound DOTA-Siglec-9 (C104H174N30O32S2, molecular weight 2420.8 g/mol), which is a cyclic CARLSLSWRGLTLCPSK peptide with intramolecular disulfide-bridged cysteines, consisting of residues 283 to 297 from Siglec-9. Additionally, the tracer has an 8-amino-3,6-dioxaoctanoyl linker (polyethylene glycol derivative) between the DOTA chelator and the peptide sequence (Figure 1). 2
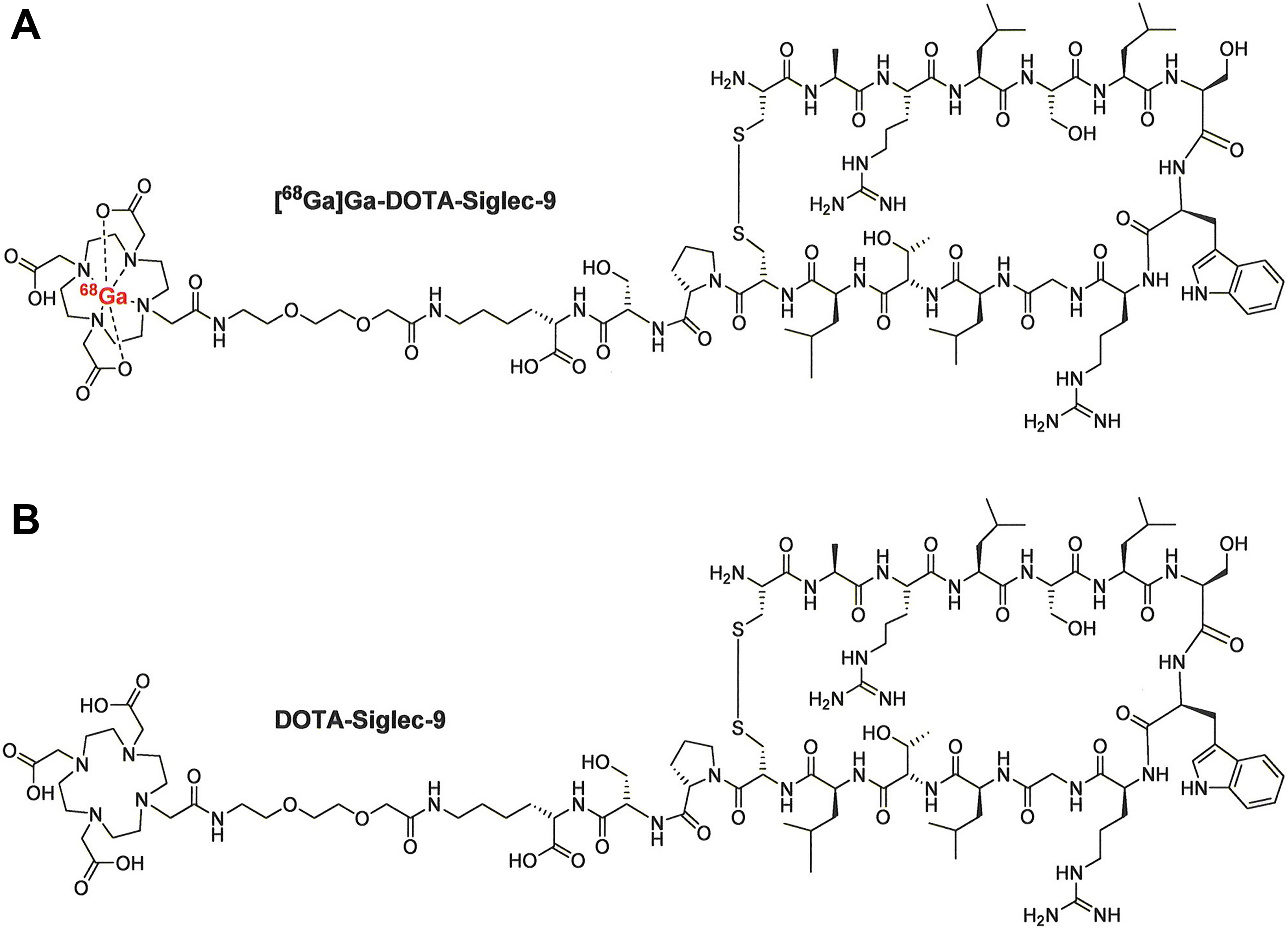
Chemical structures of [68Ga]Ga-DOTA-Siglec-9 (A) and DOTA-Siglec-9 (B).
The safety characteristics of a candidate imaging agent must be adequately evaluated before performing human studies. Preclinical studies and the resulting data analysis and reporting should be in compliance with Good Laboratory Practice (GLP). The GLP defines the quality of the research and the documentation system used; it is required by the licensing authorities and ensures high-quality and reliable test data in nonclinical studies. This extended single-dose toxicity study in rat by intravenous (iv) route of administration conforms to the Organisation for Economic Co-operation and Development (OECD) Guidelines for Testing of Chemicals, 10,11 and the dose level of 1,000-fold the clinical dose on mg/kg basis for iv was selected according to The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) guideline M3 (R2) on nonclinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals (EMA/CPMP/ICH/286/1995) – paragraph 7, and the Position paper on non-clinical safety studies to support clinical trials with a single microdose (CPMP/SWP/2599/02) – paragraph 4.1. 12,13
The estimated human equivalent dose is usually based on body surface area. Exposure in this case would be 0.582 mg/kg (rat) = 3.492 mg/m3 when referring to the Food and Drug Administration Guidance. 14 The estimated clinical dose 0.582 µg/kg would be 0.02 mg/m3, yielding a rat-to-human exposure ratio of 170 (3.49/0.02).
During the preclinical drug development, we have previously performed a pilot non-GLP acute toxicity testing of DOTA-Siglec-9 on rats (n = 12) with a dose of 300 µg/kg body weight and it was well tolerated in rats and caused no adverse clinical or anatomic pathology finding (unpublished results). We have carried out biodistribution and biokinetic evaluations in mice, rats, rabbits, and pigs with the aid of PET techniques. 2 –9 In general, this compound is rapidly excreted from urinary track and the highest radioactivity concentration is observed in urine. For example, in Sprague Dawley rats, the urine excretion of GMP-grade [68Ga]Ga-DOTA-Siglec-9 was 97% of injected dose per gram (%ID/g) at 60 minutes postinjection. In contrast, the uptake in kidneys was much less (3.7%ID/g) and even less was observed in other organs/tissues (0.023-0.76%ID/g). 5 Renal excretion is a favorable feature regarding its clinical use for PET imaging. In practice, patient is asked to void after the PET imaging to minimize radiation exposure. Regarding the plasma stability and protein binding of the corresponding radiolabeled compound [68Ga]Ga-DOTA-Siglec-9, we have performed a comparative study in vitro using plasma from different species including mouse, rat, rabbit, pig, and human. It has revealed considerable differences among species and [68Ga]Ga-DOTA-Siglec-9 is the most stable in pig and human plasma. 15
The aim of this study was to investigate the safety of DOTA-Siglec-9 after a single iv injection into a rat in compliance with GLP. It was designed to provide information on the major toxic effects of the test substance and to indicate possible target organs.
Materials and Methods
The study protocols and procedures were reviewed and approved by the National Animal Experiment Board of Finland (license number ESAVI/8609/04.10.07/2015). This study was conducted at the University of Turku Central Animal Laboratory (UTUCAL) in compliance with the “OECD Principles of Good Laboratory Practice” (ENV/MC/CHEM(98)17), as revised in 1997. 16 The GLP status of UTUCAL is based on regular inspections by the Finnish Medicines Agency.
Histopathological evaluation and statistical analyses were performed at CiToxLAB Scantox A/S, Lille Skensved, Denmark, in compliance with Executive Order No. 1245 of December 12, 2005, on GLP for Medicinal Products, as required by the Danish Health and Medicines Authority, which is in accordance with the OECD Principles of Good Laboratory Practice (as revised in 1997).
Animals, Study Design, Dose Groups, and Clinical Observations
A total of 60 Hsd: Sprague Dawley-specific pathogen-free rats (30 females and 30 males) supplied by Envigo (Horst, The Netherlands) were used in this study. The acclimatization period was 19 days, and only clinically healthy animals were accepted into the study. The animals were individually identified by tail numbering. Animals were group housed during the dosing and monitoring phase (2-4 animals per cage) in polycarbonate Macrolon III cages with Aspen bedding, nesting material, gnawing bricks (Tapvei Oy, Harjumaa, Estonia), and polycarbonate tunnels as enrichment. The temperature in the experimental animal room was 18°C to 24ºC. The relative humidity was at least 30% and did not exceed 70%. Lighting was artificial and followed a 12-hour light/dark cycle. The rats were fed ad libitum with an RM1 (E) SQC diet, batch 2622 (Special Diet Services, Witham, Essex, England). Tap water was provided ad libitum. The rats were allocated into 2 groups to receive either vehicle (saline) only or a dose 582 µg/kg of DOTA-Siglec-9 peptide, as indicated in Table 1.
Study Groups and Treatments.a

a Day of dosing defined as day 1. Study groups, necropsy days, and numbers of rats in each group are depicted in this table. Rats were administered a single dose of either saline (controls) or DOTA-Siglec-9 solution at a dose of 582 µg/kg.
The animals were weighed and randomly assigned to treatment groups. The animals were 11 weeks old at commencement of the study, and the weight variation of the animals used did not exceed ±20% of the mean weight of each sex. Blood and tissue samples were collected at 2 time points (day 2: interim sacrifice, and day 14: terminal sacrifice) for hematology, clinical chemistry, coagulation testing, and histopathological analysis. All animals terminated at days 2 and 14 were subjected to a full, detailed gross necropsy which included careful examination of the external surface of the body, all orifices, and the cranial, thoracic, and abdominal cavities and their contents. Necropsy days were scheduled so that females and males from both groups (control and treatment) were sacrificed each day. Animals were observed twice daily for general well-being and signs of morbidity and mortality. During weekends and national holidays, observations were made once daily. Detailed clinical observations were made outside the home cage just prior to dosing, before the necropsy, and once a week (for animals sacrificed on day 14). The following organs were weighed at necropsy: liver, kidneys, adrenals, testes (males), epididymides (males), uterus (females), ovaries (females), spleen, thymus, brain (the whole brain including the cerebellum but excluding the olfactory bulbs), and heart. Paired organs were weighed together. Ophthalmological examination was performed in all animals, prior to dosing with the test item and at the termination of the study.
Test Item and Dosing
Lyophilized DOTA-Siglec-9 meeting Good Manufacturing Practices (GMP) grade specifications is a white solid powder and was manufactured by ABX GmbH, Radeberg, Germany. The pH of DOTA-Siglec-9 was 5.81 as measured at 23°C and the osmolality was 288 mosm/kg H2O. Aliquots of lyophilized DOTA-Siglec-9 were stored in sealed vials in a freezer at −20°C and were protected from light. Before use, the preweighed aliquots (0.99-1.01 mg/vial) of DOTA-Siglec-9 were dissolved in the vehicle (sterile saline) to obtain test solutions with a concentration of 0.2 mg/mL ready for injection. The individual dosing volume was determined according to the weight of each rat on the day of dosing. Taking a rat of 200 g body weight as an example, the injection volume was 582.0 µL. In practice, the graduation of the syringes used for injection resulted in injection volumes of approximately 580 µL for a rat with a 200 g body weight. Mean value of the test item injection volume for females was 0.6 mL and for males 0.9 mL. The rats in the control group received sterile saline solution (mean injection volume for females 0.7 mL and for males 0.9 mL). The dissolved DOTA-Siglec-9 was administered as bolus via tail vein injection into the rats within 1 hour of the DOTA-Siglec-9 solutions being prepared. The maximum dose to be tested was 1,000-fold the clinical dose on an mg/kg basis for iv injection.
Histopathology
At necropsy, organs or representative samples of them were collected from all animals according to the guidelines for regulatory type toxicity studies. 17 –19 Organs and tissues were fixed in 10% formalin (both sides for paired organs), except for the eyes, testes, and sternums, which were first fixed in Davidson’s fixative before being transferred into 10% formalin after 48 hours. Organ samples were processed, embedded in paraffin, cut into thicknesses of approximately 5 µm, and stained with hematoxylin and eosin. Histopathological investigations on the organs and tissues of all animals were carried out by a veterinary pathologist, according to the test site’s standard operating procedures (CiToxLAB Scantox A/S). All pathological findings were entered directly into the Provantis 9.3 computer system. An internal peer review took place on selected slides.
Clinical Pathology
Blood samples for hematological, clotting, and clinical chemistry analyses were taken from animals at termination by heart puncture under isoflurane anesthesia. For hematological analyses, blood samples were collected into K2 EDTA tubes (ref. 813510; Vacutest Kima, Piove di Sacco, Italy) and analyzed with a VetScan HM5 hematology analyzer (Abaxis, Union City, California). The hematological parameters analyzed were white blood cells, red blood cells, hemoglobin, hematocrit, mean cell volume, mean cell hemoglobin, mean corpuscular hemoglobin concentration, red cell distribution width, platelets, plateletcrit, mean platelet volume, platelet distribution width, lymphocytes, monocytes, and neutrophils. Clotting time analysis for prothrombin time and activated partial thromboplastin time was performed on citrated blood samples (3.2% sodium citrate tubes, ref. 814074; Vacutest Kima) using an ACL 7000 coagulation analyzer (Instrumentation Laboratories, Bedford, Massachusetts). Blood samples from 5 animals were coagulated (2 control males, 1 female and 1 male from treatment groups); therefore, incomplete results were obtained from these animals. Blood samples for clinical chemistry were collected into lithium heparin tubes (ref. 41.1393.105; Sarstedt Micro, Nümbrecht, Germany) and analyzed using a VetScan VS2 analyzer (Abaxis). For clinical chemistry analyses, a comprehensive diagnostic profile and mammalian liver profile were performed to measure the following parameters: alkaline phosphatase, alanine aminotransferase, γ glutamyl transferase, amylase, albumin, total protein, total bilirubin, blood urea nitrogen, creatinine, total calcium, glucose, sodium, potassium, phosphate, globulins, bile acids, and total cholesterol.
Data and Statistical Analysis
Data were entered into a protected Excel file and sent as 100% quality control-checked data to the test site (CiToxLAB Scantox A/S). The following data were summarized and evaluated statistically: body weight and body weight gain, hematology, clinical chemistry, organ weights, organ weight/body weight ratios, mortality, morbidity, and clinical observations based on a summary incidence table. Data were processed to give group mean values and standard deviations where appropriate. For differences between 2 groups, each continuous variable was tested for normality by the Shapiro-Wilk method. When a normal distribution was present, intergroup differences were evaluated with a Student t test. Otherwise, they were assessed by a Wilcoxon rank-sum test. Clinical observations were not evaluated with a Fisher exact test as there were too few of them. For all tests, the level of significance (2-tailed) was defined as follows: *P < 0.05, **P < 0.01, ***P < 0.001. The statistical analyses were performed using SAS version 9.3 and SAS/STAT version 12.1 (SAS Institute Inc, Cary, North Carolina).
Results
Body Weight
The animals were weighed before the experiment, once a week during the experiment (for animals sacrificed on day 14) and at necropsy. The body weight gains of the animals from dosing day (study day 1) to necropsy are summarized in Table 2 as separate group mean values for males and females. Statistical analysis of animal body weights showed no significant differences between control and treatment group at the commencement of the study or at necropsy. Body weight gain between dosing day and necropsy did not differ significantly between any of the groups. No adverse effects were noted on the body weights of either males or females throughout the study.
Rat Body Weights (g) and Body Weight Gain (g) From Dosing Day (Study Day 1) to Necropsy.a,b
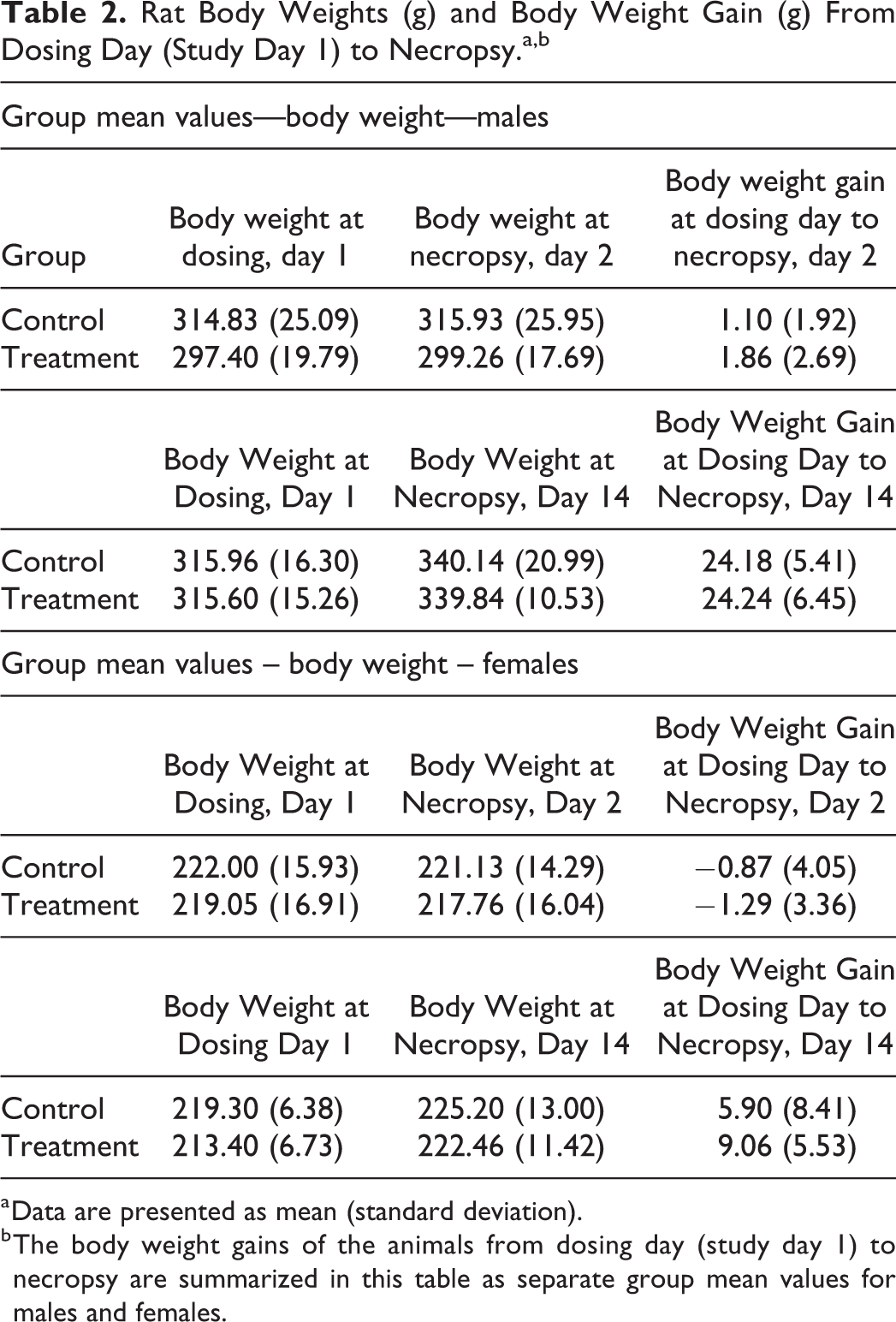
a Data are presented as mean (standard deviation).
b The body weight gains of the animals from dosing day (study day 1) to necropsy are summarized in this table as separate group mean values for males and females.
Clinical Signs, Mortality, and Morbidity
All clinical signs were recorded and no animals were found in a moribund state or showing noticeable toxic symptoms. No mortality was recorded during the course of the study. No abnormal ophthalmological findings were reported in any of the animals. Slight body weight losses and increased secretion of a red substance from the lacrimal and Harderian glands were observed in all groups during the study. Increased red porphyrin staining in rats (chromodacryorrhea) is associated with stress. 20 As these signs occurred in both the test groups and the control groups, they could possibly indicate a stress reaction related to handling. No other signs were recorded. A general summary of the clinical observations recorded (and the frequency of incidence) is presented in Table 3.
A General Summary of Clinical Observations Recorded (Frequency of Incidence) During the Study.a
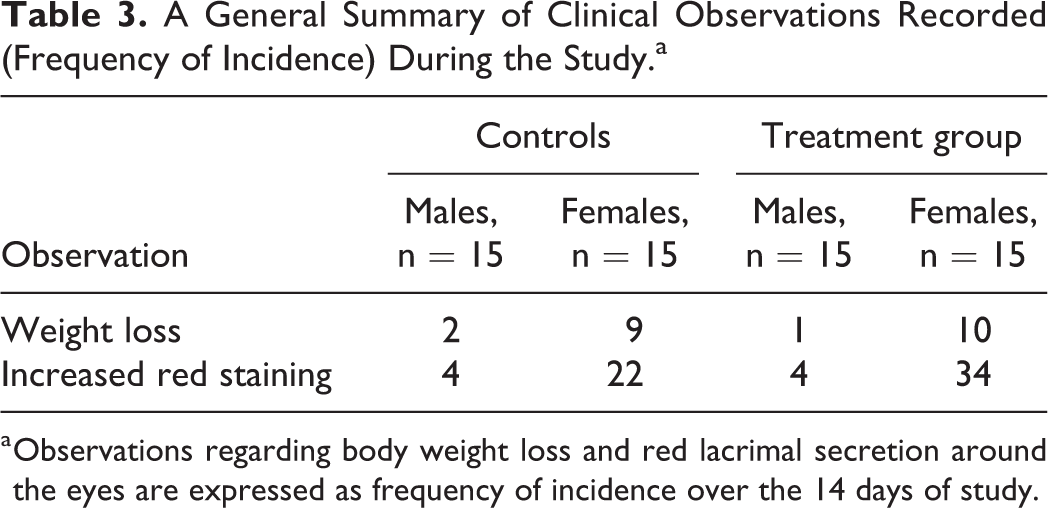
a Observations regarding body weight loss and red lacrimal secretion around the eyes are expressed as frequency of incidence over the 14 days of study.
Clinical Pathology
Interim treatment males showed a statistically significant increase in mean corpuscular hemoglobin concentration in comparison with interim control males (P < 0.01), and statistically significant decrease in hematocrit, red cell distribution width, platelets, plateletcrit (P < 0.05, vs controls), and mean cell volume (P < 0.01, vs controls). Terminal treatment females showed a statistically significant decrease in mean cell volume (P < 0.05, vs controls) in comparison with terminal control females. The clinical pathology results showing statistically significant differences are summarized in Table 4 as separate group mean values for males and females. Other relevant hematological parameters (hemoglobin, red blood cells, and white blood cells) showed no statistically significant differences between the groups.
Hematology and Clinical Pathology Results Showing Statistically Significant Differences Between Control and Treatment Groups.a
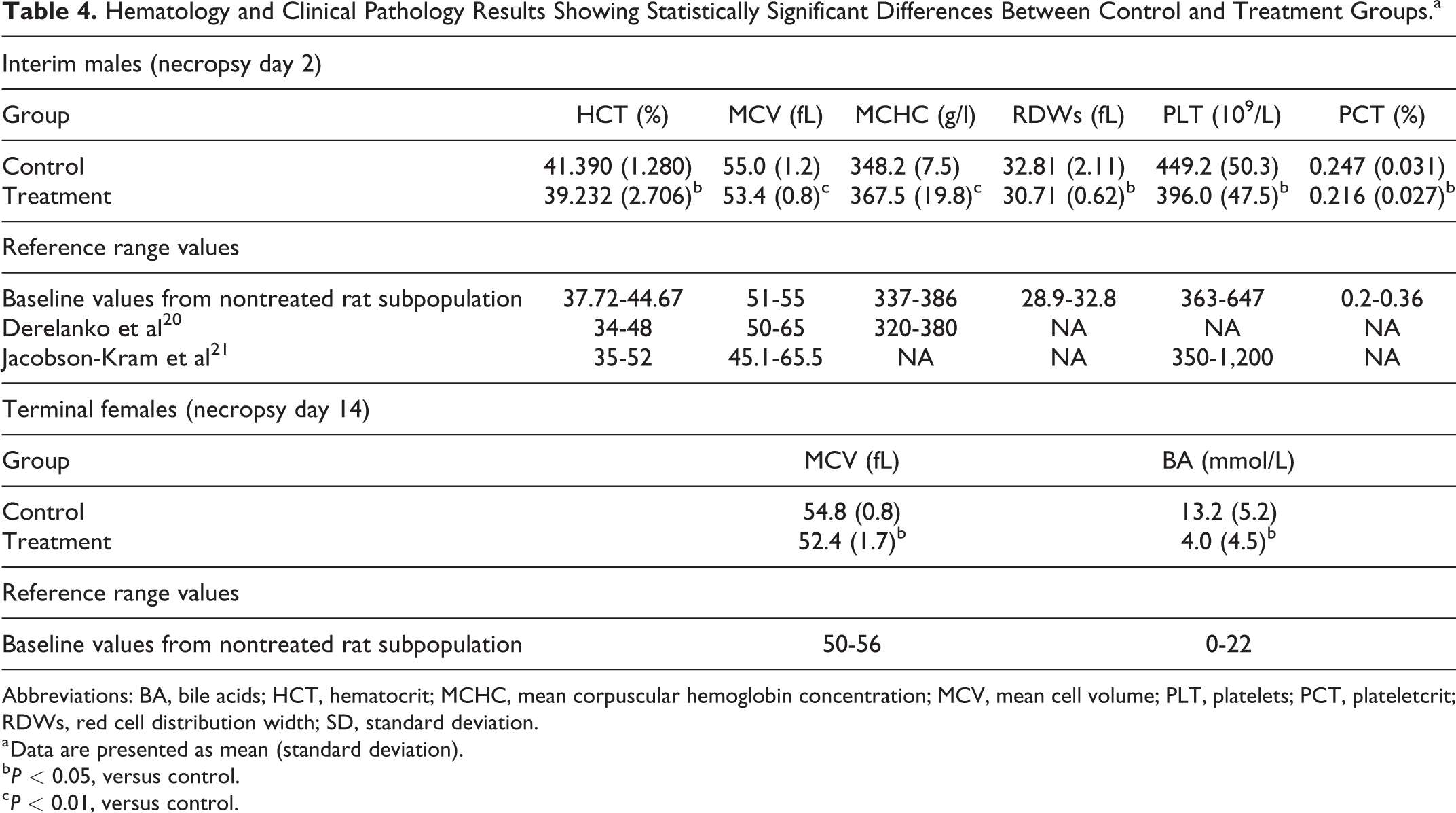
Abbreviations: BA, bile acids; HCT, hematocrit; MCHC, mean corpuscular hemoglobin concentration; MCV, mean cell volume; PLT, platelets; PCT, plateletcrit; RDWs, red cell distribution width; SD, standard deviation.
a Data are presented as mean (standard deviation).
b P < 0.05, versus control.
c P < 0.01, versus control.
The values for all these parameters are within the rat reference ranges described in the literature 20,21 and those for nontreated rat subpopulation maintained and sampled within the same study under the same experimental conditions to establish study baseline values. Reference range values are provided in Table 4. Therefore, the statistically significant between-group differences in hematological parameters are not considered of biological significance. 22
Terminal treatment females showed a statistically significant decreased level of bile acids in comparison with the control group (P < 0.05; Table 4), although the value remained within the reference ranges for a nontreated rat subpopulation. Furthermore, clinical chemistry data on the liver enzymes and histopathological evaluations did not indicate any liver or bile acid pathology
Organ Weights, Necropsy, and Histopathology Findings
A full necropsy was performed on each animal and macroscopic observations were recorded. No gross lesions were observed at injection site. Red spots were described in the thymus of one male from the treatment group, and small red subcutaneous spots were reported for one female in the treatment group. No correlating microscopic changes were reported. On the basis of the microscopic examination, these macroscopic findings were considered unremarkable.
The absolute weights of the liver and thymus were significantly lower in the interim treatment males than in the interim control males (P < 0.05), although the relative weights of these organs did not vary significantly between the groups. Statistically significant increased relative heart and testes weights were reported in the interim treatment males (P < 0.05, vs control), and statistically significant decreased relative liver weight was reported in the terminal treatment females (P < 0.05, vs control). The mean values of the absolute and relative organ weights remained within the reference ranges for nontreated rat subpopulations maintained and sampled under the same experimental conditions to establish study baseline values. The organ weights showing statistically significant differences are summarized in Table 5 as separate group mean values for males and females.
Organ Weights (mg) Showing Statistically Significant Differences Between Control and Treatment Groups.
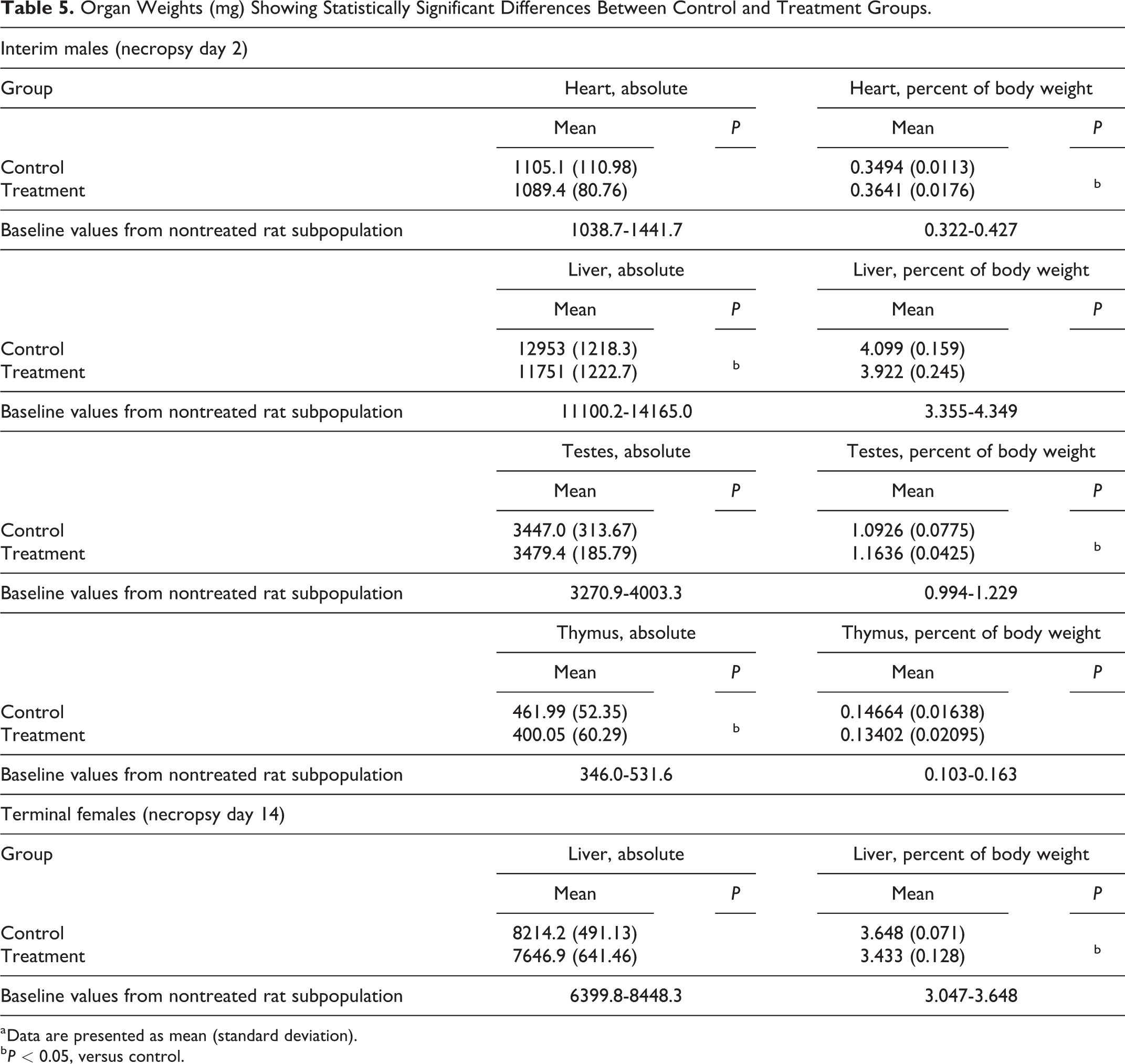
a Data are presented as mean (standard deviation).
b P < 0.05, versus control.
The absence of any abnormal findings in the clinical chemistry parameters and histopathological evaluations suggests that the statistically significant changes in organ weights are not related to the test item.
Macroscopic findings at necropsy were considered unremarkable, and no test item-related macroscopic or microscopic changes were reported in this study.
Discussion
The glucose analog 2-deoxy-2-[18F]fluoroglucose ([18F]FDG) is one of the most frequently used radiopharmaceuticals in clinical PET. Taking [18F]FDG as an example, the injected mass dose is 7 ng/kg when it is used at 4 MBq/kg of body weight at a molar activity of 100 MBq/nmol. This illustrates that PET radiopharmaceuticals are used in microdoses, which are generally considered to be <100 µg in the cases of small molecule drugs, or ≤30 nmol in the case of large molecules (eg, proteins). 23 Currently, several peptide-based 68Ga-labeled radiopharmaceuticals are routinely produced for clinical PET and in most cases the quantities of peptides are kept below 20 nmol per batch. For the production of [68Ga]Ga-DOTA-Siglec-9, we used 40 µg (16.5 nmol) of DOTA-Siglec-9 per batch to achieve a reasonably high radiolabeling efficiency and molar activity. 5 In practice, 40 µg DOTA-Siglec-9 will be used in the production of one batch, and an aliquot from one produced batch will be injected into a patient; thus, the final dose would be well below 40 µg. To leave a broad safety margin for clinical applications, we chose 40 µg as the assumed clinical dose and a 1,000-fold safety margin as the study dose in rats. This evaluation setting fulfills the requirements described in the relevant guidelines and position papers, including ICH guideline M3 (R2) on nonclinical safety studies for the conduct of human clinical trials for pharmaceuticals (EMA/CPMP/ICH/286/1995) – paragraph 7, and the Position paper on nonclinical safety studies to support clinical trials with a single microdose (CPMP/SWP/2599/02) – paragraph 4.1.
Accordingly, the solution of GMP grade DOTA-Siglec-9 compound was freshly prepared in saline and iv administered into rats. Saline was chosen as the vehicle as it will be used as the vehicle for the clinical dosing. No animals in a moribund state or having major toxic symptoms were found. Slight body weight losses and increased secretion of a red substance were observed in all groups during the study, but these signs are commonly observed in toxicological studies under stress conditions. More frequent observations in females suggest they are more sensitive to stress under conditions of acute toxicity tests. 24 As these signs occurred in both test and control groups, they probably indicate the stress reaction related to handling. No mortality or morbidity and no significant clinical symptoms were observed during the study. No test item-related effects were observed in clinical pathology, body weights, or organ weights. Statistically significant decreased level of bile acids showed in terminal treatment females might be related to the decreased relative liver weight reported in a comparison with the control group (P < 0.05, vs control). Clinical chemistry data on the liver enzymes and histopathological evaluations did not indicate any liver or bile acid pathology.
Histopathological evaluation did not reveal any abnormality related to the test item. In conclusion, iv administration of DOTA-Siglec-9 solution at a dose of 582 µg/kg was well tolerated in rats and caused no adverse clinical or anatomic pathology findings.
Footnotes
Acknowledgments
Nina Juoperi, Maija Liisa Hoffren, and Nina Kulmala are thanked for providing excellent technical assistance.
Author Contributions
P.C., E.Y., X.-G.L., U.-M.J., and A.R. conceived and designed the experiments. P.C. and E.Y. performed the experiments. P.C. and E.Y. analyzed the data. P.C., E.Y., X.-G.L., U.-M.J., J.K., S.J., and A.R. interpreted the data. P.C., E.Y., X.-G.L., and A.R. drafted the manuscript. E.Y., X.-G.L., U.-M.J., J.K., S.J., and A.R. critically revised the manuscript for important intellectual content. All authors read and approved the final the manuscript.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Sirpa Jalkanen is a shareholder in Faron Ventures Ltd. All other authors declare that they have no competing interests.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was financially supported by a grant from Business Finland (number 632/31/15TY).