
Abstract
Fine particulate matter (PM2.5) and sulfur dioxide (SO2) are 2 common air pollutants, but their toxicological effects of coexposure are still not fully clear. In this study, SO2 exposure (5.6 mg/m3) couldn’t cause obvious inflammatory responses in rat lungs. The PM2.5 exposure (1.5 mg/kg body weight) increased inflammatory cell counts in bronchoalveolar lavage fluid (BALF) and some inflammation damage. Importantly, SO2 and PM2.5 (1.5, 6.0, and 24.0 mg/kg) coexposure induced pathological and ultrastructural damage and raised inflammatory cells in BALF compared with the control. Also, they significantly elevated the levels of pro-inflammatory cytokines, adhesion molecule, and nitric oxide (NO) and promoted the gene expression of nuclear factor kappa B (NF-κB), phosphorylated p38 (p-p38), and Toll-like receptor 4 (TLR4) in rat lungs treated with higher dose of PM2.5 (6.0 and 24.0 mg/kg) plus SO2 relative to the control or SO2 group, along with the decreased inhibitor of NF-κBα and increased inhibitor of NF-κB kinase β expressions. The changes in the inflammatory markers in the presence of PM2.5 plus SO2 were not significant compared with the PM2.5 group. The results indicated that inflammatory injury and pathological and ultrastructural damage in rat lungs exposed to PM2.5 plus SO2 were involved in TLR4/p38/NF-κB pathway activation accompanied by oversecretion of pro-inflammatory cytokine, adhesion molecule, and NO. It provides more useful evidence to understand the possible toxicological mechanism that PM2.5 and SO2 copollution exacerbate lung disease.
Introduction
Fine particulate matter (PM2.5) and sulfur dioxide (SO2) are typical air pollutants mainly coexisting in coal-smoke air pollution. Epidemiological investigations have shown that short-term exposure to PM2.5 or SO2 is linked to adverse respiratory health effects, including decreased lung function and increased morbidity and mortality of respiratory diseases, such as asthma and chronic obstructive pulmonary disease (COPD), and even is related to the lung cancer. 1 -4 Many environmental studies indicated that PM2.5 or SO2 exposure alone could increase oxidative stress, inflammation, and DNA damage in lungs or lung epithelial cells, 5 -9 which contribute to clarify the pathogenesis of lung diseases induced by PM2.5 or SO2 pollution. Although most researches focused on the health risk assessment and toxicological effects of single air pollutant, there are still few studies to evaluate the potentially synergistic toxicity and possible mechanisms exposure to multiple pollutants, which should be a vital problem in estimating the joint toxicity of simultaneously coexisting air pollutants including PM2.5 and SO2. Notably, SO2 is a gas that can be transformed into sulfates in the air and carried by PM2.5, increasing the PM2.5 pollution levels. Recently, some epidemiological investigations reveal that PM2.5 and SO2 among air pollutants have more significant influences on daily admissions for respiratory diseases, 4,10 and other toxicological studies suggest that coexposure of PM10 and SO2 may induce the synergistic cytotoxicity in lung epithelial cells and coexposure of SO2, nitrogen dioxide, and PM2.5 may cause inflammatory response and endothelial dysfunction in the hearts of mice. 11,12 However, few toxicology studies of coexistence of PM2.5 and SO2 on lungs have been established so far.
Lung inflammation often occurs in the event of exposure to environmental toxins/irritants, allergens, or pathogens. During inflammation, numerous types of inflammatory cells are activated, and the levels of inflammatory mediators, for example, tumor necrosis factor α (TNF-α), interleukin 6 (IL)-6, IL-1β, and intercellular adhesion molecule 1 (ICAM-1), are increased. Orchestration of these cells and molecules leads to aggravate inflammation. 13,14 Various inflammatory stimuli may elevate nitric oxide (NO) levels, leading to oxidative toxicity. Also, the secretions of inflammatory mediators are regulated by endogenous or exogenous substances through special signal pathways, such as nuclear factor kappa B (NF-κB) and mitogen-activated protein kinase (MAPK) mainly including extracellular signal-regulated kinase (ERK)1/2, p38 kinase, c-Jun N-terminal kinase (JNK), and Toll-like receptor 4 (TLR4). 13,15 -17 Because inflammation is an important mechanism of external stimuli on lung injury and only limited data are available on the molecular mechanisms of coexposure of both PM2.5 and SO2 on inflammatory responses in lungs, elucidating that the inflammatory damage and molecular mechanisms involved in TLR4/p38/NF-κB expressions in the presence of PM2.5 plus SO2 will be useful for understanding the interaction mechanisms of the 2 air pollutants-induced toxicological effects in the lung. Hence, in this study, the pathological injuries and inflammatory responses of PM2.5 and SO2, singly and in binary mixtures, in rat lungs were investigated, followed by clarifying the possible mechanisms for their interaction by detecting the changes of key factors in the inflammation pathway involved in TLR4/p38/NF-κB/pro-inflammatory cytokines/adhesion molecule/NO.
Materials and Methods
Sampling and Preparation of Particle Samples
Particulate matter (PM2.5) sampled with a particle high-volume air sampler (Thermo Anderson, Smyrna, Georgia, USA) was collected onto quartz fiber filters from January 2014 in Taiyuan, China. The specific sampling method and preparation of PM2.5 suspensions were explained in our previous report.
18
During January 2014 in Taiyuan, China, the 24-hour mean mass concentration of PM2.5 was 69.7 μg/m3, ranging from 30 to 158 μg/m3, in which the concentrations of 35.7% samples were higher than the National Air Ambient Quality Standard (NAAQS) of China for PM2.5 (75 μg/m3). Also, the 24-hour mean mass concentration of SO2 was 0.138 mg/m3, ranging from 0.037 to 0.284 mg/m3, in which the concentrations of 42.9% samples were higher than the NAAQS of China for SO2 (0.15 mg/m3).
19
Polycyclic aromatic hydrocarbons (PAHs) in PM2.5 were detected, and the daily mean levels of benzo[a]anthracene, chrysene, benzo[b]fluoranthene, benzo[a]pyrene, benzo[g, h, i]-perylene, and benzo[k]fluoranthene were obviously higher than those of the Chinese national standards of PAHs (10 ng/m3). And the
Animal and Treatment Protocols
Healthy adult and clean grade male Wistar rats, weighing 180 to 200 g, were purchased from Animal Center of Hebei Medical University. Animals were housed in metallic cages under standard conditions (24°C ± 2°C and 50% ± 5% humidity) with a 12-hour light–dark cycle. Rats were divided randomly into 5 equal groups of 5 animals each: (1) the control (saline), (2) SO2 inhalation, (3) 1.5 mg/kg body weight (bw) PM2.5, (4) 1.5 mg/kg PM2.5 plus SO2, (5) 6.0 mg/kg PM2.5 plus SO2, and (6) 24.0 mg/kg PM2.5 plus SO2. The exposure protocol and main doses of PM2.5 and SO2 were based on our previous studies. 9,21 The PM2.5 suspensions were respectively given to rats once by the intratracheal instillation at days 1, 3, 5, 7, and 9. During PM2.5 instillation, the rats were exposed to SO2 alone or to PM2.5 plus inhaled SO2 using the technique of SO2 dynamic inhalation for 6 h/d. 22 The exposure concentration of SO2 was detected by using pararosaniline hydrochloride spectrophotometry, and the mean concentration was 5.6 ±1.2 mg/m3, which was perhaps close to doses found in most industrial environments. 23 After PM2.5 instillation, the rats in PM2.5 plus SO2 groups were exposed to SO2. Correspondingly, the control rats were treated with physiological saline.
Inflammatory Cell Counts and Hematoxylin–Eosin Staining
The rats were euthanized by sodium pentobarbital (80 mg/kg, intraperitoneally) 24 hours after the last treatment. According to the methods used by our laboratory, 9 the bronchoalveolar lavage fluids (BALFs) were extracted to analyze the changes in counts of total cells. The inflammatory cells were counted with Wright-Giemsa staining, and the histopathologic examination was performed using hematoxylin–eosin (HE) staining and Transmission Electron Microscope (TEM) technology.
Real-Time Quantitative Reverse Transcription-Polymerase Chain Reaction
Messenger RNA (mRNA) extraction, first-strand complementary DNA (cDNA), and polymerase chain reaction (PCR) amplification of TNF-α, IL-6, IL-1β, ICAM-1, inducible nitric oxide synthase (iNOS), NF-κB, inhibitor of NF-κBα (IκBα), inhibitor of NF-κB kinase β (IKKβ), TLR4, p38, and GAPDH were performed as described previously. 9,22 Toll-like receptor 4 (NM_019178) forward primer was 5′-TGCTCAGACATGGCAGTTTC-3′, while reverse primer was 5′-TCAAGGCTTTTCCATCCAAC-3′. Toll-like receptor 4 PCR product amplified fragments was 206 base pairs (bp). The p38 (NM_031020) forward primer was 5′-AAGATGCTGGTTTTGGACTCG-3′, while reverse primer was 5′-GTCAGGCTCTTCCATTCGTCT-3′. The p38 PCR product amplified fragments was 164 bp. The GenBank accession numbers and the primer sequences of other genes together with the PCR product amplified fragments are referenced in the literature. 9,22 The relative quantification of the expression of the target genes was measured using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA as an internal control.
Western Blotting
Total proteins for ICAM-1, p38, phosphorylated p38 (p-p38), TLR4, and β-actin were extracted from frozen lung tissue with protein extraction kit (Beyotime, Shanghai, China), and NF-κB nuclear and cytoplasmic protein were extracted from fresh lung tissue with a nuclear and cytoplasmic protein extraction kit (Beyotime), according to the manufacturer’s instructions. Protein concentrations were determined by Bicinchoninic acid (BCA) protein assay kit (Beyotime). Samples were mixed with loading buffer and boiled for 5 minutes. The rabbit polyclonal primary antibodies for rat NF-κB, p38, p-p38, and β-actin (Santa Cruz, California) and goat polyclonal primary antibodies for rat ICAM-1 (Santa Cruz) were incubated overnight at 4°C, while the infrared-labeled anti-rabbit and anti-goat secondary antibody (Li-COR Biosciences, Lincoln, Nebraska, USA) at a concentration of 1:5,000 were added to membranes and incubated for 1.5 hours at room temperature. The membranes were scanned and the band densities were quantified using the Odyssey Infrared Imaging System (Li-COR Biosciences, Lincoln, Nebraska, USA).
Enzyme-Linked Immunosorbent Assay
The lung tissue was weighed and homogenized in ice-cold 0.9% physiological saline. After the homogenized solutions were centrifuged for 10 minutes at 3,000 rpm, the lung supernatants were collected. Levels of TNF-α, IL-6, IL-1β, IκBα, and IKKβ in supernatants of different groups were measured using commercially available enzyme-linked immunosorbent assay kits (R&D Systems, Minneapolis, MN, USA) according to the test protocols.
Measurement of NO and iNOS Levels
Measurements of the NO content and iNOS enzymatic activity in lung supernatants were performed spectrophotometrically using the corresponding kits (Jiancheng Biochemistry, Nanjing, China), according to the manufacturer’s protocols.
Statistical Analysis
Statistical analyses were performed by the use of 1-way analysis of variance using the SPSS 19.0 package of programs for Windows. Post hoc tests were conducted to determine the difference between groups, followed by least significant difference test. A level of P < 0.05 was accepted as statistically significant.
Results
Histopathological Observation
Representative HE staining images are shown in Figure 1A to F. No histopathological abnormalities were observed in control (Figure 1A) and SO2 group animals (Figure 1B). In the PM2.5 (1.5 mg/kg bw) group, the inflammatory cells existed in the lungs (Figure 1C). In the SO2 plus PM2.5 (1.5 mg/kg bw) group, the inflammatory cell infiltration, thickened alveolar walls, diminished alveolar spaces, hyperemia, and bronchial epithelial hyperplasia were found in the lungs (Figure 1D). Exposure to SO2 plus PM2.5 (6.0 and 24.0 mg/kg bw) induced more pathological changes compared to that of other treatment groups, accompanied by the pulmonary fibrosis damage (Figure 1E, F). It suggested that PM2.5 plus SO2 induced rat lung pathological damage and inflammatory responses.
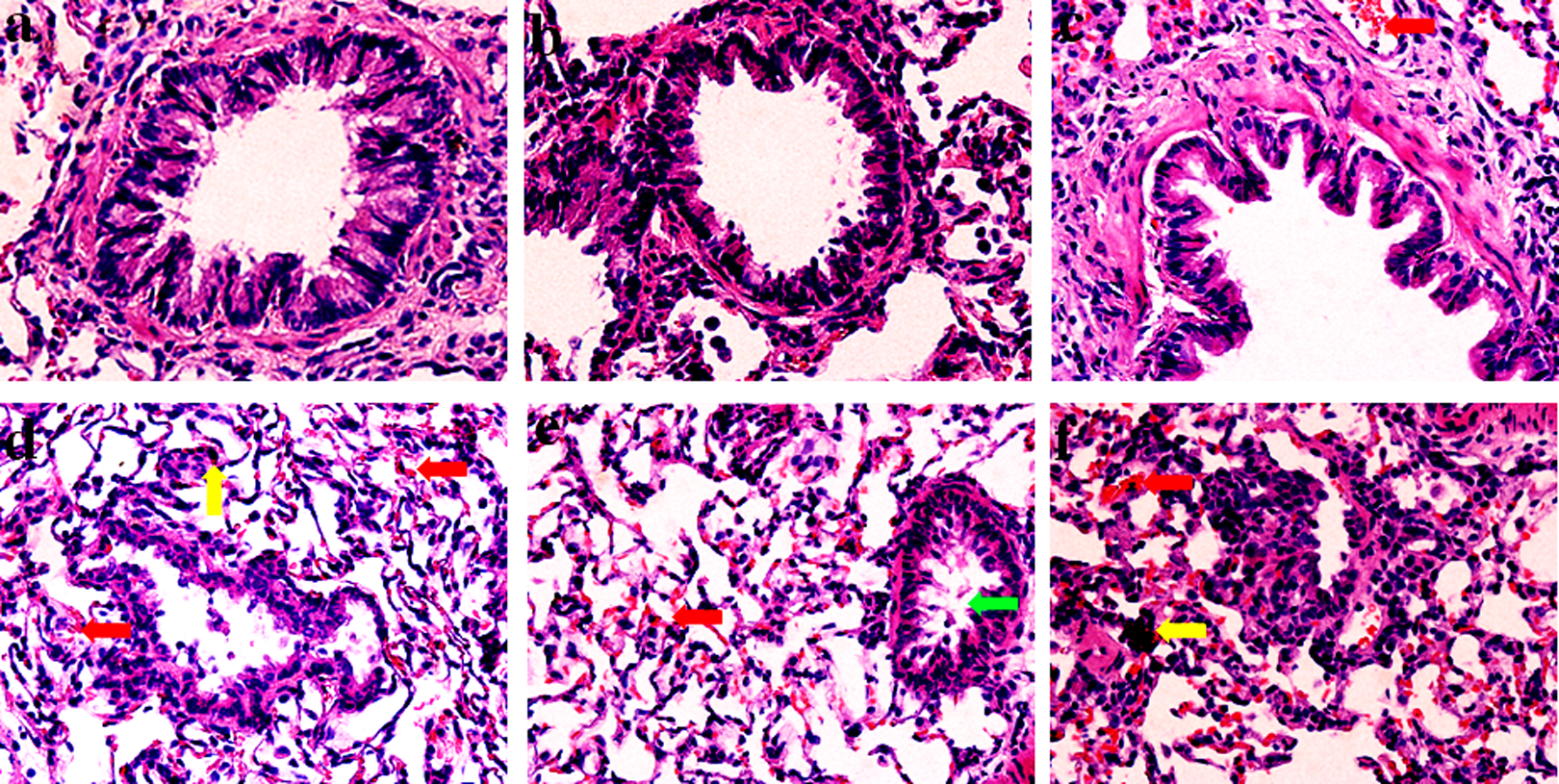
The morphological characteristics in the lungs of rats from (A) control, (B) SO2, (C) PM2.5 (1.5 mg/kg bw), (D) SO2 plus PM2.5 (1.5 mg/kg bw), (E) SO2 plus PM2.5 (6.0 mg/kg bw), and (F) SO2 plus PM2.5 (24.0 mg/kg bw). Magnification ×400. The control group was instilled with same amount of physiological saline. The red arrows indicate sites of hyperemia, the yellow arrow indicates site of inflammatory cell infiltration, and the green arrow indicates site of bronchial stenosis and mucus secretion, respectively. bw indicates body weight; PM, particulate matter; SO2, sulfur dioxide.
TEM Pathological Characters of Lung Tissue
The ultrastructure images of rat lung in different groups are shown in Figure 2. Normal lung epithelial cells (type I alveolar cell, type II alveolar cell) and tissue matrix of lung interval were observed in rats of the control group (Figure 2A). The epithelial cell morphology in single SO2-exposed animals was almost identical with the control group (Figure 2B). In PM2.5 (1.5 mg/kg bw) group, some alveolar type II epithelial (AT II) cells had obvious pathological changes, including vacuolation of osmiophilic multilamellar bodies and mitochondrial swelling (Figure 2C). The ultrastructure damage was found more obviously with increased dosage of PM2.5 in the SO2 plus PM2.5 groups. In the SO2 plus PM2.5 (1.5 mg/kg bw) group, the vacuolation of osmiophilic multilamellar bodies, mitochondrial swelling, disorder of cristae, mitochondrial vacuolation, some lysosomes, and irregularly compacted chromatin appeared in most of the AT II cells (Figure 2D). More severe damage was observed in rats exposed to SO2 plus PM2.5 (6.0 and 24.0 mg/kg bw) compared with that in the SO2 plus PM2.5 (1.5 mg/kg bw) group, along with mitochondrial membrane break, lipid droplets in the cytoplasm, and microvilli falling off in AT II cells (Figure 2E, F).
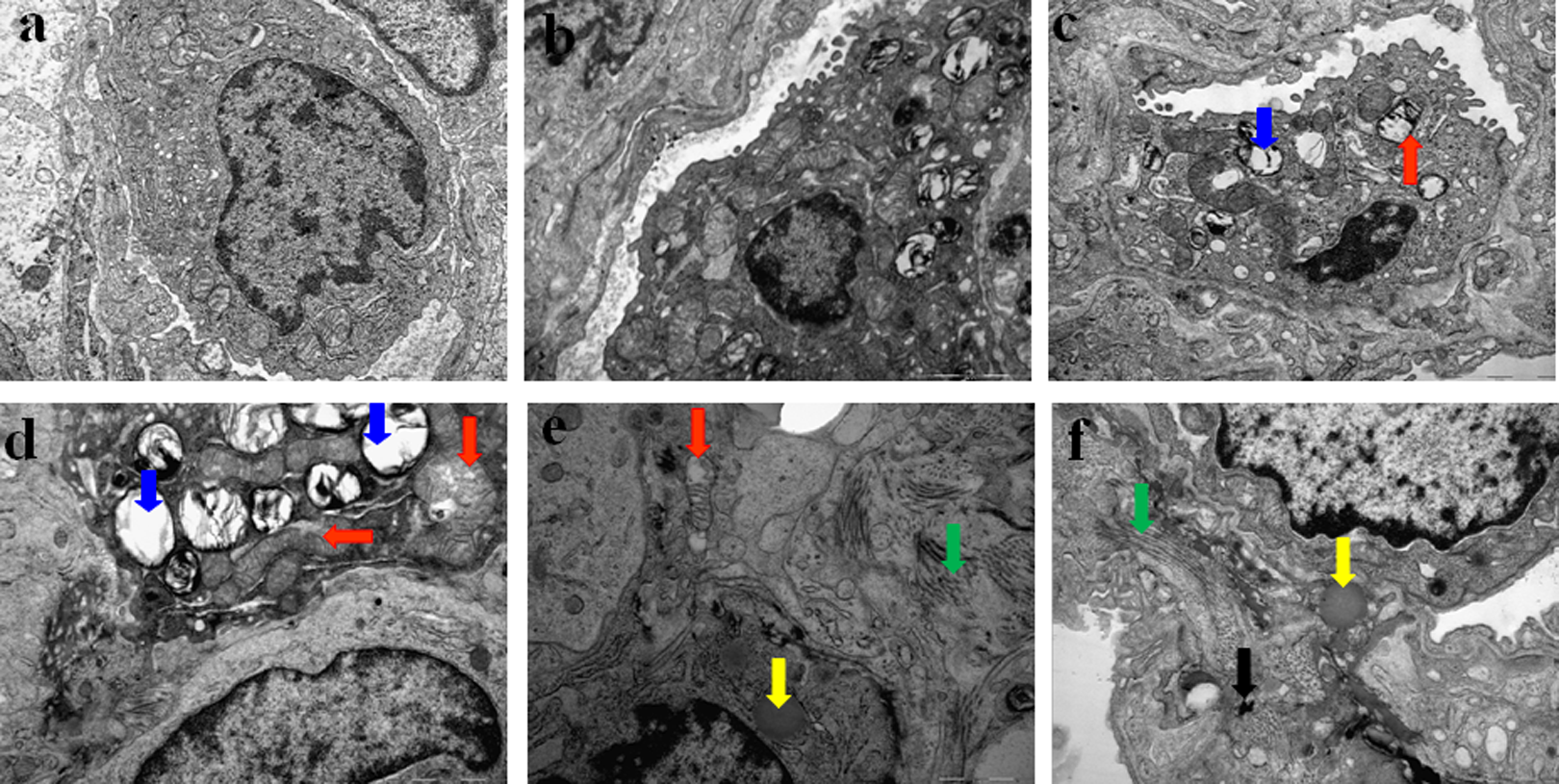
The TEM images of lung tissue. A, Control, (B) SO2, (C) PM2.5 (1.5 mg/kg bw), (D) SO2 plus PM2.5 (1.5 mg/kg bw), (E) SO2 plus PM2.5 (6.0 mg/kg bw), and (F) SO2 plus PM2.5 (24.0 mg/kg bw). Magnification ×20,000. The red arrows indicate sites of mitochondrial swelling and cristae disorder, the blue arrow indicates site of lamellar body vacuolization, the yellow arrow indicates site of lipid droplet, the green arrow indicates site of cell fibrosis, and the black arrow indicates site of particle. Alveolar type I epithelial cell (AT I) is a complex branched cell with multiple cytoplasmic plates that represent the gas-exchange surface in the alveolus, whereas alveolar type II epithelial cell (AT II) is cuboidal in shape and contains many organelles (such as mitochondria, lamellar bodies and lysosomes) that perform important biological functions. bw indicates body weight; PM, particulate matter; SO2, sulfur dioxide.
Inflammatory Cell Counts in BALF
In Table 1, the changes in the number of total cells and inflammatory cells in the SO2 group were not significantly relative to the control, except for the increase in neutrophils (NEUs) number. PM2.5 increased the numbers of total cells, alveolar macrophages, and NEUs in BALF. Further, exposure to PM2.5 at the doses of 1.5, 6.0, and 24.0 mg/kg bw plus SO2 was significantly increased compared with the control or SO2 group (P < 0.05 or <0.01), and the counts of lymphocytes were significantly increased only in the 24.0 mg/kg bw group plus SO2 (P < 0.01) versus the control or SO2 group (P < 0.01). The results revealed that SO2 plus 1.5, 6.0, and 24.0 mg/kg PM2.5 induced a significant increase in total cells and inflammatory cells in a dose-dependent manner (R 2 = 0.66-0.99). It suggested that PM2.5 alone and PM2.5 plus SO2 raised the inflammatory cell levels.
The Numbers of Total Cells and Different Inflammatory Cells in BALF of Rats Treated with SO2 and/or Different Doses of PM2.5.*
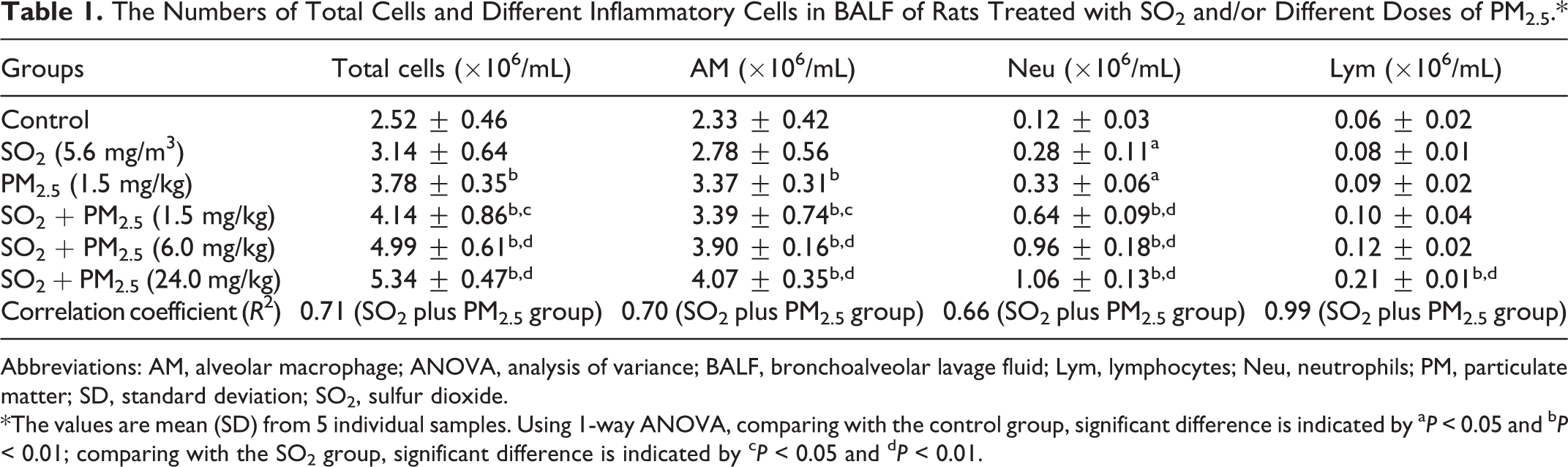
Abbreviations: AM, alveolar macrophage; ANOVA, analysis of variance; BALF, bronchoalveolar lavage fluid; Lym, lymphocytes; Neu, neutrophils; PM, particulate matter; SD, standard deviation; SO2, sulfur dioxide.
*The values are mean (SD) from 5 individual samples. Using 1-way ANOVA, comparing with the control group, significant difference is indicated by a P < 0.05 and b P < 0.01; comparing with the SO2 group, significant difference is indicated by c P < 0.05 and d P < 0.01.
Effects of PM2.5 on Expression of Inflammatory Markers in Lungs of Rats
Figure 3A, B displays the gene expression of 3 pro-inflammatory cytokines (TNF-α, IL-1β, IL-6) and adhesion factor ICAM-1 in the lungs of different groups of rats. No significant changes in the expression of genes were observed in the rats exposed to SO2 alone compared with the control. PM2.5 could markedly increase TNF-α and IL-6 mRNA expression compared with the control. Besides, the mRNA and protein levels of TNF-α, IL-1β, IL-6, and ICAM-1 showed an increase in a dose-dependent manner (R 2 = 0.60-0.99), and such increases were significantly exposed to the higher dose exposure to PM2.5 (6.0 and 24.0 mg/kg bw) plus SO2 compared to the control (P < 0.05 or <0.01). Also, TNF-α, IL-6, and IL-1β mRNA expression and IL-6 protein levels were significantly increased at the 1.5 mg/kg bw PM2.5 plus SO2 group compared with the control (P < 0.05 or <0.01). In SO2 plus PM2.5 (6.0 and 24.0 mg/kg bw) groups, TNF-α, IL-1β, IL-6, and ICAM-1 gene expressions were significantly relative to the SO2 group (P < 0.05 or <0.01), whereas they were not significantly relative to the PM2.5 group.
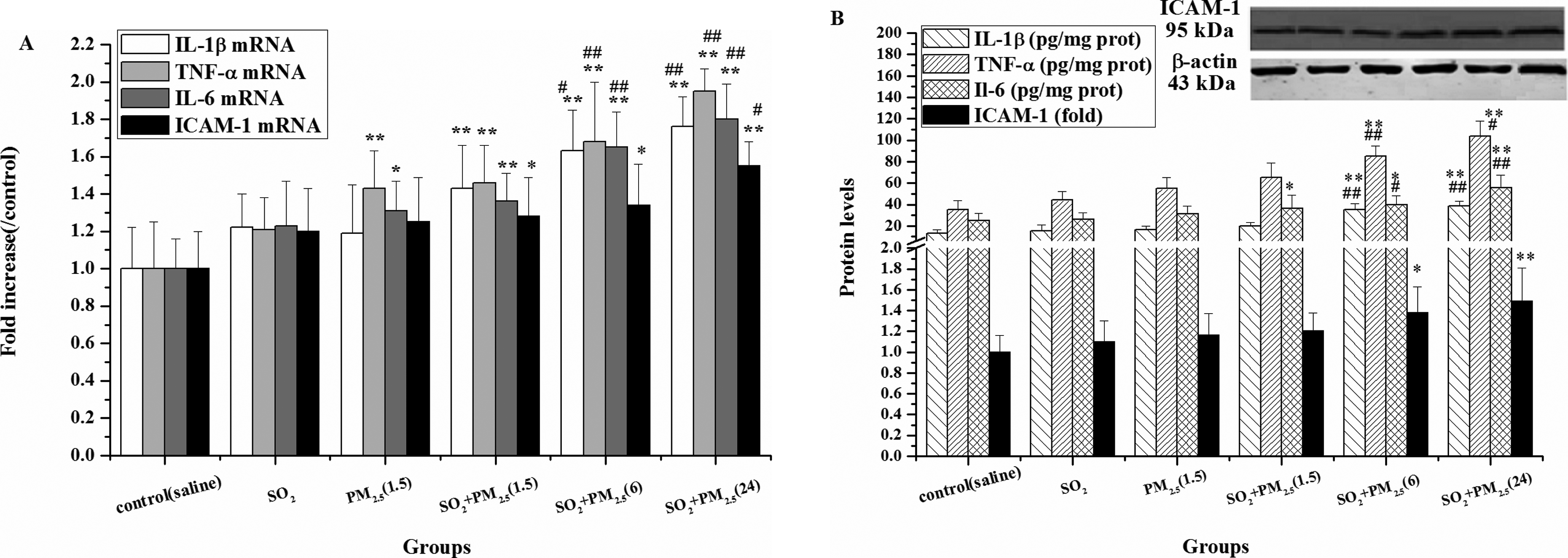
Expression of mRNA of TNF-α, IL-1β, IL-6, and ICAM-1 (A) and protein levels (B) in rat lungs in different groups. Mean expression of mRNA expression in each treated group is shown as increase compared to mean expression in the control group, which has been ascribed an arbitrary value of 1. The values are mean (SD) from 5 individual samples. Using 1-way ANOVA, comparing with the control group, significant difference is indicated by *P < 0.05 and **P < 0.01; comparing with the SO2 group, significant difference is indicated by # P < 0.05 and ## P < 0.01. ANOVA indicates analysis of variance; ICAM-1, intercellular adhesion molecule 1; IL-6, interleukin-6; IL-1β, interleukin-1β; mRNA, messenger RNA; SD, standard deviation; TNF-α, tumor necrosis factor-α.
Effects of PM2.5 on Levels of NO/iNOS in Lungs of Rats
As shown in Figure 4, no significant change in the levels of NO and iNOS in the SO2 group or PM2.5 group was found compared with the control. PM2.5 plus SO2 significantly elevated the NO content and iNOS mRNA and protein levels compared with the control (P < 0.05 or <0.01). The iNOS expression was significant in 3 SO2 plus PM2.5 groups, while NO content was increased in a dose-dependent manner (R 2 = 0.99), with a significant difference only in higher doses of PM2.5 (6.0 and 24.0 mg/kg bw) plus SO2 relative to the SO2 group (P < 0.05 or <0.01). The changes in inflammatory markers in the presence of PM2.5 plus SO2 were not significant compared with the PM2.5 group.
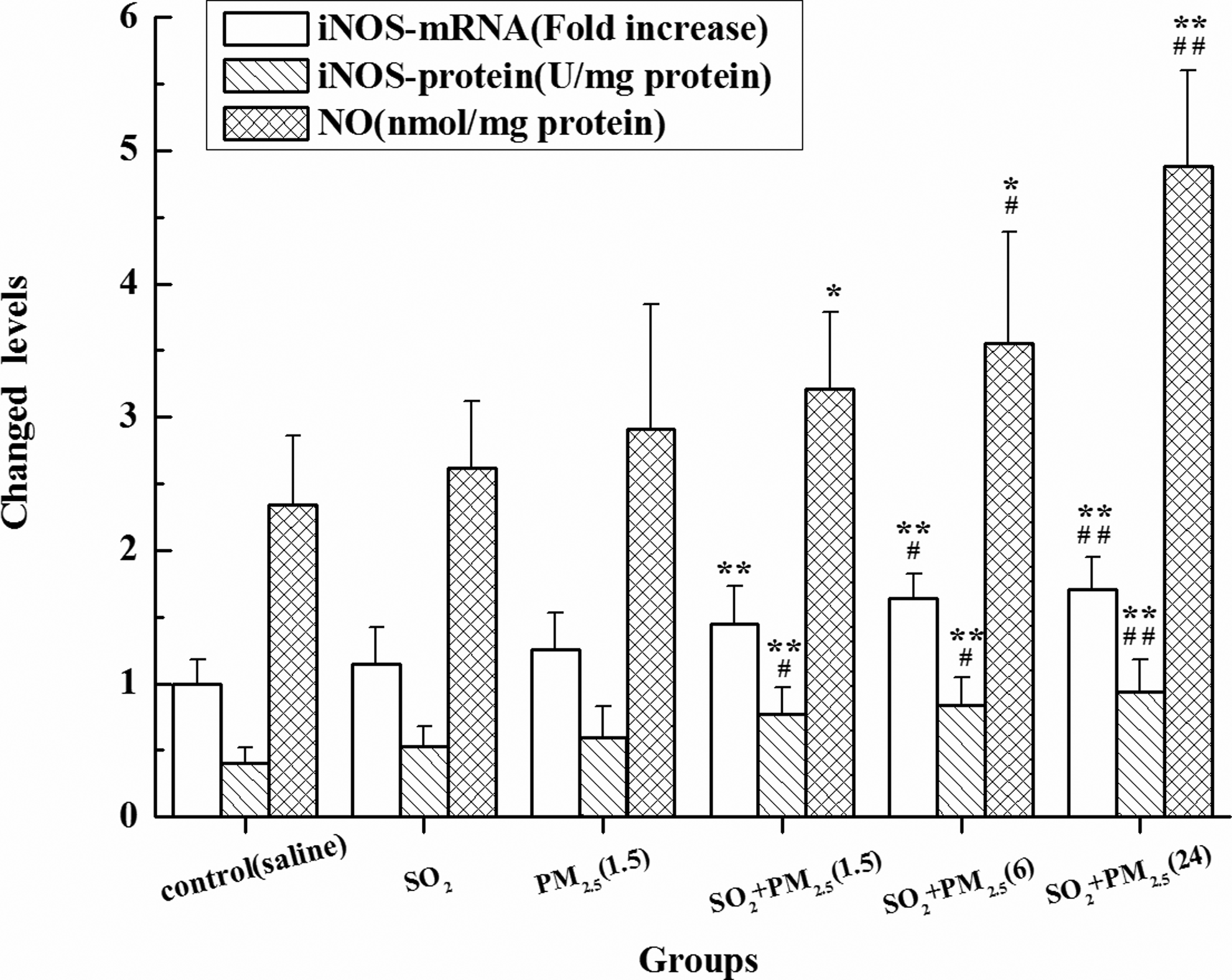
Nitric oxide content and iNOS mRNA and protein expression in rat lungs in different groups. Mean expression of mRNA expression in each treated group is shown as increase compared to mean expression in control group, which has been ascribed an arbitrary value of 1. The values are mean (SD) from 5 individual samples. Using 1-way ANOVA, comparing with the control group, significant difference is indicated by *P < 0.05 and **P < 0.01; comparing with the SO2 group, significant difference is indicated by # P < 0.05 and ## P < 0.01. ANOVA indicates analysis of variance; iNOS, inducible nitric oxide synthase; mRNA, messenger RNA; SD, standard deviation; SO2, sulfur dioxide.
Effects of SO2 Plus PM2.5 Exposure on NF-κB, IκBα, and IKKβ mRNA and Protein Expressions in the Lungs
As shown in Figure 5A, B cytoplasm NF-κB protein levels in the PM2.5 plus SO2 groups and NF-κB, IκBα, and IKKβ mRNA and protein expression in the SO2 group were not statistically significant compared with the control. PM2.5 (1.5 mg/kg) could activate NF-κB relative to the control. Exposure to PM2.5 plus SO2 obviously raised the levels of NF-κB nucleus protein and IKKβ mRNA and protein in lungs showing a concentration-dependent relationship (R 2 = 0.92-0.99), while significantly caused decrease in IκBα mRNA and protein levels compared with the control group (P < 0.05 or <0.01) in a dose-dependent manner (R 2 = 0.64 and 0.68). Meanwhile, NF-κB mRNA in the PM2.5 plus SO2 groups was increased, and mRNA and protein of IKKβ and IKKβ, and NF-κB nucleus protein levels in PM2.5 (6.0 and 24.0 mg/kg bw) plus SO2 were elevated compared with the SO2 group (P < 0.05 or <0.01). The changes in NF-κB, IκBα, and IKKβ mRNA and protein expressions in the presence of PM2.5 plus SO2 were not significant compared with the PM2.5 group.
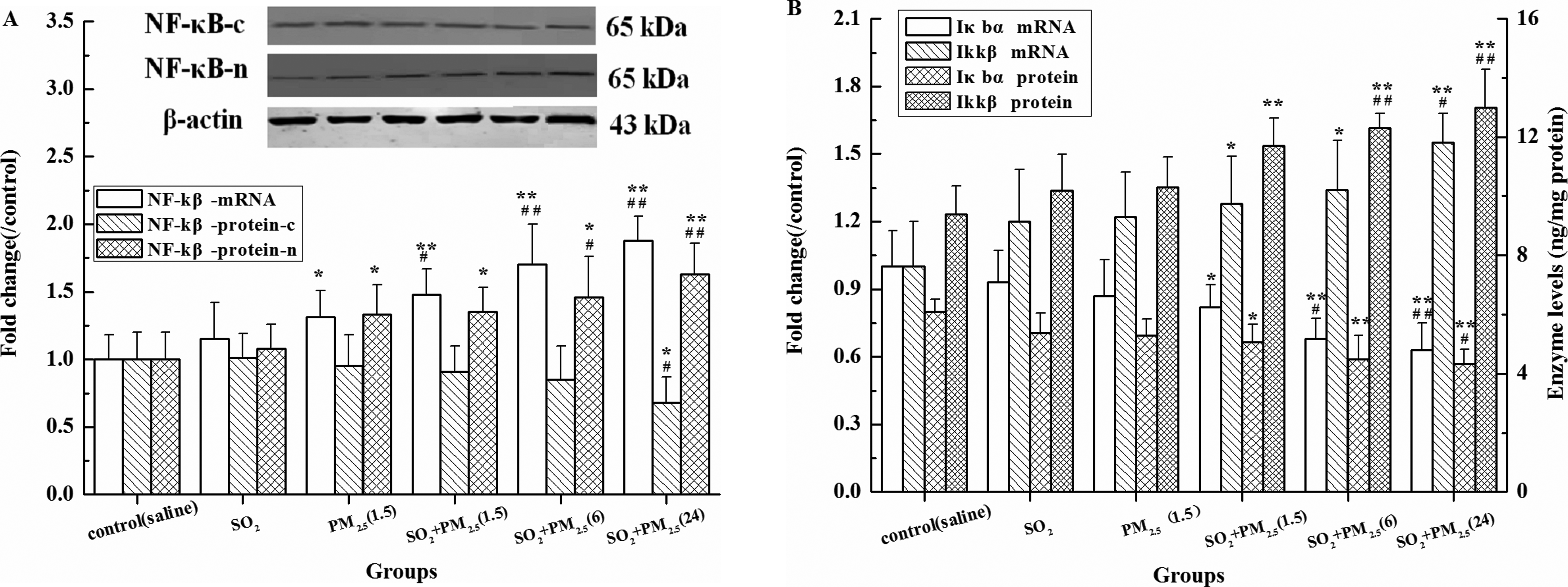
Expressions of mRNA and protein of NF-κB, IκBα, and IKKβ in lungs of rats in different groups. A, NF-κB, B, IκBα and IKKβ. Mean expression of mRNA and NF-κB protein in each treated group is shown as increase compared to mean expression in the control group, which has been ascribed an arbitrary value of 1. The values are means (SD) from 5 individual samples. Using 1-way ANOVA, comparing with the control group, significant difference is indicated by *P < 0.05 and **P < 0.01; comparing with the SO2 group, significant difference is indicated by # P < 0.05 and ## P < 0.01. ANOVA indicates analysis of variance; IκBα, inhibitor of NF-κBα; IKKβ, inhibitor of NF-κB kinase β; mRNA, messenger RNA; NF-κB, nuclear factor kappa B; SD, standard deviation; SO2, sulfur dioxide.
Effects of SO2 Plus PM2.5 Exposure on p38 and TLR4 mRNA and Protein Expressions in the Lungs
Changes in the levels of p38 and TLR4 mRNA and protein in the lungs of rats exposed to PM2.5 plus SO2 are shown in Figure 6A, B. No significant changes in p38 and TLR4 mRNA protein expressions in the SO2 group or PM2.5 group were observed compared to that of the control. The PM2.5 (1.5 mg/kg) could activate p-p38 protein relative to the control. The p38 and TLR4 mRNA expressions in lungs of 3 PM2.5 plus SO2 groups were enhanced compared with that in the control (P < 0.05 or <0.01). The p-p38 protein levels in 3 PM2.5 plus SO2 groups and TLR4 protein levels in PM2.5 (6.0 and 24.0 mg/kg bw) plus SO2 groups were significantly higher than that in the control (P < 0.05 or <0.01) in a dose-dependent manner (R 2 = 0.67-0.98). The changes in p38 and TLR4 gene expressions in the presence of PM2.5 plus SO2 were not significant compared with the PM2.5 group.
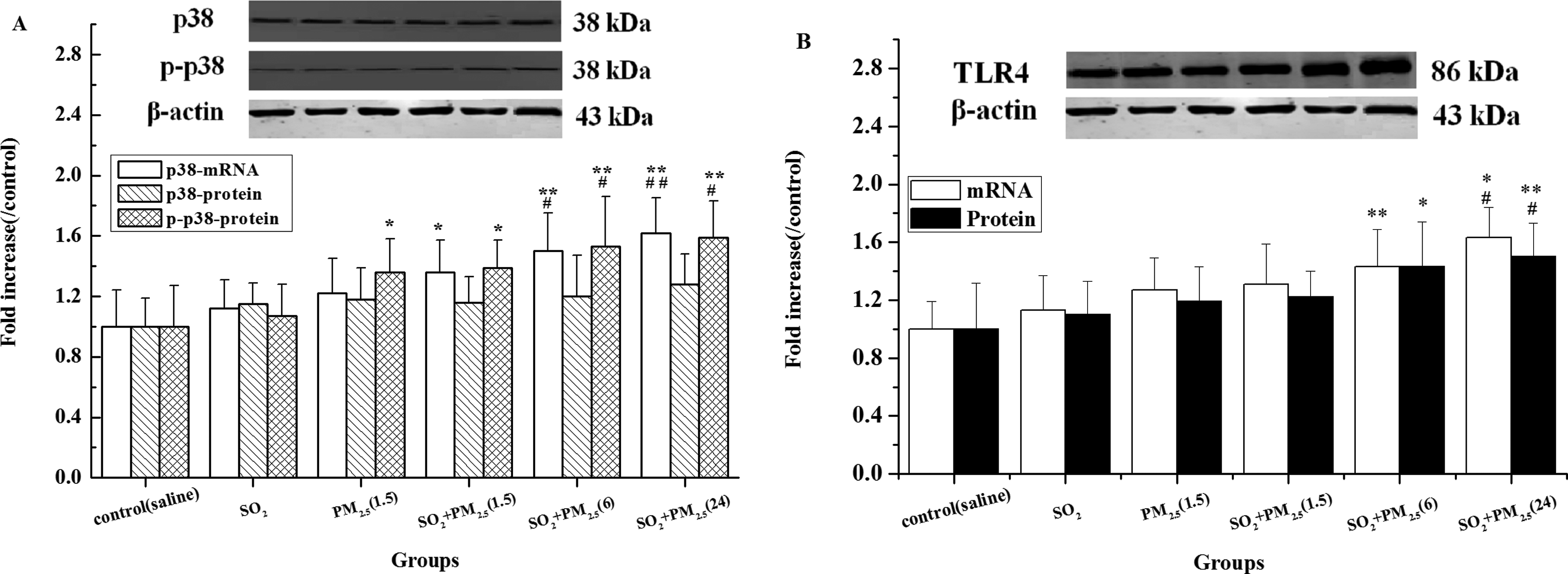
Expressions of mRNA and protein of p38 and TLR4 in lungs of rats in different groups: p38 and p-p38 (A) and TLR4 (B). Mean expression in each treated group is shown as increase compared to mean expression in the control group, which has been ascribed an arbitrary value of 1. The values are means (SD) from 5 individual samples. Using 1-way ANOVA, comparing with the control group, significant difference is indicated by *P < 0.05 and **P < 0.01; comparing with the SO2 group, significant difference is indicated by # P < 0.05 and ## P < 0.01. ANOVA indicates analysis of variance; mRNA, messenger RNA; SD, standard deviation; SO2, sulfur dioxide; TLR4, toll-like receptor 4.
Discussion
The goal of this study is to estimate the inflammatory damage effects of PM2.5 and SO2 on lungs of rats and related mechanisms to provide new evidence for setting up guidelines for evaluating the health risk of human exposure to multiple air pollutants. The data indicated SO2 short-term exposure (5.6 mg/m3) that couldn’t cause the obvious inflammatory responses and change in inflammatory factor expressions, PM2.5 (1.5 mg/kg) increased inflammatory cell numbers in BALF and pathological change. Importantly and strikingly, the concomitant exposure to PM2.5 and SO2 enhanced pathological and ultrastructural damage and inflammation occurrence followed by TLR4/p38/NF-κB pathway and overexpression of pro-inflammatory factors such as iNOS, NO, TNF-α, IL-6, IL-1β, and ICAM-1.
Inflammation is a key response under the outside stimuli, along with the increases in the inflammatory cells and the levels of pro-inflammatory factors such as TNF-α, IL-1β, IL-6, and ICAM-1, which eventually may aggravate inflammation and cause lung damage. 24 Tumor necrosis factor α, IL-6, and IL-1β are the most important pro-inflammatory cytokines and stimulate antigen presentation, adhesion molecule expression, and inflammatory cell activity. The high expression of ICAM-1 may cause the adhesion of leukocyte, NEUs, and eosinophils to the epithelial cells 25 and aggravate inflammation. Previous studies showed that PM2.5 could raise TNF-α, IL-6, IL-1β, and ICAM-1 levels in rat lungs, leading to inflammatory cell infiltration and pathological characters in rat lungs, 14,22,26 while SO2 inhalation at 28 mg/m3 increased the levels of TNF-α and IL-6. 7 On the other hand, NO, which is a gaseous signaling molecule generated by NO synthase, is enhanced by various inflammatory stimuli such as oxidants. The elevation in NO and iNOS may promote the formation of strong oxidizing reactive nitrogen species, such as peroxynitrite, leading to greater oxidative toxicity. It has been demonstrated that NO plays a damaging role in pulmonary injury 27 and exerts a key role in pathological regulation in various airway diseases including asthma and COPD. 28 As observed in the present results, coexposure of PM2.5 and SO2 significantly raised the levels of pro-inflammatory mediators including NO, iNOS, TNF-α, IL-6, IL-1β, and ICAM-1 in rat lungs exposed to higher dose of PM2.5 relative to the control or SO2 group, implying that SO2 short-term exposure at the level of 5.6 mg/m3, which is perhaps close to doses found in most industrial environments, didn’t almost contribute to the lung inflammation, whereas coexposure of SO2 and PM2.5 might enhance the lung inflammation injury in rats by the upregulation of TNF-α, IL-6, IL-1β, and ICAM-1 expression as well as by activating iNOS and augmenting NO levels.
PM2.5 plus SO2-induced inflammatory responses, elevation in inflammatory cell numbers in BALF, and abnormal alterations in lung ultrastructure, in which the main cause of SO2 and PM2.5-induced the significantly pathological change and inflammatory responses might be linked to production of free radicals. Sulfur dioxide may generate sulfur- and oxygen-centered free radicals.
29
PM2.5 may induce ROS primarily from site III of the mitochondrial electron transport chain,
30
and in vitro experiments showed that PM2.5 may produce ROS, such as superoxide (
The NF-κB, an important nuclear transcription factor, is retained in the cytoplasm in an inactive form in unstimulated cells, whereas the activation of NF-κBp65 and nuclear translocation can modulate the release of inflammatory cytokines including TNF-α and activating mediators like NO, 32,33 accompanied by IκBα degradation and IKKβ activation generally. 34 The NF-κB activation and iNOS overexpression may produce excess NO and TNF-α, in turn, excessive cytokines can further activate the NF-κB pathway, 35 thus establishing a positive autoregulatory loop that can amplify the inflammatory response and increase the duration of chronic inflammation. The experiments had proved that PM2.5 could induce NF-κB activation in human endothelial cells and SO2 plus ovalbumin could increase NF-κBp65 nuclear expression by upregulating IKKβ mRNA and protein expression and downregulating IκBα expression, aggravating inflammatory responses in lungs via raised TNF-α and IL-6 levels in BALF. 22,36 We demonstrated the changes in NF-κB, IκBα and IKKβ mRNA, and protein expression in lungs from rats exposed to SO2 and/or PM2.5. Our present study exhibited that SO2 exposure alone could not activate NF-κB pathway while coexposure to SO2 and PM2.5 could trigger IKKβ/IκBα/NF-κBp65 signal pathway activation, accompanied by the upregulation of IKKβ, NO, IL-6, and TNF-α as well as downregulation of IκBα, which may be the crucial causes that SO2 plus PM2.5 aggravate lung inflammation and damage in the rats.
In previous findings, MAPK has been shown to be a potentially important mediator in promoting bronchial hyperreactivity and lung inflammation exposed to PM2.5 or nanoparticles. 37,38 As an important member of MAPK family, the p38 MAPK is a pro-inflammatory signal transduction mediator with a rapid increase in phosphorylated p38 MAPK when the acute inflammatory response is initiated. 39 Rui et al pointed out that PM2.5 could induce phosphorylation of p38 MAPK in human endothelial cells. 36 Equally important as p38 MAPK, TLR4 exerts a significant role in the pathology of acute inflammatory lung injury induced by lipopolysaccharide. 17,40 Shoenfelt et al reported a TLR regulation mechanism of PM2.5 on inflammatory immune responses in peritoneal macrophages of mice. 41 It’s worth noting that TLR4 may mediate activation of p38 MAPK and NF-κB signaling pathways 42 and then cause a series of cascade signal responses and enhance the expressions of pro-inflammatory cytokines like IL-6, IL-1β, and TNF-α, 15 provoking inflammation.
Exposure to SO2 plus PM2.5 increased TLR4 expression along with phosphorylation of p38 MAPK, activation of NF-κBp65, and overexpressions of IL-6, IL-1β, and TNF-α in rat lungs (Figures 3 –5). It showed that the TLR4 upregulation seemed to parallel p38 MAPK and NF-κBp65 activation and high expression of pro-inflammatory cytokine. As an upstream regulation factor, TLR activation may promote p38 MAPK phosphorylation and NF-κB nuclear translocation. 42 It was also noticed that p38 MAPK activity could regulate the transcriptional activation of NF-κB, 43 resulting in NO, TNF-α, IL-6, and ICAM-1 excessive expression. In contrast, NF-κB also may mediate TLR expression. 44 We speculate that the TLR4 and NF-κB or p38 MAPK play a key regulation role in the initiation and development of lung inflammation, while they probably constitute a complex loop network, in which each gene has a own specific function and may interact with other genes, jointly modulating inflammatory mechanism of coexposure of SO2 and PM2.5 along with pro-inflammatory factors. It is not simple upstream/downstream relationships among TLR4 and NF-κB or p38 MAPK in this regulating network. It appears that the combined effect of SO2 and PM2.5 on lung inflammatory injury is complex and how they regulate each other via TLR4/NF-κB/p38 MAPK pathway need to be further studied.
In conclusion, this study presents novel results on the lung inflammatory injury effects of a binary mixture of PM2.5 and SO2 on rats. The data indicated that SO2 short-term exposure (5.6 mg/m3) did not cause the obvious inflammatory responses and change in inflammatory factor expressions, PM2.5 (1.5 mg/kg) could induce a little lung inflammation along with the gene expressions of several inflammatory factors including TNF-α, IL-6, NF-κB, and p-p38. What we focused on and the novel finding were that concomitant exposure to PM2.5 and SO2 enhanced pathological and ultrastructural damage and inflammation occurrence followed by TLR4/p38/NF-κB pathway and overexpression of pro-inflammatory factors such as iNOS, NO, TNF-α, IL-6, IL-1β, and ICAM-1. These results provide more useful evidences to understand the possible mechanism that PM2.5 and SO2 co-pollution exacerbate lung disease and suggest that health risk assessment should consider the interactions between ambient PM2.5 and gaseous coexposure.
Footnotes
Author Contributions
Ruijin Li contributed to conception and design, acquisition, analysis, and interpretation of data, and critically revised the manuscript. Lifang Zhao contributed to design, acquisition, analysis, and interpretation of data, and drafted the manuscript. Jinlong Tong contributed to acquisition and analysis and drafted the manuscript. Yuchao Yan contributed to acquisition and analysis of data and drafted the manuscript. Chong Xu contributed to acquisition and analysis of data and drafted the manuscript. All authors gave final approval and agreed to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by Shanxi Scholarship Council of China (No. 2013-16), Nature Science Foundation of Shanxi Province in China (No. 2014011036-2), and Foundation of Educational Committee of Shanxi Province in China (No. 2014110).