
Abstract
Developing inhaled drugs requires knowledge of lung anatomy, cell biology, respiratory physiology, particle physics, and some plumbing. Although dose makes the poison, in the context of an inhaled drug, the “dose” is not easily defined. This lack of clarity around dose poses issues and challenges in the design of inhalation toxicology programs. To better understand dose, the influence of ventilation is discussed as are the perturbations in pulmonary function observed with inhalation exposure that can affect dose. Methods for determining inhaled drug deposition to arrive at an estimate of lung dose are examined. Equally important to understanding dose are the techniques used to deliver aerosols to animals. With a better understanding of dose and inhalation exposure, species-specific histopathologic lesions, both common background and toxicologically significant lesions, are reviewed. Finally, insight into how regulators synthesize and evaluate these complex findings to assess clinical safety risks is presented.
Keywords
Introduction
Developing inhaled drug products is a challenging and complex process. Understanding of the area requires knowledge of lung anatomy, cell biology, respiratory physiology, particle physics, and some plumbing. Although Paracelsus informed us that the dose makes the poison and thus is of paramount importance, in the context of an inhaled drug, the “dose” is not easily defined. This symposium, presented at the 35th annual American College of Toxicology meeting, Orlando, Florida, November 11, 2014, was designed to provide background information enabling drug developers to understand the issues and challenges associated with the assessment of inhaled drugs including the design of inhalation toxicology studies. First, an overview of the key questions and issues in inhalation toxicology was followed by a presentation covering ventilation, how to measure it, its influence on dose, and the perturbations observed with inhalation exposure. The topic of dose was further explored to better understand the assumptions and methods associated with determining drug deposition and clearance to arrive at lung dose. Next, the techniques used to deliver aerosols to animals in a physiologically appropriate manner, as well as the devices used to conserve test article (TA), were examined. With dose and inhalation exposure covered, species-specific histopathologic lesions, both common background and toxicologically significant lesions, were discussed. Finally, insight into how regulators synthesize and evaluate these complex findings to assess clinical safety risks was presented.
Delivery of drugs through the respiratory system has some unique advantages. For topical therapy, especially for pulmonary disease, inhalation dosing allows high local dose and rapid drug action at a fraction of the oral dose that would be required to achieve the same lung concentration. Inhalation delivery also may limit systemic exposure and related toxicity and is a non-, or at least minimally, invasive route of administration. Inhalation delivery is also suitable for systemic delivery via the lung. Again the advantages, compared to parenteral administration, are that it is relatively noninvasive, there is a large absorptive surface (lung ∼140 m2, nose ∼180 cm2), hepatic, first-pass metabolism is avoided, and drugs may achieve a rapid systemic peak exposure after inhalation delivery.
There are many similarities and a few differences in the development of orally inhaled drug products compared to other routes of administration. One of the primary differences is the extra time that is required to develop and characterize the exposure atmosphere. Another difference is the large quantity of drug that is required for nonclinical development compared to other routes of exposure. Additionally, often 2 types of controls are used, sham/air and vehicle control, especially when the vehicle is novel. As it is difficult to have animals breathe directly from a clinical inhalation device, the inhalation exposure system will likely be different from that used in the clinic. For example, rodents are generally exposed nose-only to avoid skin/fur deposition, thus decreasing the role of gastrointestinal exposure due to preening. For nonrodents, generally a face mask, helmet, or oropharyngeal (OP) tube is used to deliver the drug to the respiratory tract.
Nomenclature
Prior to discussion of the factors important in particle deposition and response to inhaled drugs, the generally accepted method of computing dose and some of the nomenclature surrounding inhalation dosing need to be discussed. First, in an inhalation study, the term dose can refer to many different measurements. This reality generally results in confusion to those unfamiliar (and even those familiar) with the field. The terminology is not standardized, thus the terms used here may be used differently in other publications. The problem of defining dose occurs because there are multiple points during the delivery of an inhaled drug product where dose can be measured. Various losses occur from the time a drug is added to a device until it deposits in the lung or enters the systemic circulation. Figure 1 illustrates these losses. The actual amount of loss is highly dependent on many factors, and thus, the values listed are for illustrative purposes only.

Amount of loss at each stage is for illustration only. The actual amount of loss is highly dependent on many factors. For example, a dry powder soluble small molecule might have systemic bioavailability approaching 90%, whereas a nebulized monoclonal antibody or particularly insoluble drug might have <1% systemic bioavailability.
Dose Equation
From Figure 1, the delivered and deposited dose are those typically of interest in an inhalation toxicology study. The Association of Inhalation Toxicologists clearly called out the equation that is the standard for calculation of inhaled dose across preclinical species and specifies the underlying assumptions when calculating the deposited dose.
1
The estimation of inhaled dose is calculated using the formula:
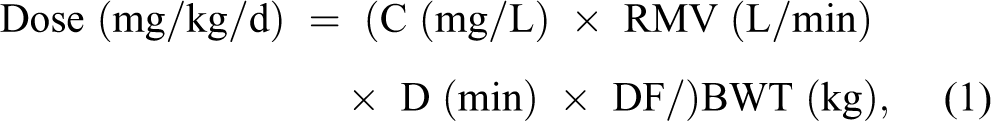
Each of these components of the dose equation will be discussed in greater detail in subsequent sections. As described previously, the estimate of the dose that is available to enter the respiratory system is termed the delivered dose (DD) but is also sometimes reported as the inhaled, presented, or nominal dose. 2 When dosing animals in an inhalation toxicology study, the protocol will base the estimated DD on an expected average animal weight (BWT), aerosol concentration (C), and duration of exposure (D). Respiratory minute volume, the product of the amount of air inhaled in 1 breath, and the number of breaths in 1 minute will typically be calculated from an allometric equation based on the anticipated average BWT for the species being tested.
For DD, the DF is considered 1 (100% is available for deposition). This estimate is referred to as the targeted DD. When C, D, BWT, and sometimes RMV have been measured, actual values are substituted in for each animal, and an average inhalation dose is obtained that is generally called the achieved DD. This DD is generally used by many non-US regulatory agencies because it can be easily measured as an estimate of dose. Of course, it is not an estimate of the dose that is deposited in the airways and lung. This is where the DF is applied to estimate a deposited dose. The deposited dose is the measurement of the dose used by the Food and Drug Administration (FDA) in regulatory decisions. Although deposited dose is theoretically more likely to be related to a response to the drug, particle deposition in animals and humans can only be estimated based on limited studies of deposition under specific conditions that may not represent the conditions in the inhalation toxicology study under investigation. Thus, both delivered and deposited dose have shortcomings in terms of relating dose to exposure and response.
Estimated Parameters
Because there are 2 factors in the dose equation that are estimates (DF and RMV), a better understanding of the assumptions underlying these parameters is required. Table 1 provides standard values for some of these estimated values in different species.
Standard Values for Inhalation Toxicology Calculations.
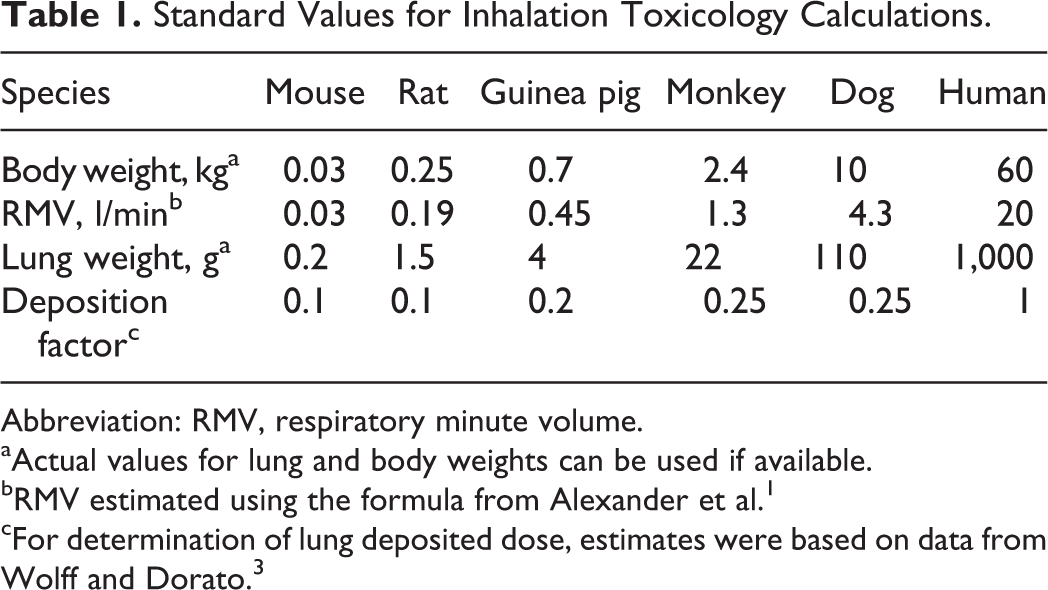
Abbreviation: RMV, respiratory minute volume.
aActual values for lung and body weights can be used if available.
bRMV estimated using the formula from Alexander et al. 1
cFor determination of lung deposited dose, estimates were based on data from Wolff and Dorato. 3
Primarily, lung deposition is determined by the characteristics of the inhaled particles and formulation as well as a number of host factors. Particle characteristics include the size, shape, density, electrical charge, and hygroscopicity. Formulation characteristics include pH, tonicity, viscosity, solubility, and the physical/chemical characteristics of the drug and any added excipients. The key host factors that influence particle deposition include any unique respiratory tract anatomy of the species and the breathing rate, depth, and pattern. These factors are discussed subsequently.
The Role of Ventilation in Determining Dose
Jeffrey S. Tepper
Allometry
As per Equation 1, the RMV is a primary determinant of dose. In most cases, this factor is estimated from an allometric equation using BWT. Typically, the RMV is calculated from the formula
1
:
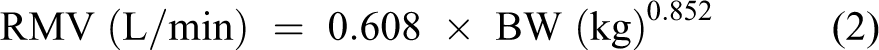
Although there are a number of allometric scaling equations that attempt to scale RMV across all mammalian species, 4 Equation 2 was fit from data in commonly used inhalation toxicology species under conditions similar to those used in such studies and is therefore believed to better approximate RMV for most inhalation studies. As RMV scales with BWT, different size animals receive similar doses, and thus exposures, on a dose/weight basis.
Functional Changes in the Lung With Toxic Insult
For inhalation studies, the respiratory tract is generally the site most likely to show evidence of toxicity, but not all toxicity manifests as morphologically observable injury. Functional changes, which can occur with inhalation exposure, can range from acute life threatening to subtler changes in ventilation and breathing pattern that can alter the dose and distribution of an inhaled drug. Since functional changes, particularly acute types, may occur without concomitant changes in morphology, it is important to carefully measure pulmonary function for inhaled drugs as part of the ICH-S7A safety pharmacology assessment. 5 As per ICH-S7A, “clinical observation of animals is generally not adequate to assess respiratory function.”
Functional changes that may manifest with inhaled products include changes in ventilation, irritant reflexes, bronchoconstriction, and restrictive, obstructive, and diffusional changes. As an initial assessment, frequency of breathing and tidal volume, the components of RMV, are usually screened. To measure RMV, head-out or whole-body plethysmography is typically used in rodents. From one primary signal of volume (or flow), both tidal volume and respiratory rate and their product, RMV, can be obtained in unanesthetized restrained animals. Similar methods including pneumotachography can be applied to nonrodent species. Besides using a pneumotach or a plethysmograph for the measurement of RMV in animals, new telemetric methods have been developed to obtain such measurements, primarily in nonrodent species, while animals are minimally confined in their home cages. Such techniques are now commonly used in safety pharmacology studies but could also be used to get a more accurate evaluation of inhaled dose. Tidal volume and respiratory rate are controlled independently, and as such, analyses of both components as well as other derived measures from this signal (inspiratory and expiratory times and flows) are useful. Such analyses can help differentiate sensory from pulmonary irritancy, bronchoconstriction/obstruction from fibrosis/restriction, and altered respiratory drive from respiratory muscle abnormalities.
However, measurements of RMV and its components alone may not uncover lung functional impairments or adequately detect changes in the small airways and lung parenchyma. Murphy reported that 3-fold increases in airway resistance, produced by intravenous infusion of the bronchoconstrictor methacholine, did not change RMV in the rat, dog, or monkey (unpublished results). 6 If volume or flow rate is measured while simultaneously measuring driving pressure, additional respiratory mechanical variables, such as compliance and resistance, can also be derived, allowing a more comprehensive evaluation of pulmonary function. Additional techniques have been developed allowing the measurement of static lung volumes (eg, total lung capacity and functional residual capacity), forced expiration (forced expiratory volume over a specified period of time and forced vital capacity), homogeneity of airflow (nitrogen washout), and diffusion. However, these measurements are typically made in anesthetized animals. These techniques allow a complete evaluation of pulmonary function analogous to the tests performed in humans. 7,8 Although not typically used for initial safety pharmacology assessments, such supplemental studies may be needed when adverse effects are suspected that are related to compound class or when initial assessment, clinical studies, pharmacovigilance, or literature indicates more in-depth evaluation is required.
Safety Pharmacology
Although currently most safety pharmacology pulmonary function assessments are done as stand-alone acute studies, there are cases where incorporation of these tests into toxicology studies may be acceptable and even encouraged. For example, if there is low or no concern for primary or secondary pharmacology in the lung, effects on the peripheral/central nervous system, or muscle, then looking acutely and after repeated dosing may enable the investigator to better detect off-target responses. Similarly, if cumulative effects are expected based on primary or secondary pharmacology of the therapeutic, then repeat dose assessment may make sense. Another appropriate reason for evaluation during a repeat dose study may be when the drug accumulates in the lung, so that Cmax at steady state is much higher than what can be achieved with acute dosing. However, when high levels of acute primary or secondary pharmacology are expected with target expression in the lung, it may be better to investigate these acute, potentially life-threatening changes in a stand-alone study. Furthermore, if the functional measurements interfere with toxicology-related activities or end points, or the appropriately trained staff cannot collect functional measurements due to time/equipment/space constraints, a stand-alone study is advised.
Dose is Dose, Right? Determining Pulmonary Dose in Clinical and Nonclinical Studies
Philip J. Kuehl
When conducting clinical and nonclinical studies, the term dose is used to define the amount of a compound delivered. In nearly all cases, the calculation of dose is straightforward and without room for interpretation. However, as described previously, in inhalation drug delivery, the representation and understanding of the meaning of dose vary greatly.
Why Does Dose Matter?
With the lack of standardization around inhalation dose, a reasonable question is to ask “why does it matter?” The answer is not entirely simple and lies in the need for the representation of dose for each study. A different representation of dose may be required based on the type of study (clinical/nonclinical, efficacy/safety), for example, the same formulation/active pharmaceutical ingredient (API) combination may use a different assumption within the calculation of dose for efficacy studies compared to safety studies. Further complicating the question of “why does it matter?” for clinical studies is the issue of compliance caused by the complexity of using some inhalation devices and the differences among patients in how they operate the device. One might consider that the differences in delivery of an oral pill vary little based on the formulation and packaging, however, the differences in delivery between how a patient uses a dry powder inhaler and a pressurized metered dose inhaler (pMDI) can result in large differences in dose. Finally, during drug development, there is a need to calculate the dose in both nonclinical (typically a rodent and a nonrodent species) and clinical studies, which introduces differences in inhalation parameters that ultimately affect the understanding of dose.
With all of these differences, there is clearly a need for standardization in the calculation and presentation of inhaled dose. As discussed previously, the Association of Inhalation Toxicologists has recommended Equation 1 as the standard for calculation of inhaled dose across preclinical species and discusses the assumptions for calculating deposited dose. 1
Deposition Fraction
Deposition fraction, also called inhaled fraction or inhalable fraction, is the value that is modulated based on the variables of the study and the desire to estimate the pulmonary deposited dose (PDD). If a DF value of 1 or 100% is used, then the inhaled dose that is calculated is called a DD. Often for nonclinical safety studies designed to support FDA regulatory submissions, DF values, as presented previously in Table 1, are used to estimate the PDD.
These DF values were based on previous research attempting to validate DF for different combinations of species, study types, and aerosols. 3,9,10 However, many of these studies were conducted prior to the invention of current state-of-the-art techniques that enable increased quantitative assessment of deposition and regional deposition. Thus, these validation studies need to be reassessed in light of new techniques and data. Contributing to the need for better understanding of DF is the fact that much of the historical literature was conducted with inhalation materials that no longer represent current pharmaceutical aerosols in terms of density (high density vs unit density vs very low density) and geometric standard deviation (GSD; monodisperse vs polydisperse aerosols).
Dose Quantification
There are typically 2 methods used to quantify the dose to the lungs following inhalation: pharmacokinetics (PKs) and deposition imaging. Each of these carries with its advantages and disadvantages highlighted in Table 2. Regardless of the advantages/disadvantages, both methods have been utilized in an attempt to quantify the drug delivered and deposited in the lung.
Summary of Methods to Quantify Dose to the Lungs and Associated Advantages/Disadvantages With Each Method.

Abbreviation: API, active pharmaceutical ingredient.
Pharmacokinetics
Pharmacokinetic study design considerations to quantify dose to the lungs are similar to the concerns for a systemic PK study. One should specifically define the species of interest, determine the need for animals to be fasted, estimate time points based on what is known about the compound clearance, understand the assay limit of detection, and be able to model the data to quantify systemic exposure via pulmonary dose. For compounds that are known to have oral bioavailability, often a charcoal block can be utilized to eliminate or reduce oral absorption; however, the dose(s) of charcoal to be delivered must be tested and confirmed to block oral absorption in order to interpret the data.
An example of the application of PKs to quantify dose to the lungs has been published recently with a biological compound (PYY[3-36]). 11 The utility of a biological compound in the utilization of the PK method is that the oral bioavailability is generally accepted to be 0, which enables the assumption that the systemic exposure is the result of the pulmonary dose. This study utilized subcutaneous (0.1 mg/kg), intraperitoneal (0.1 mg/kg), and inhalation dose delivery. The original assumed DF for this aerosol was 2.5% based on the particle size of 2 μm mass median aerosol diameter (MMAD) and published DF data for mice. 12 This resulted in a calculated PDD of 0.21 mg/kg. Pharmacokinetic analysis would have predicted a DF to be ∼1.25% based on the overlay of the concentration–time profile (Figure 2). The difference between 2.5% and 1.25% DF could either be that they are similar (less than 2% different) or that they are different (a factor of 2 different), which highlights the need to understand and call out the DF used.
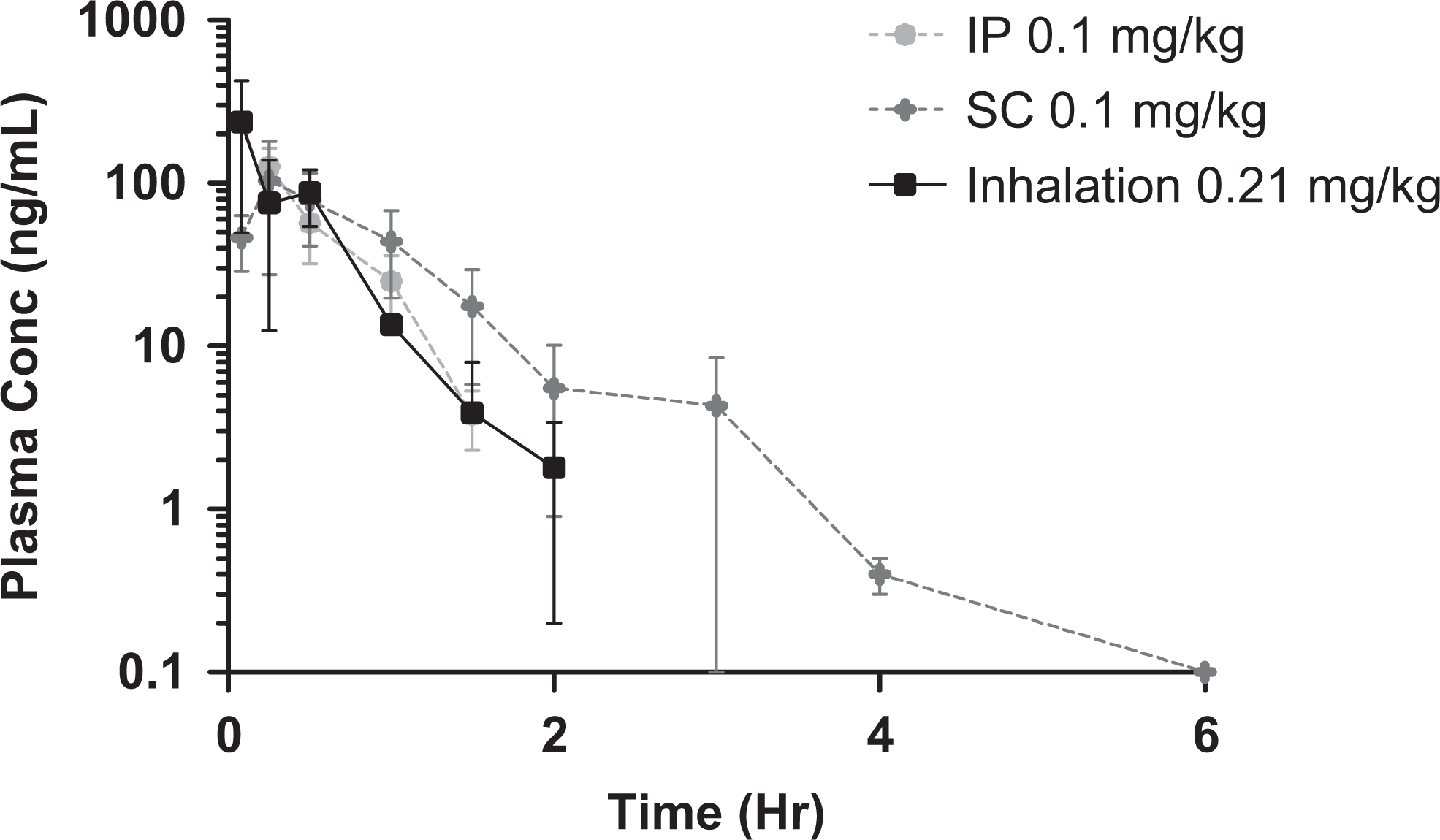
PYY systemic concentration versus time profile following intraperitoneal (IP), subcutaneous (SC), and inhalation delivery. Republished with permission from Taylor & Francis Ltd (www.tandfonline.com). 11
Deposition imaging
The other method for estimating aerosol exposure is deposition imaging of the lung. Deposition imaging utilizes a radiolabeled aerosol (typically technetium-99m) coupled with 2-dimensional and/or 3-dimensional image acquisition using a gamma camera. The study design(s) for deposition imaging studies have been well defined and should be considered prior to conduct. 13 Although these considerations are focused for clinical deposition imaging studies, similar points should be considered for nonclinical studies. For example, as the gamma camera quantifies radioactivity and not the drug, the ability to interpret the imaging data are based on confirming that the radiolabel is homogeneously distributed throughout the aerosol. In order to validate the radiolabel, standardized methods must be followed prior to utilization in vivo. 14 Although methods have been published for radiolabeling nebulized formulations, dry powders, and pMDIs for inhalation, the methods must be developed and validated for each formulation to be tested. This validation work is often resource intensive and can be a mitigating factor in the selection of deposition imaging as the appropriate tool to answer the question of dose to the lung. When these studies are conducted as per standardization guidelines, they enable the quantification of DF via the analysis of the imaging data in nonclinical and clinical inhalation settings. 12,15
In Silico Modeling
Regardless of the method utilized to quantify DF, the resultant published data are typically mined and utilized to build/design/refine in silico methods to calculate DF. A common example of software utilized to predict rat and human DF based on the aerosol characteristics is the multiple-path particle dosimetry (MPPD) model (current version 2.11). The current version was utilized to compare and contrast the rat data acquired via deposition imaging and is shown in Figure 3. Interestingly, the empirical data and the MPPD model are in good agreement for aerosols between 1.0 and 5.0 μm MMAD. However, there is a marked difference for aerosols at 0.5 μm MMAD. Similarly, when recent human deposition data 16 are input into the MPPD software, the correlation is good between 2.0 and 3.0 μm MMAD with increased difference at 1.0 μm MMAD particle size. One potential cause for the difference at the smaller particle size could be the estimation of the amount of the aerosol that is exhaled. However, clinical deposition imaging data indicate that only a small fraction is exhaled even at 1.0 μm MMAD. 15
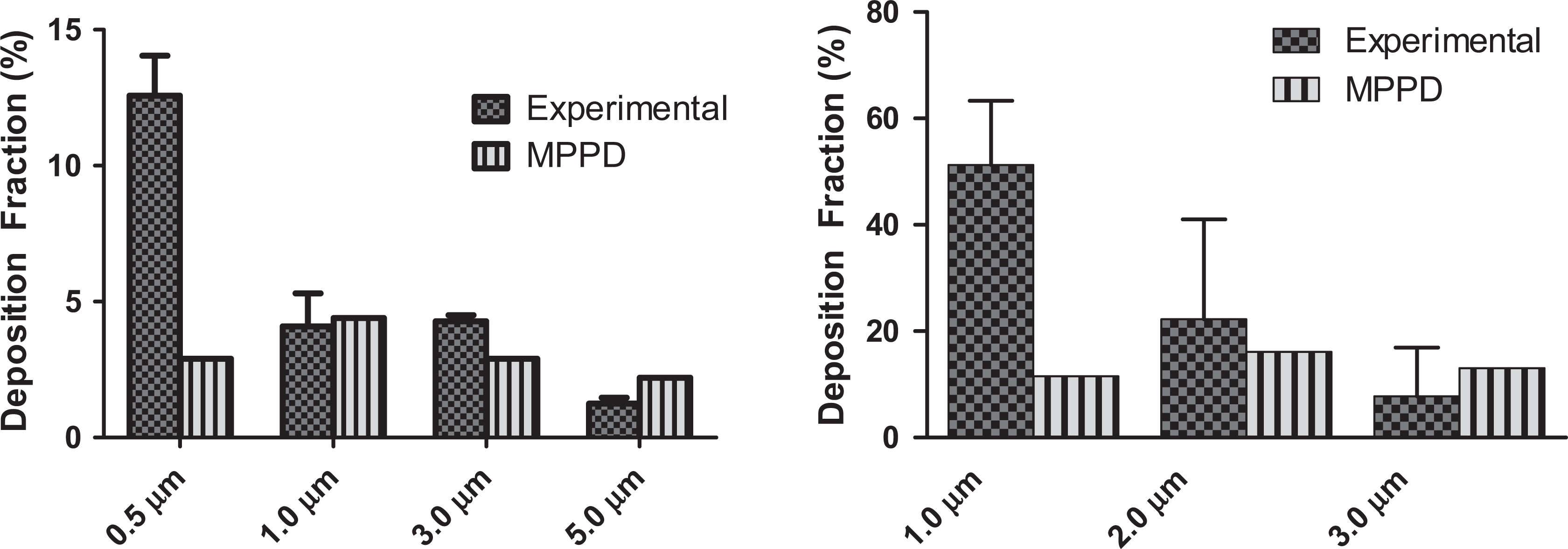
Comparison of multiple-path particle dosimetry (MPPD) deposition fraction with those measured by deposition imaging in rats (left) and humans (right).
Section Conclusion
Overall, these data indicate that both the PK and deposition imaging methods have utility in the quantification of pulmonary dose. The quantification of pulmonary dose enables the calculation of DF that can be used to calculate the pulmonary dose in a wide variety of different settings. The selection of the DF can be based on specific studies designed to quantify DF for a formulation or can be selected based on literature or modeled values. Regardless, the appropriate DF is not always clear and is affected by many variables including species, type of study, and the study objectives. Irrespective of the DF selected, it is important to state what and why the DF was selected whenever pulmonary dose is presented.
You Need How Much!!! TA Conservation During In Vivo Inhalation Studies
Stuart Cracknell
Inhaled drug delivery to conscious animal models has always used many times more TA than is actually delivered to the respiratory tract. Half of this effect is associated with the inconvenient reality that animals breathe out as well as in. The magnitude of systematic losses is typically greatest for dry powder formulations, but poor efficiency can also be a feature of liquid droplet deliveries. Losses and inefficiencies occur in both the initial aerosol generation process and in the aerosol delivery systems used to bring the atmosphere product to the breathing zone (Figure 1). Enhancing the efficiency of TA usage can enable the completion of critical proof-of-concept and investigational new drug-enabling inhalation investigations with significantly smaller quantities of an active moiety than would be necessary when using standard laboratory techniques. This is especially important early in TA development when the quantities available are commonly both limited and produced at the highest unit cost due to laboratory-scale manufacture.
Test article manufacturing costs associated with inhalation delivery may in some instances exceed the cost of running the in-life studies that drive the consumption. As a consequence, even small percentages of reduction in usage may achieve worthwhile savings.
Reducing TA consumption is a high priority for the contract research sector as the high cost of material supply offers opportunities in terms of competitive edge and the safe disposal of wasted material can be both challenging and costly. The costs associated with disposal are indirectly transferred to the pharmaceutical industry and therefore act to drive up testing costs.
Estimation of TA Requirements
Test article requirement predictions to conduct a program by the inhaled route often result in a degree of sticker shock when presented to an inhalation-naive sponsor representative. Relative to alternative parenteral routes, inhalation will commonly require between 6 and 10 times greater consumption of TA to achieve the same assumed dose. This is due to the combination of losses from the aerosol generation process, generation at airflows that necessarily exceed the calculated RMV of the individual animals that will be placed on a system, and the assumption of poor delivery to the respiratory tract that will be made by the regulatory agencies. In the case of the FDA, only 10% of aerosol delivered to rodents will be assumed to reach the pulmonary system, with a 25% assumption of lung deposition made for nonrodent species (see Table 1). This compares to the conservative assumption of 100% deposition for clinical delivery by the FDA. The final losses that may be incurred are not easily predicted but can significantly reduce delivery efficiency. To achieve high multiples of the clinical dose target, one option is to use high aerosol concentrations, but these could drive up systematic losses due to proximity and induced electrostatic charge effects on the particles, especially when using dry powders, but can also affect delivery efficiency for some liquid droplet atmospheres. Alternatively, to reduce these systematic losses, the aerosol concentrations would need to be lower, but the exposures would necessarily be longer to achieve the same target dose.
Pharmaceutical company representatives are often frustrated by the inaccuracy of the estimates of TA consumption provided by the contract research sector. The process of predicting TA needs for any given study or program is based on the combination of understanding the methods of generation that will be used and the inclusion of a sufficient overage that aims to eliminate the risk of last minute, secondary TA requests. These estimated overages will typically be increased in the absence of experience with a particular formulation and are sometimes influenced by the predictions of contract research organization competitors.
This subsection will focus on (1) methods of aerosol generation and delivery, (2) exposure system design, and (3) study and program design and logistics, all factors that can impact TA consumption. Case examples of methods that affected TA usage are provided.
Methods of aerosol generation and delivery
Inhaled delivery in nonclinical toxicity studies can be by direct intratracheal instillation or insufflation, whole-body exposure, and snout-only exposure by either a directed flow to individual animals or a plenum arrangement that exposes all animals to a shared volume of aerosol (flow through). Large animal (rabbits, dogs, and primates) exposure is commonly via facemask delivery and generally uses a directed flow aerosol. Masks may permit nasal breathing or incorporate a tongue depressor tube to encourage oral respiration. Oral exposure can be assured by delivering the aerosol via a cannula inserted into the oropharynx, but exposure duration is limited by the tolerance of the animal to the procedure. An additional factor limiting the widespread use of this technique is the additional cost associated with individual handling of the animals for the procedure.
Whole-body exposure
This method is a multiple exposure route technique and only rarely applicable for pharmaceutical products. Relevance is largely confined to some medical gas and vapors and when exposure duration exceeds recommended standards for confinement of animals.
Intratracheal methods
Intratracheal instillation is a highly efficient delivery technique that introduces TA directly into the upper airways. However, the method requires anesthesia and therefore in most circumstances is unsuitable for daily repeat administrations due to the risk and potential ancillary toxicities associated with anesthetic administrations and tracheal damage. The technique involves significant direct impaction of material on the tracheal walls and bifurcations and cannot be considered truly representative of a passively inhaled aerosol but can be used as an adequate surrogate for early proof-of-concept and PK studies (Figure 4).
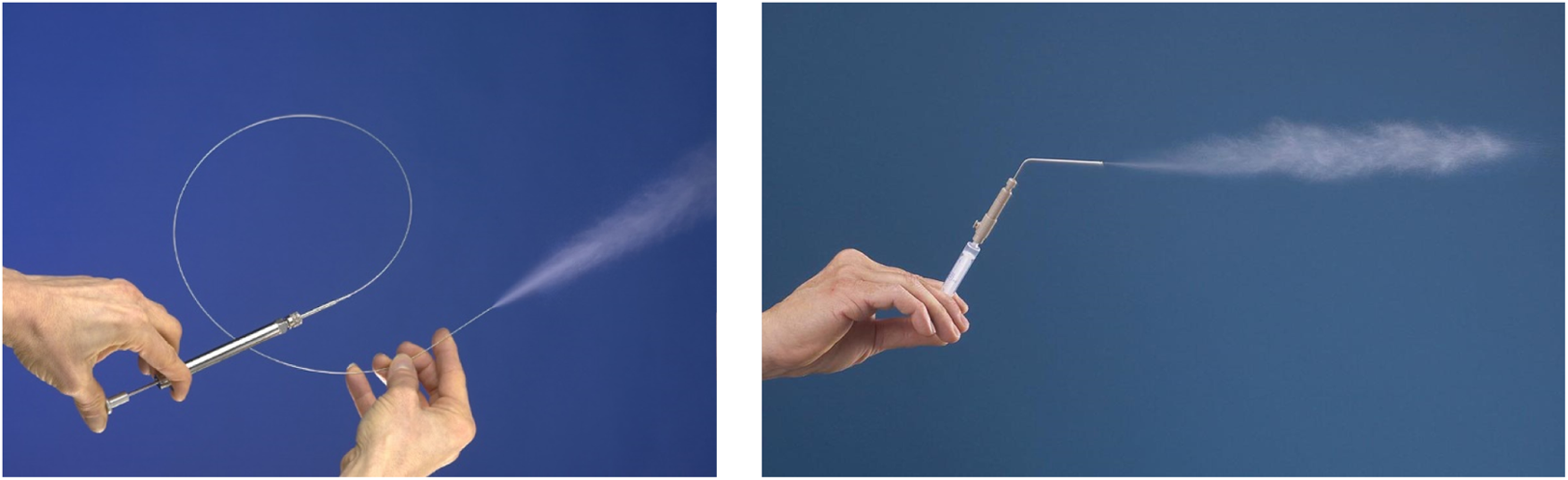
Direct administration into the trachea of anesthetized animals provides a surrogate for inhalation delivery with the benefit of high delivery efficiency. Losses onto the tracheal walls and at bifurcations are typically greater than during passive inhalation exposure. ©Photos of the MicroSprayer® Aerosolizer and Dry Powder Insufflator™ reproduced with permission of Penn-Century, Inc.
Snout-Only Exposures—Rodents
Two approaches to delivery are available (Figure 5): flow through and directed flow (also referred to as flow past). Flow-through chambers are designed such that all animals are exposed to a shared volume of aerosol. When used for the exposure of large numbers of animals, flow-through exposure system designs require higher airflow to overcome stratification of delivered atmospheres and to minimize the effects of humidity, oxygen, and carbon dioxide content from the test model (animal) on the aerosol. Directed flow systems deliver atmosphere to individual animals and take exhaled air directly to waste, largely eliminating the impact of test model respiration on the delivered aerosols. One important distinction is that directed flow systems can be used at significantly lower airflows per animal exposed than their equivalent flow-through alternative.
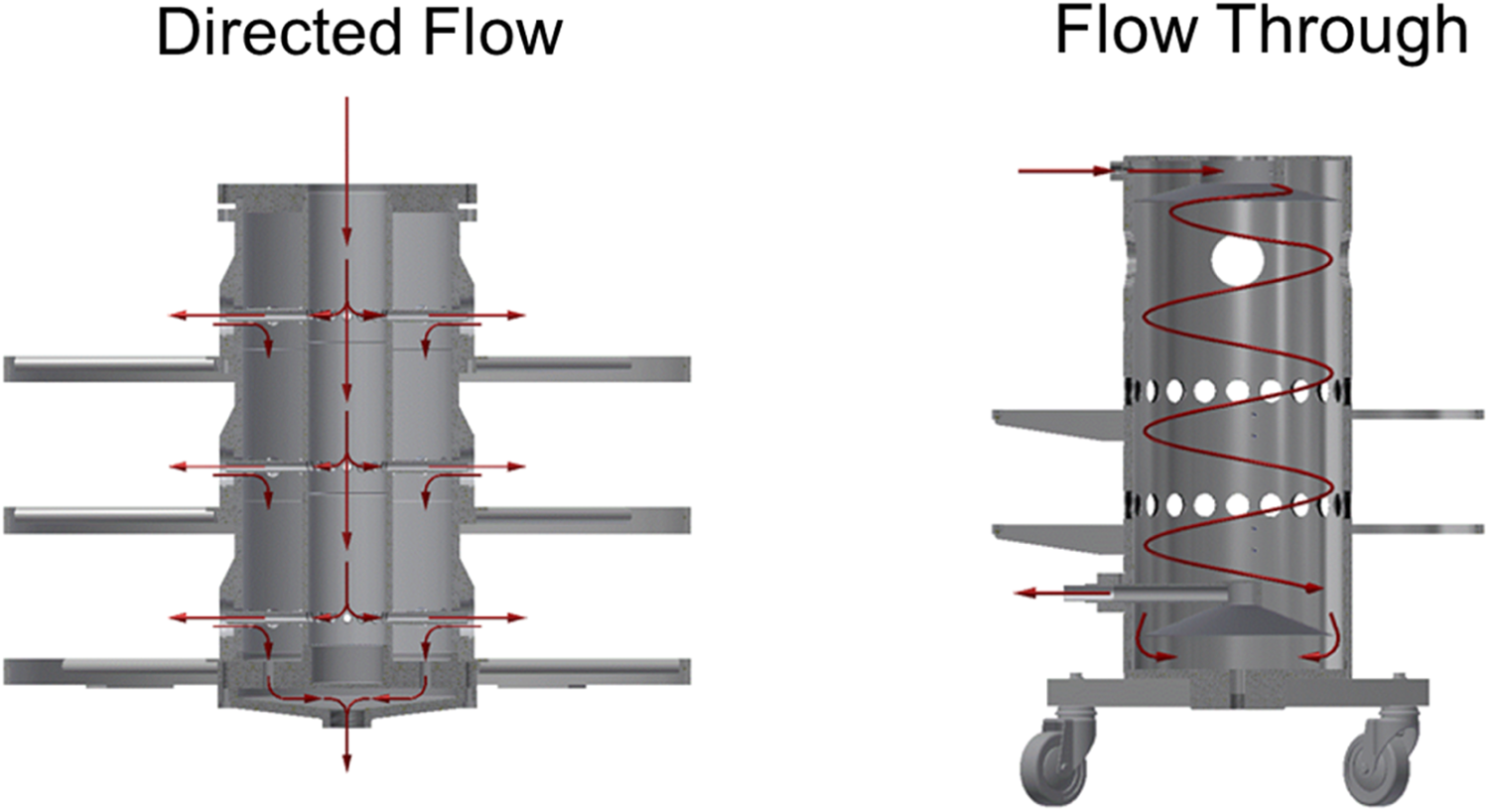
Directed flow exposure provides fresh aerosol to each exposure port, largely eliminating carbon dioxide accumulation, oxygen depletion, and minimizing moisture content changes that can be a feature of flow-through systems unless high airflows are used. The greater internal volume of flow-through systems provides superior buffering of aerosol concentration, relative to similarly scaled directed flow platforms, and their structural simplicity facilities both operation and decontamination. The ability of directed flow systems to operate at lower airflow per exposure port than flow-through platforms, without jeopardizing animal welfare, can be used to reduce test article consumption.
Oronasal exposure methods in dogs, primates, or swine
These exposure systems typically use a directed-flow approach that allows minimization of the airflow and therefore TA consumption. Practically, a mask or helmet is fitted to the animal to deliver the aerosol (Figure 6). The long delivery path in plenum systems can be associated with losses in TA aerosol transfer efficiency; however, this is commonly balanced by reduced systematic losses attributable to the use of individual animal generation systems.

Facemask delivery is commonly used when long exposures to high test article concentrations are required to achieve the necessary clinical overage. Exposure durations can range from 10 minutes to 2 hours in primates and up to 4 hours in dogs and swine. During exposure, animals may be restrained in chairs (nonhuman primates) and slings (dogs and swine), and mini pigs and dogs can also be trained to accept facemask exposures under conditions of minimal body harness restraint.
Oropharyngeal methods—delivering TA only as the animals inhale
Although requiring greater manpower to run, exposures to large animal species can be performed using OP delivery methods (Figure 7). These methods provide the option of direct individual administrations to large animal species (dogs and primates) while bypassing the nasal turbinates and buccal cavity. Aerosol is delivered and exhaled air exhausted through a flexible tube that is located in the OP region directly above the larynx. The aerosol delivery tubes have to be customized to individual animals, and tolerance of the procedure may be limited. Where test aerosol is delivered coincident with inspiration, the technique may provide more efficient TA delivery than the corresponding mask exposure system, while at the same time mimicking clinical delivery methods more closely. Although this method can be an effective TA conservation technique in some situations, as a consequence of the individual handling required to conduct the exposures, OP methods are costlier to perform particularly at the upper extremes of animal model tolerance (10 minutes in primates and 15 minutes in dogs).
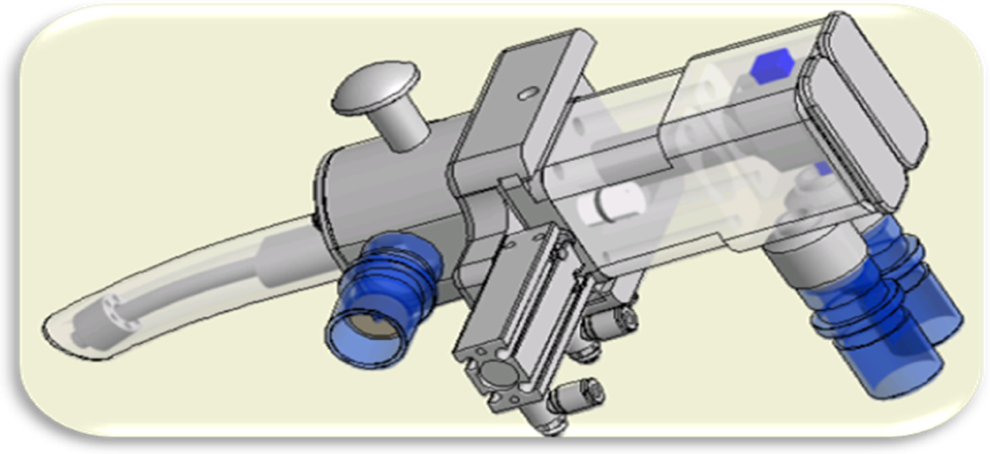
Oropharyngeal (OP) delivery in dogs and primates permits delivery from encapsulated clinical platforms and more closely mimics clinical administration. Systems can be constructed to deliver liquid droplet or particulate aerosol, and the route eliminates the filtration in the nasal turbinates that occurs during the nasal breathing that is normal for mask restrained laboratory animals. The duration of deliveries by the OP route is commonly limited to a maximum of 10 minutes.
Oropharyngeal dosage is usually assessed by a combination of aerosol samples collected from the exposure platform during simulated breathing and mass balance determinations based on the analytical determination of residual material in the systems after animal dosing.
Exposure system design
The origins of TA consumption are both readily identifiable and subject to a measure of control. The single most important factor is system airflow requirement, which is largely driven by all the other study design decisions and requirements including the generation efficiency that can be achieved by the selected platform, the exposure platform selected, and the number and body mass of the animals to be exposed. In most instances, the aerosol for inhalation exposures is also delivered dynamically throughout the period, leading to waste associated with the proportion delivered during test model exhalation. Even this waste can be controlled in some circumstances by delivering aerosol only as the animals inhale.
System airflow
Test article usage in inhalation deliveries is always expressed in terms of Units/L of system airflow, therefore, flow in excess of that needed by the animals receiving exposure will usually unnecessarily consume additional material. Consequently, one of the earliest questions that must be raised whenever limitation of TA consumption is critical is “how much air is really needed?”
Airflow and generation efficiency
It is possible to confuse the efficiency of aerosol production with the efficiency of TA usage. While related, they can be independent of each other. A capsule-based aerosol generation system may have a “raw” efficiency that is substantially inferior to a laboratory generator such as a Wright dust feed or rotating brush generator. Despite this, the ability of capsule-based systems to operate at very low airflows can mean that they are the logical choice to conserve material if only a low flow is needed. Where aerosol is produced at high initial efficiency but at an unnecessarily high flow rate, the excess can only be passed directly to waste. For example, 25% generation efficiency at 2 L/min where 2 L/min of aerosol is required = 25% overall; however, 50% generation efficiency at 10 L/min where 2 L/min of aerosol is required = 50% × 20% or 10% efficiency overall. The ability of a low flow but only moderate efficiency platform to conserve material only declines as the system airflow requirement increases.
Systematic delivery platform losses
The losses within the internal construction of aerosol generators must also be taken into account. Nebulizers with reservoir volumes that result in high daily discard of formulation and particulate generators that have canisters with a large dead space will all result in losses that influence overall TA utilization efficiency. Compressed air nebulizers, in particular, come in a wide variety of designs, each of which has its own characteristic volume of systematic reservoir residue that in most instances is not suitable for reuse due to concentration effects during reflux and exposure to the compressed air used in generation.
Systematic delivery platform losses
For particulate generation systems, any systematic losses associated with priming of delivery pipes to normalize losses attributable to static charge and sedimentation, together with any device-specific consumption in the platform containers (canisters, hoppers, capsules, etc), should also to be included in loss calculations. This material has to be considered when estimating the overall efficiency of TA usage rather than a simplistic comparison of nominal to achieved aerosol concentration.
Study and program design and logistics
Number of animals
One consumer of TA that may provide an opportunity to reduce TA usage that is commonly missed is the number and strain of the animals used. With the exception of instillation and insufflation methods, the TA use for all inhalation deliveries is determined in units of weight per unit volume of air. As a consequence, one of the most important conservation decisions is determining the flow of air that is really required.
Animal strain decisions
Taking the example of the beagle dog, differences in mean body mass at the end of a 39- or 52-week chronic toxicity can be as much as 6 kg. As airflow is typically standardized throughout a study based on the average body mass that is predicted to occur at the end of the treatment, delivery of aerosol at a flow rate suitable for the greater RMV of the large strains can impact TA consumption in a study or program by as much as 35% of the total. For rodents, the differences in body mass are smaller, but the same considerations will apply with the system airflow at the start of a study established to ensure adequate ventilation per animal throughout the treatment period. Consequently, the use of a smaller strain or fewer animals provides an opportunity to reduce TA consumption.
Satellite groups
The increased sensitivity of modern analytical equipment has significantly reduced the volume of blood that is needed to provide the plasma or serum for toxicokinetic (TK) assessments. The application of microsampling techniques and the use of main study animals for TK blood sampling allow the elimination of additional cohorts of animals that are included in study designs solely to permit TK sampling. In parallel with the elimination of additional animals, the system airflow can be reduced with a corresponding impact on TA needs.
Case studies
Animal numbers—case study 1
The application of microsampling techniques to a 28-day inhalation toxicity study (ITS) eliminated the animals for TK, and antidrug antibody assays reduced the number of animals in the high-dose group from 52 to 30. The exposure system airflow resulting from this change in sampling technique reduced TA needs by over 40%.
Efficiency—case study 2
The apparent efficiency of generators may change based on the circumstances of use. In this case study, the measured efficiency of nebulizer A in one location was significantly higher than that for nebulizer B. This situation was reversed when the same 2 nebulizers were used at a second location for the same TA. The real difference in performance was associated with the required system airflow as the operating flow in the first location was lower than needed to optimize delivery for nebulizer B. When the system airflow was increased to allow the exposure of dogs in the second location, nebulizer B airflow was brought into its optimal range with corresponding improvement in aerosol production efficiency.
Efficiency—case study 3
A nebulizer with a large reservoir volume was specified by a sponsor for the majority of studies in a program and required a large volume (30 mL) of discard per unit for a generation system using 3 or 4 nebulizers per system. One nonpivotal study was performed using an alternative low reservoir volume (1 mL) high pressure unit. Nebulizer selection was based on the perception that the large volume unit exerted lower shear forces on the TA and that shear was responsible for the dissociation of a carrier structure from the TA. The sponsor also expected to specify the use of a large-volume nebulizer for clinical use. Early in the program, it was established that TA damage was attributable to desiccation of the carrier and could be resolved by humidification of the supply air. The TA discard resulting from the decision to use the large-volume nebulizer was over 250 L at a manufacturing cost in excess of US$10,000,00. This total was 30 times greater than would have been possible with the low-volume unit.
The final irony
When the product was marketed, a mesh micropump delivery for the TA was recommended!
Section conclusions—to design TA efficient studies
Test article needs should not be consumed by unnecessarily high airflows—except where gains in efficiency exceed the dilution effect; TA losses into dead space should be minimized where possible and should be considered in efficiency calculations; TA subdivision for use and packaging into the exposure platform containers should be considered in usage calculations; TA consumption due to delivery during animal model exhalation is usually unavoidable; however, in some instances, OP delivery options for large animal species may be considered; TA consumption is impacted by the combined body mass of the animals exposed—consider both the number of animals on study and the strain selected; and TA need not be consumed by a fear of changing aerosol generators to achieve economy—particle size and achieved concentration matter, not the delivery platform.
There is no single best way to deliver inhalation atmospheres but recognizing that the most important factor in minimizing usage is controlling total system airflow and accepting that delivery methods can be altered in line with system scale. Attention to these factors provides the greatest opportunity for TA usage efficiency.
Respiratory Tract Functional Anatomy and Common Microscopic Findings
Kristen J. Nikula
Introduction
The respiratory system is the largest mucosal surface in the body and is constantly exposed to the external environment. Additionally, total cardiac output circulates through the pulmonary vasculature. Therefore, the respiratory system may be unintentionally exposed to air pollutants and environmental toxicants or may be intentionally exposed to pharmaceutical agents instilled or sprayed into the nasal cavity, inhaled, or delivered systemically.
The respiratory system can be divided into 2 parts: the conducting airways and a respiratory portion. The main purpose of the conducting airways is to provide a passage for moving air in and out of the body. Secondary functions include (1) warming, moistening, and filtering inspired air, (2) host defense against infectious agents, inhaled particles, and water soluble or reactive gases and vapors, (3) olfaction, and (4) xenobiotic metabolism, which occurs in specialized cells in the nose, Bowman glands, and bronchioles. The respiratory portion functions to take oxygen in and eliminate carbon dioxide. Secondary functions include activation and inactivation of circulating hormones and xenobiotic metabolism.
Several factors affect the location and type of injury that occurs when a toxicant is inhaled. These include the physiochemical characteristics of the agent; dose, which affects the severity of injury; and the metabolic capabilities or sensitivity to injury of the exposed cells. 17 Differences among species in respiratory tract structure and cell types may lead to differences in sites of injury or vulnerability. Reversibility of injury is determined by the severity of injury and the characteristics of the host tissue response.
This section provides an overview of respiratory tract functional anatomy and common microscopic findings. Greater detail regarding the structure, function, and pathology of the respiratory system can be found in the publication by Harkema et al, 17 and detailed information on common respiratory tract findings can be found in the publication by Chamanza et al, 18 McInnes, 19 Renne et al, 20 Renne and Gideon, 21 and Sato et al. 22,23
Nose and Nasopharynx
The nose is a complex structure with 5 major types of epithelia lining different portions of the nasal passages and nasopharynx. These epithelia are squamous, transitional, respiratory (mucociliary), olfactory, and lymphoepithelium. There are significant differences among species in nasal architecture and breathing patterns. Noses of humans and nonhuman primates are simple structures, and breathing is the primary function. In contrast, noses of mice, rats, rabbits, and dogs are complex structures and, in addition to breathing, olfaction is extremely important. Humans and most nonhuman primates breathe through the nose and mouth, whereas several common laboratory species, such as mice, rats, and rabbits, are obligate nasal breathers. Differences in nasal structure, particularly the number and shape of turbinates, affect airflow patterns and sites of particle impaction or influence the site of exposure to gases and vapors. Complex structures such as those in dogs, rabbits, and rodents provide greater filtration and absorption of potential toxicants and thus provide more protection to the lower respiratory tract.
Regardless of species differences in nasal architecture and differences in epithelial cell types lining various portions of the nasal passages, certain responses to injury are common across species. For toxicology studies, these responses result in microscopic findings that are the same whether the injury is due to an administered TA or an “incidental” injury resulting in a “background” finding. The context of the finding, particularly the relationship of incidence and severity to dose, is used to differentiate TA-related findings from incidental, background findings. Common, nonneoplastic, TA-related or background findings in nasal mucosae include: shortening (resorption) or loss of cilia; epithelial cell dedifferentiation; mucous hyperplasia and/or metaplasia; epithelial hyperplasia; squamous metaplasia; respiratory metaplasia of olfactory epithelium; degeneration, atrophy, necrosis, and ulceration; eosinophilic globules (hyaline droplets): this finding occurs in rodents as a response to irritants and as a background finding with an age-related increase in incidence and severity; inflammation; and hyperostosis of turbinates.
Larynx
Like the nose, the larynx is a complex structure, various locations within the larynx are lined by different types of epithelia, and there are species differences in structure, location of these epithelial cell types, and vulnerability to injury. Rodent laryngi contain a ventral pouch lined by respiratory epithelium, whereas dog and primate laryngi have lateral diverticuli that are lined by thick stratified epithelium. In rodent laryngi, stratified squamous epithelium lines the epiglottis and vocal processes, there is a transitional zone lined by nonciliated, transitional epithelium located at the base of the epiglottis, and most of the remaining larynx is lined by pseudostratified, ciliated, respiratory epithelium. The stratified squamous epithelium is resistant to irritant injury, whereas the transitional epithelium at the base of the epiglottis is susceptible to injury. Laryngeal findings are common in inhalation studies using rodents, and the sites most commonly affected are the base of the epiglottis, the vocal processes of the arytenoids, and the area located dorsolateral to the ventral pouch. In contrast, stratified squamous epithelium extends further caudally in dog and nonhuman primate laryngi and the normal, squamous epithelium is thicker in dogs and nonhuman primates than in rodents. As a result, laryngeal lesions due to inhaled TAs are relatively rare in dogs and nonhuman primates.
As in the nose, the same types of responses occur regardless of the source of injury and the relationship of incidence and severity to dose differentiates spontaneous from TA-related findings. Common, nonneoplastic findings in laryngeal mucosa include: loss of cilia and dedifferentiation; squamous metaplasia; hyperplasia; inflammation; and degeneration, necrosis, and ulceration.
Tracheobronchial Airways
The trachea extends distally from the larynx and bifurcates at the carina to form 2 extrapulmonary bronchi. The trachea and bronchi contain intramural cartilage and submucosal glands. Bronchioles, which are the airways distal to the bronchi, lack intramural cartilage and submucosal glands. The bronchi and bronchioles branch in a dichotomous (equal diameter daughter branches with equal branching angles) pattern in humans and in a monopodial (unequal diameter and branching angle) pattern in rodents, dogs, and most nonhuman primates. The terminal bronchioles are the most distal nonrespiratory bronchioles. Intrapulmonary bronchi are present in humans, nonhuman primates, and dogs, and they are not present in rodents as the intrapulmonary airways lack intramural cartilage. The tracheobronchial airways are lined by pseudostratified respiratory epithelium that is tall, columnar proximally and becomes low, cuboidal distally. The relative abundance of ciliated versus secretory cells and the types of secretory cells (mucous, serous, or club cells) vary by proximal to distal location and species. Ciliated cells are the most common epithelial cells lining the conducting airways, and beating cilia propel the movement of epithelial lining fluid. Mucous cells are the main secretory cells throughout the tracheobronchial airways of humans, nonhuman primates, and dogs. Mucous cells are located only in the trachea and extrapulmonary bronchi in specific pathogen-free rats and mice. Serous cells are the main secretory cells lining the conducting airways from the trachea to the proximal bronchioles in rats, and club cells become abundant distally. Club cells are the main secretory cells throughout the tracheobronchial airways of mice.
Common responses of mucociliary mucosa to irritants include: ciliostasis, resorption, and loss of cilia; hypersecretion of mucosubstances; mucous cell hyperplasia; mucous metaplasia; squamous metaplasia; and inflammation
Additional common, background findings in tracheobronchial airways include: thickening of the tracheal basement membrane in nonhuman primates; bronchial and bronchiolar lymphohistiocytic or mononuclear cell infiltrates (nonhuman primates and dogs); these findings are also known as peribronchial and peribronchiolar lymphohistiocytic or mononuclear cell infiltrates; and squamous epithelium is a normal finding in dog tracheas; it occurs in the zone where the membranous and cartilaginous portions of the trachea meet.
Lung
The junction of the conducting and alveolarized airways is known as the centriacinar region of the lung. In humans, nonhuman primates, dogs, and cats, the centriacinar region consists of several generations of respiratory bronchioles with widely scattered alveolar outpocketings. Rabbits, hamsters, mice, and rats have single or no respiratory bronchioles. In mice and rats, the terminal bronchioles join directly with alveolar ducts, which are completely lined by alveoli. The centriacinar region is a common site of injury from inhaled particles and gases that reach the lung.
Common TA-related, nonneoplastic findings in the centriacinar region include: increased alveolar macrophages; inflammation; epithelial cell degeneration or necrosis; type II cell hyperplasia; and secretory cell metaplasia, also known as bronchiolization.
Common background findings in the centriacinar region include: bronchiolar (peribronchiolar) lymphohistiocytic or mononuclear cell infiltrates in nonhuman primates and dogs; and increased alveolar macrophages in rodents, dogs, and nonhuman primates.
The gas-exchange region of the lung consists of the respiratory bronchioles and alveolar parenchyma. In those species with respiratory bronchioles, the airway wall between alveolar outpocketings is lined by ciliated and club cells. The alveolar parenchyma, which comprises 90% of the lung volume, consists primarily of alveolar ducts and alveoli lined by type I and type II epithelial cells, capillaries lined by endothelial cells, and the extracellular matrix of collagen and elastin intermixed with interstitial cells (ie, fibroblasts and myofibroblasts), which are located in the thick regions of alveolar septa. Gas exchange occurs across the thin portions of alveolar septa where type I and endothelial cells are separated by a single, fused basement membrane.
Common TA-related, nonneoplastic findings in the gas-exchange region of the lung include: increased alveolar macrophages; inflammation; epithelial cell degeneration or necrosis; and type II cell hyperplasia.
Common background findings in the lung include: increased alveolar macrophages or chronic inflammation characterized by increased alveolar macrophages, interstitial inflammation, fibrosis, and type II cell hyperplasia in some cases. This finding is often located in the peripheral lung in alveoli adjacent to the pleura and is also known as chronic histiocytosis. Although best described in rats, it also occurs in dogs and nonhuman primates; acute alveolar inflammation; and chronic interstitial inflammation. In dogs, this finding is characterized by septal fibrosis, variable numbers of inflammatory cells, and often type II cell hyperplasia. In the past, it has sometimes been called fibrosing alveolitis.
Nonadverse Versus Adverse Findings
It is difficult to generate a list of findings that are always considered nonadverse or adverse when observed in inhalation studies because both context and severity affect the determination of adversity for most findings. Many respiratory tract findings occur as spontaneous, incidental, and background findings, and when observed in that context, they are not relevant to the determination of an adverse dose level. Additionally, adaptive findings are unlikely to be adverse. Findings that are specific to the experimental model or mode of exposure, such as eosinophilic globules in rodents exposed by nose-only inhalation, are unlikely to be relevant for human risk assessment. Findings that are commonly observed in inhalation studies using rodents and that are typically considered nonadverse, adaptive changes include: eosinophilic globules in nasal mucosae; mucous cell hyperplasia in nasal respiratory epithelium or minimal to moderate mucus cell metaplasia in respiratory transitional epithelium after exposure to irritants; minimal to slight, nondiffuse squamous metaplasia in the respiratory tract; and increased alveolar macrophages, without concomitant findings of inflammation or structural alteration of the parenchyma, after exposure to poorly soluble particles.
24
However, as in any study, the study director and study pathologist must consider the context of the findings and should take a weight-of-the-evidence approach in determining whether a finding is considered nonadverse or adverse when observed in a particular study.
Regulatory Nonclinical Safety Evaluations of Inhalation Drug Products
Luqi Pei
Regulatory nonclinical safety evaluations (RNSEs) involve complex decision-making processes. Regulators use RNSEs to determine whether there are adequate nonclinical data to support the safe use of drug product candidates in humans. Regulatory nonclinical safety evaluations require a comprehensive and sound evaluation and interpretation of pharmacological, PK, and toxicological findings of laboratory studies, as well as an understanding of the relevance of nonclinical findings to humans. Inhalation drug product (IDP) RNSEs require specialized knowledge of the unique features of IDPs and ITSs in animals. This presentation focused on the following: evaluation of the design and conduct of animal ITS, the evaluation and interpretation of study results, and safety margin (SM) calculations.
Inhalation Drug Products
Inhalation drug products are a special category of products indicated for various diseases. Most IDPs have fixed drug-device combinations, but some may have separate drug products and delivery devices. Inhalation drug products vary remarkably in product characteristics and indications. Most IDPs are indicated for respiratory diseases, but an increasing number of IDPs are being developed for nonrespiratory indications.
Inhalation drug products generally consist of 2 parts: the device and the formulation. Examples of devices include inhalers, nebulizers, sprays, and intratracheal tubing. Development of devices requires complex knowledge of aerosol science and engineering and delivery mechanisms. Inhalation drug products may vary in dosage forms and formulations. Dosage forms include dry powders, solutions, suspensions, gases, and nanotechnology products. Formulations include the API with or without excipients. Active pharmaceutical ingredients may be small molecular entities, proteins, or oligonucleotides. Compared to orally administered drug products, there are fewer established excipients in IDPs. 25
Scope of RNSE
Inhalation drug products RNSEs evaluate the API, excipients, impurities, degradants, leachables, and extractables of IDPs. Potential API formulation interactions are also evaluated. Regulatory scientists critically evaluate the pharmacologic, PK, and toxicological profiles of compounds of interest. Toxicology data evaluations are the most critical and resource-consuming component of RNSEs. Toxicology data review is comprised of general toxicity, genetic toxicity, carcinogenicity, reproductive toxicity, and developmental toxicity evaluations, as well as assessment of other relevant toxicological end points. Toxicological end points could be assessed by studies of inhalation and noninhalation routes of administration (eg, reproductive toxicity studies), but ITS are the only studies that fully characterize the toxicological profiles of inhaled drugs on the respiratory system. Because the intended clinical use and available nonclinical data of drugs vary with IDPs, different IDPs may merit different nonclinical programs. Regulations and formal guidance are available to define a nonclinical development program.
Considerations for ITSs
Inhalation toxicity studies have many special considerations. They include but are not limited to the following: (1) the design and conduct of ITS require special knowledge, skills, facility, and equipment; (2) the mode of exposure and exposure apparatus may vary among studies and species; (3) exposures of animals to the TA are theoretic estimates and are subject to many parameters; and (4) species differences may exist for certain responses to inhaled drugs.
Inhalation toxicity studies require special facilities and equipment to ensure proper exposure of animals to the TA and protection of laboratory personnel and environment. Because of the involvement of complex aerosol science and engineering, all equipment and procedures should be adequately validated before use. Adequate staff training is essential to ensure high-quality data. Pivotal ITS must comply with Good Laboratory Practice regulations. Facility procedures and equipment may introduce artifacts that affect the study results. For example, novel rodent restraint tubing was reported to cause excessive mortality in several studies. 26
Modes of exposure vary significantly among ITS. The mode of exposure may include inhalation, spray, and instillation. Further, equipment and species-specific considerations may influence whether TA is administered via nose-only, head-only, or oral (or OP tubing) modes. Pivotal ITS in support of the safety of IDPs should use a proper mode of exposure. For example, inhalation studies would typically be expected for drugs acting in the lung. Nasal spray studies are likely inadequate to assess the effects of inhaled drugs on the lungs. Similarly, ITSs conducted by oral inhalation in nonrodent species are inadequate to evaluate the effect of drugs on the upper respiratory tract, especially the nose.
Dosimetry
Dosimetry refers to an estimate of the exposure of animals to TA. Accurate assessments of exposures of animals to TA in ITS are essential for sound RNSEs, yet there are often inconsistencies in exposure assessments done by regulatory and research scientists due to differences in dosimetry and SM calculations, as well as differences in interpretation of toxicological end points. Inconsistency in dosimetry-related terminology may also contribute to disparate assessment. Study reports may use the DD, achieved dose, or inhaled dose to describe the same information. A recent publication by the Association of Inhalation Toxicologists Working Group provided recommendations with the goal of harmonizing some of these factors. 1 However, the importance of the PDD in the overall safety assessment of IDPs has not been fully appreciated globally.
Exposures of animals to TA in ITS have been traditionally estimated using experimental and empirical data. The DD refers to the amount of inhalable TA present in a volume of air which an animal breathes during a defined treatment period each day. The DD is calculated using Equation 1. The inhalable fraction is the fraction of particles with particle aerodynamic diameter of 1 to 5 μm. Pulmonary deposited dose, a fraction of DD, is obtained by multiplying the DD by a DF for a given species. As shown in Table 1, Division of Pulmonary, Allergy, and Rheumatology Products (DPARP)-accepted DFs are as follows: 0.1 in rodent species, 0.25 in dogs, and 0.25 in monkeys.
Both DD and PDD are affected by many parameters, including minute volume, aerosol drug concentrations, duration of exposure, particle size, and DF. The minute volume varies with animal species and BWT. Aerosol drug concentrations may be determined by gravimetric or analytical assays. There may be differences in DDs and PDDs derived from gravimetric and analytical data in some cases because the TA is unstable or due to other factors. Dosimetry calculations in such cases should be based on analytical results.
Particle size is a critical parameter in dosimetry evaluation because the potential for variability in ITS is high. Mass median aerosol diameter and GSD are 2 terms used to describe particle size and distribution patterns. The MMAD in ITS should be between 1 and 5 μm. The GSD should be in a reasonable range (generally under 3). Inhalation toxicity study with unusually large MMAD and GSD may be unacceptable for regulatory purposes due to lack of adequate toxicological assessment of drug product that is respirable.
To achieve adequate exposure and SMs, some studies may need to expose animals to the maximum feasible dose (MFD). The MFD is defined as a dose achieved using both the maximum feasible aerosol drug concentration and the maximum treatment duration per day. The DPARP of the US FDA recommends a maximum daily treatment duration up to 3, 4, 2, and 1.5 hours per day in mice, rats, dogs (2 separate exposures of 1 hour each), and monkeys, respectively. The nonclinical testing of IDP rarely requires a 6-hour daily exposure duration, as defined by the US Environmental Protection Agency. 27
Species-Specific Findings
Inhalation toxicology studies may produce species-specific findings that are not relevant to humans. Laryngeal squamous metaplasia in rodents is one example. 28,29 Arguments to support species specificity should be based on adequate justification from sources including the scientific literature.
Safety Margin
A key step in RNSEs is to determine whether there are sufficient SMs for the intended clinical doses of IDP. Because of the special characteristics of IDP in humans and ITS in animals discussed previously, DPARP does not follow approaches for calculating SMs recommended by the FDA Guidance for Industry: Estimating the Maximum Safety Staring Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers (2005).
Safety margins for IDP are calculated by dividing the no observed adverse effect level (NOAEL) by the intended nominal clinical dose using the same units. The NOAEL unit may be mg/kg/d, mg/g lung weight, or the plasma drug area under the curve (AUC), depending upon the available data and the toxicity end points. The toxicity end points can be summarized into local (ie, organ and tissues in the respiratory system) and systemic effects (ie, all organs/system besides the respiratory system). Adequate SMs are needed for both local (ie, lung) and systemic effects.
Safety margins for systemic effects
Safety margins for systemic effects may be calculated either on a mg/kg/d basis or on an AUC basis. Safety margins on an AUC basis are appropriate only to address concerns for systemic effects; margins based on AUC comparisons are recommended when that data are available.
Safety margins for local effects
Safety margins for local effects can be calculated either on a mg/kg/d basis or on a mg/g lung weight basis. Consideration of SMs on mg/g lung weight basis alone may be used only when there is no safety concern for the systemic effects of the drug.
The DPARP uses the PDD as the actual exposure in animals when mg/kg/d or mg/g lung is used to calculate SM. This practice differs from some other regulatory agencies that use achieved DD for SM calculations. The human exposure is the maximum intended nominal clinical dose divided by a BWT of 60 kg, a lung weight of 1,000 g, or the observed systemic exposure (AUC) reported in clinical trials.
The criteria for adequate SMs may vary with species and toxicology end points. The DPARP considers the following SMs adequate to support the maximum allowable clinical dose of drugs in humans in most cases: SMs based on mg/kg BWT or mg/g lung weight should be at least 10, 10, 6, and 5 in mice, rats, dogs, and monkeys, respectively. An SM of at least 1 based on AUC is generally acceptable, but an SM of 2 or greater may be needed in cases of severe, nonmonitorable toxicity.
Example SM calculations for an IDP
Scenario: Company A proposed to give asthmatic adults 1 mg/d MiracleDrug for 2 weeks.
The company conducted pivotal 4-week ITSs with MiracleDrug in rats and dogs to support the proposed clinical study. The rat was determined to be the most sensitive species. The NOAELs (achieved doses) for local and systemic effects in the rat study are 1 and 5 mg/kg/d, respectively. No human AUC data are available yet. Question: Do the NOAELs in the 4-week study in rats provide adequate nonclinical coverage (SM) for the proposed clinical dose? Answer: Yes, the NOAELs in the 4-week rat ITS provided sufficient SM for the proposed human dose. These NOAELs provide SM of 30 and 17 for systemic and local effects of the drug, respectively. As these values are greater than 10, the minimum required margin for rats, there is adequate nonclinical support for the proposed clinical dose.
Safety margin calculations and evaluations can be performed in 4 steps: (1) determining the PDDs in rats and humans, (2) determining the lung doses in rats and humans, (3) calculating SM for both systemic and local effects for human doses, and (4) evaluating the adequacy of the SM.
Determining PDDs in rats and humans:
Determining lung doses in rats and humans:
Calculating SM for systemic and local effects:
Calculating SM for systemic effects (note: AUC is used if available):
Calculating SM for local effects:
On a mg/kg basis:
On a mg/g lung/day basis:
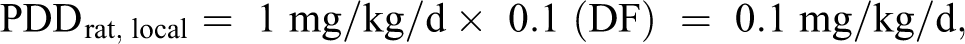
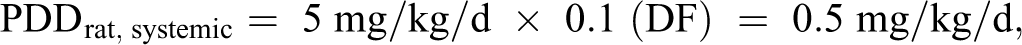
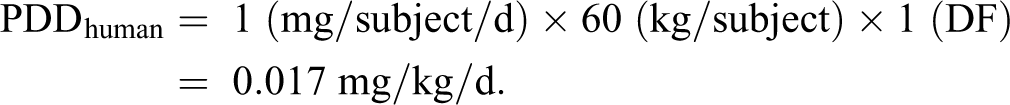
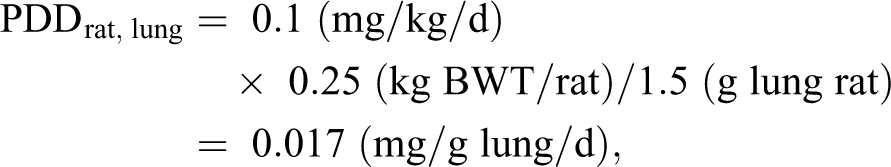
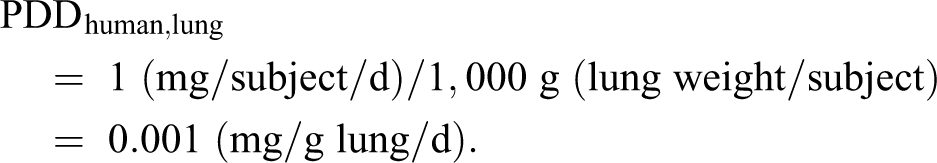
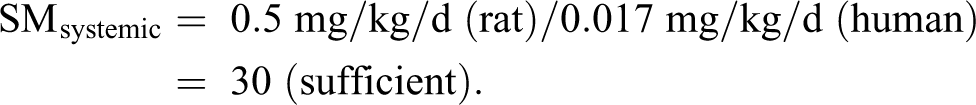
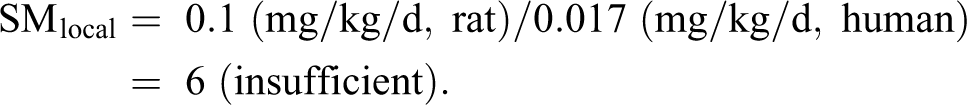
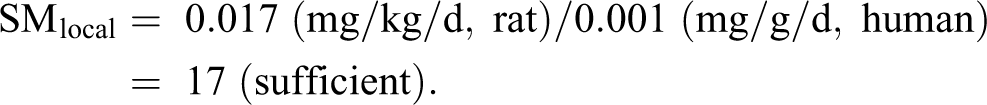
In conclusion, RNSEs of IDPs involve unique, complex, and challenging evaluations and decision-making processes because of the special characteristics of IDP, the route of administration, and ITS in animals. These unique issues should be taken into consideration by regulatory and research scientists when conducting nonclinical safety evaluations of IDPs.
Symposia Summary and Conclusion
Overall, there are real advantages of delivering drugs into and via the lung, such as less drug is used, there can be a rapid onset of effect, systemic toxicity may be reduced, and patient convenience and compliance may be increased compared to parenterally administered drugs. However, inhalation exposure does require additional planning and preparation to acquire sufficient drug, develop a formulation that allows for efficient drug delivery, and acclimation of the animals to the exposure system. Strategies to reduce these efforts and costs are available to keep development on a similar timeline as for other routes of exposure.
Although improvements have been made in understanding both dose and response after inhalation exposure, there is still too much uncertainty for the first-in-human inhalation dosing that results in increased risk to volunteers or unnecessary precaution in setting the first inhuman dose, resulting in increased time and cost to companies developing inhaled therapies. Ultimately, the concept of dose for inhalation studies requires more accuracy in the estimation or measurement of both RMV and lung deposition with such estimates validated against data. Fortunately, although there are differences in lung anatomy and physiology between humans and laboratory animals resulting in differences in inhaled drug deposition, the consequences of inhaled drug exposure are generally well conserved across species; as such, inhalation toxicology studies and respiratory safety pharmacology studies have proven useful in the development of inhaled drugs.
Footnotes
Authors’ Note
The section entitled “Regulatory Nonclinical Safety Evaluations of Inhalation Drug Products” reflects the views of the author, Luqi Pei, and should not be construed to represent FDA’s views and policies.
Acknowledgments
The authors wish to thank The American College of Toxicology for allowing the presentation of this symposium at the annual meeting, Matthew Reed for speaker suggestions, as well as the generous support of Lovelace Respiratory Research Institute, Huntingdon Life Sciences Inc, and Seventh Wave Laboratories, LLC.
Author Contributions
J. Tepper contributed to conception and design, contributed to analysis and interpretation, drafted the manuscript, and critically revised the manuscript. P. Kuehl contributed to conception, contributed to analysis and interpretation, drafted the manuscript, and critically revised the manuscript. S. Cracknell, K. Nikula, L. Pei, and Blanchard, J contributed to design, contributed to analysis and interpretation, drafted the manuscript, and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Philip J. Kuehl was supported by Lovelace Respiratory Research Institute, Stuart Cracknell by Huntingdon Life Sciences Inc, and Kristen J. Nikula by Seventh Wave Laboratories, LLC.