
Abstract
Vascular endothelium plays a vital role in the organization and function of the blood vessel and maintains homeostasis of the circulatory system and normal arterial function. Functional disruption of the endothelium is recognized as the beginning event that triggers the development of consequent cardiovascular disease (CVD) including atherosclerosis and coronary heart disease. There is a growing data associating mercury exposure with endothelial dysfunction and higher risk of CVD. This review explores and evaluates the impact of mercury exposure on CVD and endothelial function, highlighting the interplay of nitric oxide and oxidative stress.
Cardiovascular risk factors or disorders such as hypercholesterolemia, chronic smoking, hypertension, or chronic heart failure alter the regulatory function of endothelium in coronary, peripheral conduit, and resistance vessels. In addition to these risk factors, attention has recently been given to toxic effects of mercury in the cardiovascular system. 1 –3 A growing mass of data now strongly indicates an association of cardiovascular disease (CVD) with mercury exposure.
Mercury, a heavy metal belonging to the transition element series of the periodic table, is widespread and persistent as a pollutant in the environment. Mercury occurs naturally in the environment, anthropogenic activities cause the release of this element into the environment, leading to the pollution of air, water, and soil. 4,5 This element has been in commercial (compact fluorescent lightbulbs, batteries, etc) and medical use (laxative, dental amalgam, etc) for centuries, and human beings are exposed to it. 4
Mercury exists in 3 forms: metallic mercury (also known as elemental mercury), inorganic mercury, and organic mercury, and its different forms and duration of exposure have different health effects. Exposure to mercury is the second most common cause of heavy metal poisoning. Blood vessels are the primary site of exposure to the toxic effects of mercury, because when mercury enters the body, it reaches the various organs through the circulatory system. Toxicity from mercury is associated with in vivo oxidative stress. Mercury exposure induces the generation of reactive oxygen species (ROS), with subsequent oxidative damage in several organs and systems as well as alters the antioxidant defense system in the cells. 6 –10 Oxidative stress that results in endothelial dysfunction and loss of endothelium-dependent vasorelaxation is one of the most commonly observed cardiovascular effects of mercury exposure. 11 –14
The aim of this review is to explore the relationship between mercury exposure, CVD, and endothelial cell (EC) function/dysfunction, focusing predominantly on interaction/balance between bioavailability of nitric oxide (NO) and oxidative stress.
Mercury and Its Association With Cardiovascular Disorders
Population Studies
An association between the environmental and occupational exposure to mercury and the risk of CVDs was established after follow-up studies of severe cases of poisoning, and studies also showed cardiovascular abnormalities in Iraq and in Minamata, Japan. 15,16 Since then several studies have associated cardiovascular disorders with mercury exposure. 17 –31 Numerous population studies have reported tachycardia, high blood pressure (BP), decreased heart rate variability (HRV), increase in the risk of stroke, and increased risk of total cardiovascular mortality. 17,19 –25 In a native population of Quebec (Canada), organic mercury exposure has been associated with increased resting HR but with no significant association with BP. 26 In a study conducted on 274 schoolchildren, an increased level of mercury in urine has been associated with elevated cholesterol level, which is a known risk factor for myocardial infarction, coronary disease, and CVD. 27 It has always been considered that consumption of fish is beneficial for the health of heart because they are rich in the potent antioxidant and long chain n-3 polyunsaturated fatty acids. However, a number of studies indicate that the consumption of fish contaminated with mercury causes high mercury levels in blood, hair, urine, and toenails, which diminish the cardioprotective effect of n-3 polyunsaturated fatty acids. 28 –30
In a study of 236 healthy people, mercury content in hair due to smoking (cigarettes contain a slight amount of mercury), which in itself is a major risk factor for the development and progression of CVD, was found to be positively associated with increased BP and abnormal lipid metabolism. 31 Mercury is also associated with decreased HRV among French Polynesian teenagers, with no significant association with resting HR, BP, or Pulse Pressure (PP) among teenagers or adults. 32 In 2014, a total of 2114 healthy adults who were not exposed to mercury occupationally were sampled, and it was observed that mercury was associated with metabolic syndrome and parasympathetic dysfunction, indicating that mercury exposure may play a role as a possible risk factor for CVDs. 33
However, data from the Kuopio Ischaemic Heart Disease Risk Factor Study of 848 men and 909 women suggest that hair mercury is not associated with high BP. 34 A recent study has demonstrated that there is no association between childhood BP and prenatal mercury exposure. 35 The Seychelles Child Development Study has negated the hypothesis that prenatal or recent postnatal methyl mercury exposure from fish consumption is associated with impaired autonomic HR control. 36 In another population study, toenail and hair mercury levels in Faeroese whaling men were significantly associated with increased carotid intima–media thickness (IMT) and hypertension, 18 suggesting a greater role of vascular system in the mercury exposure–associated CVDs.
Animal Studies
In animal models of mercury exposure, various cardiovascular effects have been observed. The route, dose, and period of mercury exposure determine the kind of cardiovascular effect caused. A decrease in HR accompanied with an increase in systolic BP has been observed in male rats chronically treated with methyl mercury chloride. 37 Chronic exposure of rats to mercuric chloride also causes an increase in BP and decrease in cardiac contractility with no change in HR. 38 In a different longer chronic study, positive inotropic response, with increase in BP and cardiac contractility and decrease in baroreceptor reflex sensitivity, was observed. The investigators suggested that the mechanism for the cardiac effects in the chronic study involved the release of norepinephrine from presynaptic nerve terminals. 39 Rossoni and coworkers observed that after acute mercuric chloride administration, there was a decrease in arterial BP in anesthetized rats. 40 da Cunha and coworkers have also observed that in situ administration of mercuric chloride (2 µmol/L) produced a significant increase in the perfusion pressure mediated by the formation of superoxide anions in isolated perfused rat tail vascular bed. 41
Chronic exposure to inorganic mercury also causes a significant increase in left ventricular end diastolic pressure, blood and cardiac tissue mercury content, and myocardial lipid peroxides and attenuates baroreflex sensitivity. 42 In a similar experimental study, the mice treated for 21 days with a drinking solution of methyl mercury (40 mg/L) showed increased total and non-HDL plasma cholesterol levels, supporting the concept of mercury-induced cardiovascular toxicity. 43
Both population and animal studies signify that mercury exposure leads to CVDs, like hypertension, CHD, myocardial infarction, reduction in HRV, increase in sudden cardiac death, increase in carotid IMT and carotid obstruction, generalized atherosclerosis, renal dysfunction, renal failure and proteinuria, and an overall increase in the total cardiovascular mortality. 1,2,4,28,44 –53
The beginning event that triggers the development of consequent CVD is the functional disruption of the endothelium or endothelial dysfunction, and there is a growing data associating mercury exposure with endothelial dysfunction and higher risk of CVD.
Physiological Role of Endothelium
The vascular endothelium is a single layer of cells lining the inner wall of the vasculature and functions as a regulator of vascular tone by releasing various vasoactive substances: vasodilators (NO, prostacyclin [PGl2], endothelium-derived hyperpolarizing factor [EDHF], bradykinin, adrenomedullin, C-natriuretic peptide) and vasoconstrictors (endothelin 1 [ET-1], angiotensin II, thromboxane A2 [TXA2], prostaglandins, hydrogen peroxide [H2O2], and free radicals).
54
The
The ECs constitutively express a NO synthase (NOS) that generates NO using Endothelium-derived vasoactive factors. In response to shear stress and chemical stimuli, such as acetylcholine (ACh), the constitutively expressed endothelial nitric oxide synthase (eNOS) cleaves the substrate 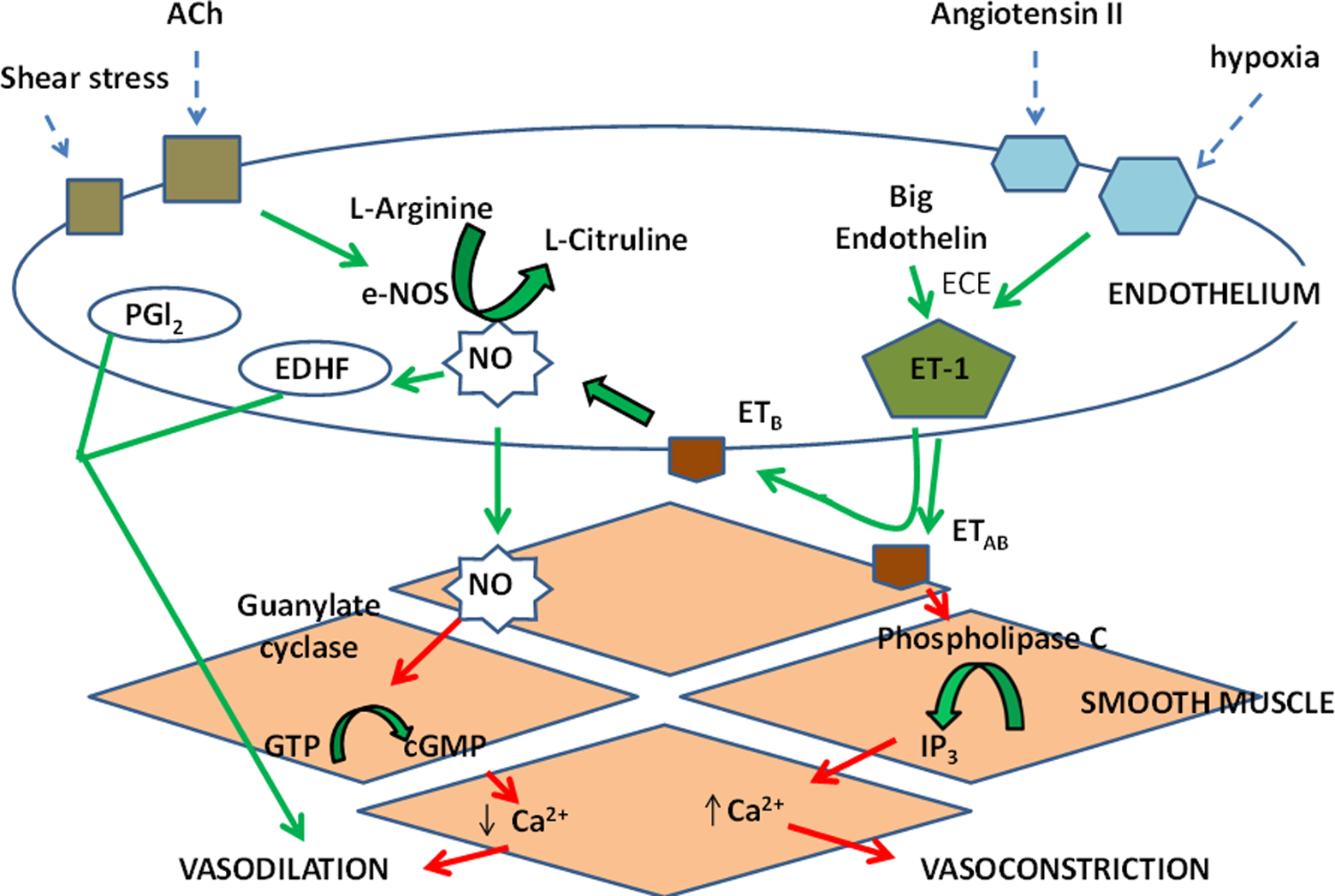
In addition to its function as a vasodilator, the NO released from ECs is also a potential inhibitor of platelet aggregation and causes VSM proliferation and nuclear transcription of leukocyte-adhesion molecules including vascular cell adhesion molecule and intercellular adhesion molecule. 63 Conversely, the endothelium is able to secrete the potent vasoconstrictor ET-1, which through its proinflammatory and mitogenic effects augments the pathogenesis of CVD. 64 Other endothelial-derived vasoconstrictors include prostaglandin H2, TXA2, and ROS.
Phospholipids of cellular membranes play an important role as the structural and functional entities in the ECs. Phospholipases are enzymes that specifically hydrolyze the membrane phospholipids and generate bioactive lipid second messengers, which play a vital role in cell signaling. 65 Phospholipase D (PLD) is one such signaling enzyme, ubiquitously present in all mammalian cells that preferentially hydrolyzes phosphatidylcholine, generating phosphatidic acid and choline. 65 The PA is further metabolized to either 1,2-diacylglycerol (DAG) or lysophosphatidic acid. 66,67 The formation of DAG activates protein kinase C, which can also contribute to VSM contraction via protein phosphorylation.
Endothelial Dysfunction
Endothelial dysfunction is characterized by a shift in the actions of the endothelium toward reduced vasodilation, a proinflammatory state, and prothrombic properties. 68 It is associated with most forms of CVD, such as hypertension, coronary artery disease, chronic heart failure, peripheral artery disease, diabetes, and chronic renal failure.
Although several processes may lead to endothelial damage, the generation of oxygen-derived free radicals and subsequent lipid peroxidation may be key components in it. Additionally, a fast interaction between NO and superoxide anions results in the formation of peroxynitrite, 69 which is a less potent vasodilator and possesses the ability to initiate lipid peroxidation. 70 The accelerated degradation of NO and increased formation of peroxynitrite reduce the availability of endothelium-derived NO. Hence, the endothelium-dependent vasodilatation is maintained at a reduced level. By doing so, there may also be some contribution from the K+ ATP channel pathway. 71
Mercury Exposure, Oxidative Stress, and Vascular Endothelium
Mercury and its derivatives are known to constrict VSM cells. However, only limited information is available on the role of ECs in mercury-induced vasoreactivity. Chronic exposure to mercury is reported to increase vascular resistance and induce hypertension. 17,72 Despite many proposed mechanisms for this relationship, the definitive pathogenesis remains unclear, possibly because individuals exposed to mercury usually display multiple systemic disorders. The proposed mechanisms are typically described by animal model studies on VSMs. Contrary to previous reports that mercury and its derivatives constrict VSM cells, Golpon and coworkers for the first time demonstrated that in the isolated aortic tissue, mercury had a dual effect in the vasculature. 12 At low concentrations, mercury caused an endothelium-dependent vasorelaxation. At higher concentrations, mercury altered the structure and function of vascular endothelium and vasoconstriction was observed. Similar observations were reported by our group ascertaining that mercury exposure produces a biphasic response—vasorelaxation at lower concentrations and vasoconstriction at higher concentrations. 73
The ECs secrete oxygen-derived free radicals and H2O2 in response to stress. 74 Superoxide anion inactivates NO, 69 resulting in vasoconstriction of arteries. 75 Reactive oxygen species may also facilitate the mobilization of cytosolic Ca2+ in VSM cells 76 or promote Ca2+ sensitization of the contractile elements. 77 The vascular endothelium is very sensitive to metal toxicity–induced oxidative stress. 78 –80 Increase in ROS at high concentrations of mercury chloride results in the cytotoxicity of the EC monolayers, leading to severe EC dysfunction. 81 Correspondingly, Park and Park 82 have also reported that mercury chloride increases ROS and apoptosis in bronchial epithelial cells.
In bovine pulmonary artery ECs, it has been reported that mercury ions induce oxidative stress through depletion of GSH and inactivation of thiol enzymes.
81
Oxidative stress in vascular ECs on mercury exposure induces the activation of PLD due to the generation of ROS, which results in the generation of DAG, a second messenger for vasoconstriction.
83
It has been previously documented that vascular ROS production, plasma malondialdehyde levels, and total antioxidant status increase after both acute and chronic mercury exposure in rats. 11,14,42,73,85 ,87 Low-dose mercury exposure alters the coronary vascular reactivity because of endothelial dysfunction. This at least in part is because of increased ROS production, which is due to the increase in both nicotinamide sdenine dinucleotide phosphate oxidase (NOX): NOX-1 and NOX-4 subunits, suggesting the involvement of NADPH oxidase. A decrease in antioxidant defenses would also contribute to the increased superoxide production observed after mercury treatment. 86 Contrarily, several authors have reported augmented antioxidant defenses after acute and chronic mercury exposure. 3,14,79 It is therefore possible that antioxidant mechanisms might become activated in mercury-exposed rats to protect cells against the increased oxidative stress. The evidence available suggests that antioxidants may play an important role in abating some health hazards of heavy metals.
Mercury Exposure, Oxidative Stress, and NO Signaling
Furchgott and Zawadzki (1980) discovered that the endothelium releases a substance that relaxes the underlying VSM, which was later shown to be NO or a related compound. 55,63,87–89
Studies on isolated aortic rings as previously mentioned indicate that mercury produces a dual/bipasic response. Mercury at higher concentrations alters the structure and function of vascular endothelium and produces vasoconstriction, which is ameliorated by antioxidants super oxide dismutase (SOD), catalase, and
In another in vitro mercury-exposure study, mercury modulated the vascular reactivity. An increase in vascular response to phenylephrine was observed as a result of reduced NO bioavailability due to increased release of ROS. 85
The interplay between NO and oxidative stress was further validated by an acute exposure study of methyl mercury chloride (5 mg/kg, by mouth) in rats. Oxidative stress was produced along with an increase in serum NO levels. A significant increase in the acetylcholine vasodilator response was observed. This effect was mediated by increased production of NO because of the stimulation of eNOS. It is interesting to note that this increase occurred even when there was oxidative stress, suggesting a state of inclination toward NO
90
(Figure 2).
Effect of low/high mercury exposure on endothelium-derived vasoactive factors. In response to low mercury exposure (↑), an increase in the nitric oxide synthase (eNOS) activity/expression causes an increase in nitric oxide (NO) with no effect on prostacyclin (PGl2) and endothelium-derived hyperpolarizing factor (EDHF). Low mercury exposure also causes oxidative stress, and a fast interaction between NO and superoxide anions results in the formation of peroxynitrite, which causes vasoconstriction. In response to high mercury exposure (↑), decrease in the eNOS activity/expression causes decrease in NO with persistence or upregulation of EDHF. High mercury exposure also causes oxidative stress and a fast interaction between NO and superoxide anions, resulting in the formation of peroxynitrite, which causes increase in vasoconstriction.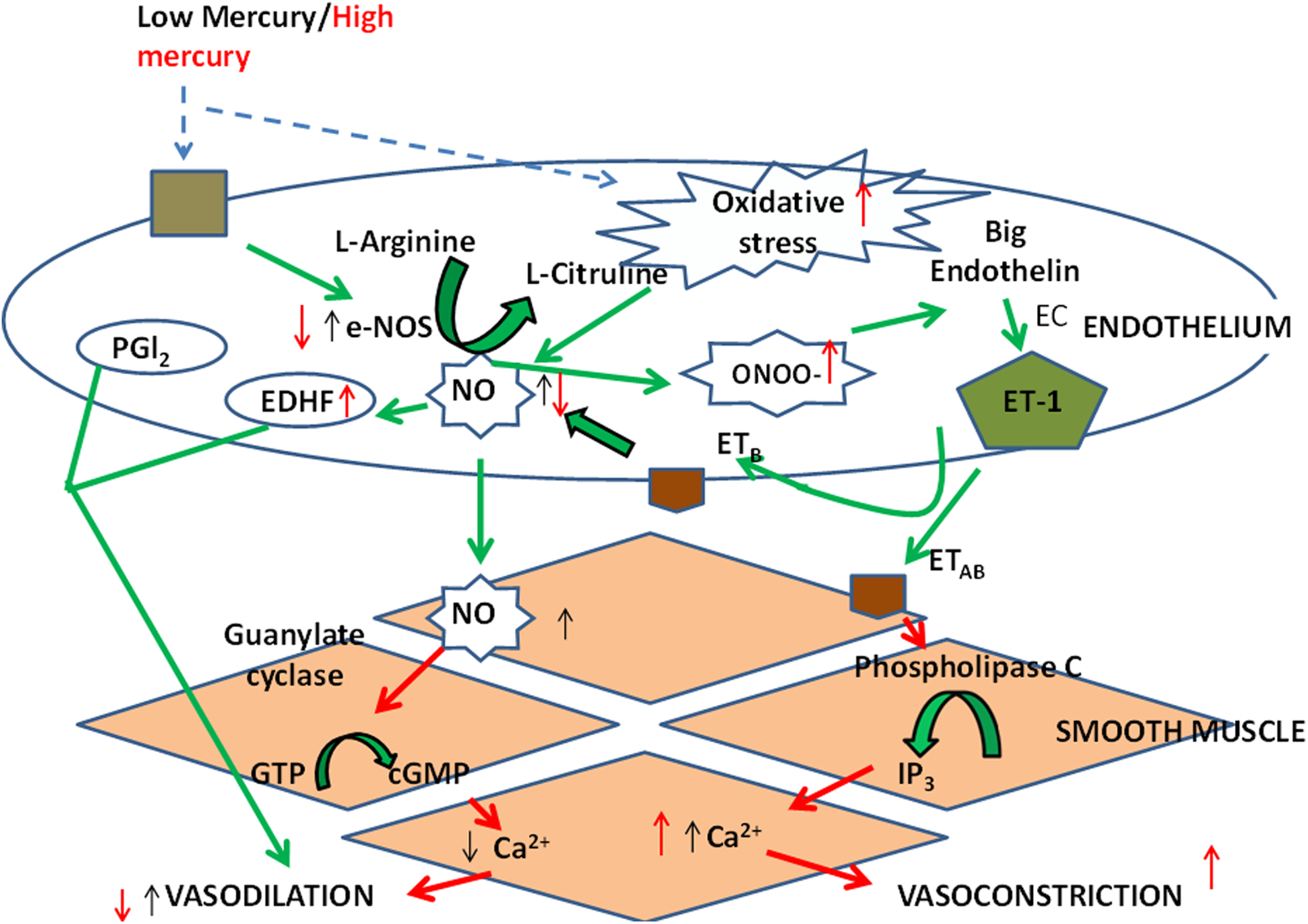
Contrary to above, an opposite interaction between NO and oxidative stress was observed in a chronic study of healthy Wistar rats exposed to inorganic mercuric chloride in drinking water for 30 days. Oxidative stress accompanied with increased NO levels and endothelial dysfunction was observed. Although there was an increase in the serum NO level, endothelial dysfunction was detected. As there was a significant increase in free radical production, the free radicals must have interacted with NO and reduced the bioavailability of NO. Reduced NO bioavailability and oxidative stress resulted in endothelial dysfunction. The EDHF pathway was relatively resistant to mercury exposure, suggesting that there may be an upregulation of K+ ATP channels in order to maintain circulation to compensate the attenuated NO-mediated vasodilatation (Figure 2). 11
Conclusion
Vascular endothelium is highly sensitive to oxidative stress, and this stress is the main cause of the endothelial dysfunction observed in CVDs such as hypertension and atherosclerosis. 88 The route, dose, and period of mercury exposure play an important role in determining its harmful effect on the vascular endothelium. Mercury produces a biphasic response in the VSM—vasorelaxation and vasoconstriction. It appears that a delicate balance exists between free radicals and NO released by ECs on mercury exposure. When there is an increase in NO release, enhanced endothelial function is observed. When the balance is tipped in favor of oxidative stress, endothelial dysfunction is observed. Therefore, NO-signaling mechanism and oxidative stress play an important function in the mercury-induced CVDs in the populations exposed to mercury.
Footnotes
Author Contribution
Swati Omanwar contributed to conception and design; contributed to acquisition and interpretation; drafted manuscript; and critically revised the manuscript. M. Fahim contributed to conception and revised the manuscript for important intellectual content. Both authors gave final approval and agreed to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.