
Abstract
Myocardial infarction, commonly known as heart attack, evolves from the rupture of unstable atherosclerotic plaques to coronary thrombosis and myocardial ischemia–reperfusion injury. A body of evidence supports a close relationship between the alterations following an ischemia–reperfusion injury-induced oxidative stress and platelet activity. Through their critical role in thrombogenesis and inflammatory responses, platelets are fully (totally) implicated from atherothrombotic plaque formation to myocardial infarction onset and expansion. However, mere platelet aggregation prevention does not offer full protection, suggesting that other antiplatelet therapy mechanisms may also be involved. Thus, the present review discusses the integrative role of platelets, oxidative stress, and antiplatelet therapy in triggering myocardial infarction pathophysiology.
Introduction
Cardiovascular diseases (CVDs) consist of heart and blood vessel disorders, which include, but not limited to, thrombosis, peripheral arterial disease, cerebrovascular disease, and coronary heart disease (CHD). 1 In addition to being the leading cause of death worldwide, the prevalence of CVDs is increased by other risk factors, such as increased life expectancy, a sedentary lifestyle, and unhealthy diets and associated metabolic diseases. 2,3 A myocardial infarction (MI) is a common acute event in CVDs that occurs when the tissue is damaged due to the arrest of blood perfusion with the consequent cessation of oxygen and nutrient supply, as well as the accumulation of toxic substances. 4 In general, MI evolves from the rupture of unstable atherosclerotic plaques to coronary thrombosis and myocardial ischemia–reperfusion (I/R) injury. 5 During acute myocardial ischemia, the lack of oxygen switches cell metabolism to anaerobic respiration, contributing to increased intracellular and mitochondrial calcium levels. Moreover, cell swelling and rupture as well as cell death by necrotic, necroptotic, apoptotic, and autophagic mechanisms occur. 6 When oxygen levels are restored, reperfusion results in reactivation of the electron transport chain and reactive oxygen species (ROS) over generation in cardiomyocytes, in addition to pro-inflammatory neutrophils infiltration in the injured tissue, exacerbating the damage. 7 In this context, platelets play a critical role in the thrombogenic and inflammatory responses to myocardial I/R injury. Platelet activation leads to the release of pro-inflammatory, mitogenic, and proapoptotic molecules and cytotoxic agents, in addition to the interaction with leukocytes and endothelial cells triggering myocardial I/R injury onset and expansion. 8 –12 Alterations in platelet hemostatic properties, as well as their role in the immune response, may favor the expansion of damage at the infarct zone besides well-known activities on the development of atherothrombotic plaques. 13 Thus, the current review revises the role of platelets, oxidative stress, and antiplatelet therapy in the context of MI pathophysiology.
Cardiovascular Diseases and MI
Cardiovascular diseases are the leading cause of death worldwide and a major barrier to human development. 2,3 Globally, the number of deaths due to CVDs was 17.9 million people, representing 31% of all global deaths in 2016. 3,14,15 Among them, CHD was the leading cause of CVDs. The effects of CHD are usually attributable to the detrimental effects of acute I/R injury. Ischemia–reperfusion injury typically arises in patients presenting an acute ST-segment elevation myocardial infarction (STEMI). 6,16,17 Heart failure occurs in about 22.1% of all patients with acute MI and the number of cases increases in patients with prior acute MI, worse renal function, and no stenting. 18 In addition, a ventricular wall rupture is a fatal acute MI complication with a mortality rate of 70% to 90%. 19,20 Myocardial infarction is usually due to alterations in the arterial wall or thrombotic occlusion of a coronary vessel caused by the rupture of a vulnerable plaque. 4 Acute MI indicates irreversible myocardial injury, resulting in necrosis in a significant portion of myocardium (generally >1 cm). 21 The size of the infarction is a determinant for the physiopathological events that occur after MI. 22
Oxidative Stress and MI Expansion
Reactive oxygen species are generated as aerobic metabolism byproducts that can physiologically act as signaling molecules in the control of cellular and tissue homeostasis, cell division, migration, contraction, and the production of both lipid and protein mediators. 23 Several cardiovascular enzymatic ROS-generating systems have been reported, such as the phagocytic and nonphagocytic NADPH oxidase (NOX) enzymatic complex and its homologues, uncoupled endothelial nitric oxide synthase, and xanthine oxidase (XO). 24 Excessive production of ROS is generally compensated by plasmatic or cellular antioxidant systems, such as the ascorbate/α-tocopherol pair, glutathione, glutathione peroxidase, heme oxygenase 1, superoxide dismutase 1 and 2 (SOD1 and SOD2), and catalase, among others. 25 –28 However, several cardiovascular risk factors favor the imbalance between pro-oxidant and antioxidant systems in favor of the former, leading to a state called “oxidative stress,” 29 which plays a capital role in the pathogenesis of CVDs. 8,9
Attenuation of increased myocardial ROS may have, at least in part, beneficial effects on left ventricular remodeling after failure. 30 –32 In this regard, hydrogen peroxide (H2O2) can directly induce myocardial tumor necrosis factor α (TNF-α) production via the p38 mitogen-activated protein kinase (MAPK) pathway and, in turn, mediate myocardial dysfunction and apoptosis. 33 Similarly, aortic superoxide anion (O2 ·−) generation via the p38-MAPK pathway was significantly enhanced in rats with heart failure accompanied by the elevation in the protein levels of the NOX subunit p47phox. 34 Meanwhile, persistent increased hydroxyl radical (·OH) production has been observed in post-MI patients showing progression to heart failure. 35 The described oxidant species are able to oxidize the low-density lipoprotein (LDL), thus increasing the levels of circulating oxidized LDL (oxLDL), a marker which has been reported to be associated with the development of CVDs. 36 Reactive oxygen species participates as second messengers in myocardial signaling events by MAPKs, small GTP-binding proteins, the Src family of tyrosine kinases, and cytokines that activate the nuclear transcription factors related to the stress, which leads to cell hypertrophy and apoptosis. 37 In addition, the activation of the antioxidant signaling pathway Nuclear factor erythroid 2-related factor 2 (Nrf 2) by adenosine monophosphate-activated protein kinase in cardiomyocytes protects against hypoxia/reoxygenation injury by reducing ROS production, apoptosis, and production of pro-inflammatory molecules, thus decreasing hypoxia/reoxygenation injury. 25,26
Role of Platelet Oxidative Stress in MI
Platelets are nuclear pieces of megakaryocyte cytoplasm approximately 1.5 to 3 μm in diameter. 38 The normal function of circulating platelets during the hemostatic process is to stop blood loss after a tissue trauma. 39,40 In response to a vascular injury, platelets present a sequence of events, including adhesion, activation, secretion, and aggregation. 41 However, normal platelet function is altered under pathological conditions when prothrombotic stimuli are present, thus increasing the risk of suffering acute events associated with CVDs. 42 –45 During MI, platelet adhesion to the compromised endothelium is exacerbated due to increased local oxidative stress and platelet reactivity. This contributes to the progression of damage in conjunction with the release of oxidizing species and prothrombotic mediators derived from both endothelial cells and platelets. 46,47
NADPH oxidase isoforms are the primary source of ROS and have been consistently implicated in different CVDs. 48,49 Active NOX complexes are composed of multiple subunits comprising of catalytic (NOX 1-5) and regulatory (p22phox, p40phox, p47phox, p67phox, NoxO1, NoxA1, and the small GTPases Rac 1 and Rac 2) components, whose expression may vary according to cell type. 50 In platelets, NOX2 was identified through the detection of the gp91phox and p22phox membrane subunits as well as the cytosolic p47phox. 51,52 Similarly, NOX1 is also expressed in human platelets, although to a lesser extent when compared to NOX2. 53 Delaney and colleagues 54 compared the differential functions of the NOX1 and NOX2 isoforms in platelet activation and thrombosis in a mouse model. This study shows that NOX2, but not NOX1, is needed for thrombi formation without showing alterations in the bleeding time of the tail for any of the enzymes. 54 More recently, the differential response and ROS generation by NOX1 and NOX2 was determined when platelets were activated with thrombin or collagen. 55 Although intracellular-generated ROS by NOX1 is important for platelet activation by collagen, dismutation of NOX2-derived O2 ·− to H2O2 is the main stimulus for platelet activation by thrombin. Moreover, the platelet response to oxLDL requires preactivation of platelets through both NOX1 and NOX2. 55
In addition to NOX enzymes, other enzymatic and nonenzymatic pathways are involved in the formation of ROS in platelets that can contribute to oxidative stress, as mentioned above. 56,57 Overall, the mentioned mechanisms participate in the cellular damage associated with MI are also associated with increasing oxidative stress and boosting the platelet response by different agonists. 8,58 –60 Moreover, O2 ·− or H2O2 also acts as second messenger in platelet activation via calcium mobilization, nitric oxide (·NO) inactivation, and isoprostane formation from nonenzymatic oxidation of arachidonic acid. 61,62 Under ischemic heart conditions, an increase in platelet XO activity and lipid peroxidation have been reported. 63 In addition, there is a decrease in antioxidant enzyme activities that are able to eliminate or decrease the levels of free radicals: SOD, catalase, glutathione peroxidase, and glutathione reductase. 64 Plasma and platelet antioxidant activity decrease in patients with CAD and associated with increased platelet aggregability. 65 –67
Thioredoxin and thioredoxin-like proteins, such as those pertaining to the protein disulfide isomerase (PDI) family, are also of relevance for oxidative milieu during MI. In platelets, PDI has a capital role for platelet aggregation through the isomerization of a disulfide bond in the αIIbβ3 integrin, which is considered the most important component and final convergent pathway in virtually all platelet aggregation mechanisms. 68 Moreover, PDI is also implicated in the function of other integrins, such as the collagen receptor α2β1 69 and the von Willebrand factor receptor glycoprotein 1bα. 70 Notwithstanding, it has been shown that PDI acts as a modulator for distinct members of NOX enzymes in the vascular system. 71 There are close associations between PDI and NOX1 71 –73 and phagocytic NOX2 74,75 and NOX4. 72,76 Specifically to NOX2, PDI regulates its function possibly through mechanisms involving thiol groups on its various subunits and thereby contributing to ROS generation. 71
Having said that, it is clear that the oxidative stress generated during MI induces a reactive and activated state in circulating platelets participating in the atherothrombotic plaque formation, MI, and MI expansion (Figure 1).

Role of platelets and oxidative stress in the evolution of myocardial infarction (MI). Different ways by which platelet activation promotes infarct expansion: (i) Platelet oxidative stress. In an MI mouse model, the physiologically relevant platelet agonist thrombin, thromboxane-A2, and exogenous ROS have significant pathologic roles. Extracellular signal-regulated kinase 5 (ERK5, a member of mitogen-activated protein kinase family) is activated in platelets, which sense the changes in the levels of these mediators and especially in the persistent cardiac ischemia. 147 Reactive oxygen species are usually produced during the reperfusion phase. An in vitro study in human platelets showed that when exposed to anoxia and reoxygenation, an induction of ROS occurs related to spontaneous platelet aggregation. 148 NADPH oxidase 2 (NOX2) would be behind the increase in platelet ROS levels induced by reoxygenation after anoxia. 149 Platelet activation would be given by the ability of ROS to activate phospholipase A2 and the cyclooxygenase pathway, which are involved in the synthesis of the proaggregant thromboxane A2 from arachidonic acid oxidation. 150 Considering these facts, both increased antioxidant defenses and, using NOX2 or phospholipase A2 inhibitors, reverse the effects induced by anoxia–reperfusion. 148,149 In this sense, in patients under percutaneous coronary intervention (PCI) that is a typical in vivo model of ischemia–reperfusion, there is an increase in thromboxane B2 (the detectable product derived from thromboxane A2 decay), isoprostanes, and soluble NOX2-derived peptide, a marker of systemic NOX activation after PCI. Moreover, an infusion of antioxidant (SOD and vitamin C) induces a reversion on the observed proaggregant mediator increase. 148 (ii) Matrix metalloproteinases (MMPs): MMPs are a family of proteases responsible for degrading extracellular matrix molecules as well other bioactive molecules like receptors and cytokines. 151 Matrix metalloproteinase 9 is a collagenase that has implications in tissue remodeling before infarction, which is involved in adjacent tissue damage contributing to infarct expansion. 152 High levels of MMP-9 have been related to heart failure in patients with MI 153 and selective silencing of MMP-9 has been proposed for myocardial protection. 154 Although neutrophils are recognized as the main source of MMP-9 at infarcted areas, recent studies suggest that platelets may also contribute significantly. Matrix metalloproteinase 9 content was decreased in mice platelets after 3 days of artery ligation-induced MI, indicating that platelets release MMP-9 post-MI. Furthermore, inhibition of platelet MMP-9 release reduces the protease presence and activity at the injured tissue, reducing myocardial remodeling and contributing to achieve a protective phenotype. 147 Overall, considering that platelets are able to release all these mediators, blocking platelet aggregation in combination with the secretion of granules and proteins could be a promising therapeutic strategy in acute MI. 155 (iii) Platelet–leukocyte interaction: In an in vitro model of ischemia and reperfusion, platelets mediate leukocyte infiltration in reperfused vessels, acting as a bridge between leukocytes and endothelium. 156,157 The mechanisms by which platelet thrombus control leukocytes migration to vascular injury sites involves a gradient of platelet-derived chemokine CXCL7 (also known as NAP-2), a chemokine that binds to CXC chemokine receptor I/2 (CXCRI/2) at leukocytes promoting cells’ intravascular migration through platelet thrombi. 158
Atherothrombotic Plaque Formation
Platelets play a critical role during the development of a cardiac event. Firstly, they participate in the formation of atherothrombotic plaques, whose rupture is capable of generating arterial thrombosis and infarction. 77 In conditions that represent a risk factor for CVD development, there is an increase in platelet activation markers. There is also an increase in P-selectin and αIIbβ3 integrin membrane expression, among others. These modifications are associated with oxidative stress in addition to a platelet hyperreactivity state. 62,78 In obese patients, there are an increase in P-selectin and sCD40L plasma levels associated with an increased NOX2 activity, 79 oxLDL triggers foam cell formation and accumulation in atherosclerotic plaques, which in turn may favor platelet activation, stimulating a loop of oxidative damage. 80 This, in addition to the observed effects of platelets on the endothelial cells via CD40L, strengthens the nexus between oxidative stress of platelets and atherosclerosis. 81
Myocardial Infarction
Myocardial infarction is the result of destabilization of an atherosclerotic plaque in a coronary artery. 82 Plaque rupture exposes the necrotic core to the circulation (primary thrombogenic substrate) and initiates platelet activation, adhesion and aggregation on the exposed vascular surface, and activation of the clotting cascade, leading to the so-called atherothrombotic stroke. 5,77,83 –86 Thus, platelet activation-dependent thrombus formation plays a crucial role in the pathogenesis of MI by adhering to a ruptured atherosclerotic plaque site. 87 –91 Platelets also actively participate in the early stages of MI: (a) thrombotic occlusion of an epicardial coronary artery at a disrupted/eroded atherosclerotic plaque, (b) microembolization of atherothrombotic platelet-rich aggregates, (c) platelet-mediated vasoconstriction, (d) enhanced intravasal thrombus formation in the microcirculation, and (e) platelet-mediated inflammatory reactions in the ischemic myocardium. These events determine the degree of myocardial ischemia and cardiac contractile dysfunction, where platelets play a key role in the initiation of thrombotic occlusion, promoting tissue damage within the microcirculation of the ischemic myocardium and impair cardiac recovery. 92,93
Plasma thioredoxin levels and platelet aggregability increased concomitantly in patients with acute MI, which was associated with a lower left ventricular ejection fraction. 94 Patients with more extended coronary atherosclerosis have a higher platelet reactivity, which might partly account for a higher risk of periprocedural MI. 95 Approximately one-third of patients suffering an STEMI, even with coronary stenting, develop a “no reflow” phenomenon that is associated with enhanced platelet activity or inadequate platelet inhibition at the time of MI. 96 In addition, these patients with STEMI have less time from diagnosis to angiography and are more likely to undergo percutaneous coronary intervention (PCI) after angiography. 97 Platelet P-selectin expression is substantially enhanced in patients with acute coronary syndrome (ACS), particularly in those with STEMI, and correlates with myocardial necrosis markers (troponin I and creatine kinase-MB). 98 –101
Myocardial Infarct Expansion
Ischemia is a relevant event that occurs during infarction when there is a lack of oxygenation and nutrients in the tissue that causes damage in the affected area. 11 Reperfusion of the area, in contrast to what one would expect, is an additional stress that enhances tissue damage. Platelets accumulate in the reperfused myocardium preferentially at the most damaged areas, hindering blood flow and contractile recovery or increasing myocardial injury. 93,102 –104 The deposition of platelet aggregates in intramyocardial vessels has been associated with unstable angina, a clinical symptom of hypoxia. 105 Studies using animal models have made possible to determine that circulating platelets become activated early during reperfusion and their activation depends on the duration of the preceding coronary occlusion and is proportional to the extent of myocardial injury, 106 while platelets exposed to ischemic conditions induced necrotic injury in an isolated heart. 107
Notwithstanding, reperfusion also stimulates calcium elevation in immune cells, generating the release of phospholipids-derived products and ROS. 108 Platelet-activating factor (PAF) is a mediator derived from phospholipids that increases differentially in the early reperfusion phase compared to the ischemic and preischemic stages. 109 Platelet-activating factor receptor antagonism protects myocardium against reperfusion injury by reversion of platelet hyperaggregability and neutrophils oxidative burst. 110 Platelet-activating factor has been proposed as an important player in reperfusion injury, since its infusion in a nonischemic model allows the platelet–neutrophil interaction that is capable of inducing a dysfunction compared to that caused in ischemic conditions. 111 Deletion of PAF receptor has also exhibited a reverse in the infarction size in a cerebral ischemia model. 112
Response to Antiplatelet Drugs
Antiplatelet therapy is essential in the prevention of MI. 113 One of the gold standards for oral treatment of CVDs is antiplatelet therapy with aspirin 114 that is dependent on the inhibition of the constitutive platelet isoenzyme prostaglandin endoperoxide H synthase 1. 115 However, a significant number of patients experience recurrent cardiovascular events despite being on antiplatelet therapy. 116,117 This could be associated with a poor response to antiplatelet drugs, an event commonly known as resistance to antiplatelet drugs. 116,118 This term is used when a drug cannot exert its pharmacological action due to the inability to reach its target action as a result of the reduction in the bioavailability of the drug, its inactivation in vivo, or its negative interaction with other substances as well as alterations that the target may suffer. 117,119 Oxidative stress may be associated with increased platelet aggregation due to a decreased response to antiplatelet therapy. 120 –122 Aspirin resistance is estimated to be present in 5% to 75% of patients and is related to increased cardiovascular mortality. It has been reported that aspirin resistance in coronary artery disease correlates with an increase in oxidative stress markers (eg, isoprostanes and lipid peroxidation products), but not to the expression of enzymes targeted by their therapeutic action. 123,124
Dual antiplatelet therapy with aspirin and clopidogrel has been the main treatment for platelet inhibition used in patients with ACS. In reperfused myocardium platelets, it promotes the development of an inflammatory environment at the infarcted myocardium, while inhibition with clopidogrel decreases the expression of inflammatory cytokines such as interleukin 1 and TNF-α in infarcted tissue. 102 However, a prospective study has only shown a 20% reduction in adverse vascular events and an increase in hemorrhagic complications with the addition of clopidogrel to aspirin. 125 –128 In addition, the response to clopidogrel and aspirin varies widely among individuals and high platelet reactivity on both aspirin and clopidogrel has been associated with adverse cardiovascular events, including cardiovascular death, nonfatal MI, stent thrombosis, stroke, and sustained ventricular tachycardia. 97,129,130 The data discussed above have clinical limitations, considering the absence, in a significant portion of the patient population (around 11%), of an active CYP2C19 enzyme. 131 This enzyme is required to activate the prodrug clopidogrel, thus those carrying the defect are insensitive to the use of clopidogrel. 131
Experimental studies have shown that platelet aggregation is associated with considerable oxidative status, which may be implicated in platelet nonresponsiveness to dual antiplatelet treatment. 120 –122 In a rabbit model of myocardial I/R injury, both cangrelor and clopidogrel were evaluated. 132 Clopidogrel exhibited a very slow onset of protective action requiring 2 days of treatment before platelets were inhibited and then the hearts were protected. Meanwhile, cangrelor was administered intravenously 10 minutes before reperfusion and reduced the ischemic zone from 38% to 19%. In this context, cangrelor protection was prevented by inhibitors of the postconditioning pathway. Among others, inhibitors of adenosine A2B receptors, extracellular signal-regulated kinase, Akt, redox signaling, or mitochondrial adenosine triphosphate–sensitive potassium channel channels were used. 132 However, in thrombocytopenic rat models, cangrelor did not exert protection. These data suggest that preventing aggregation alone is not protective; thus, the protective mechanism involves the interaction with a specific factor in the platelet. Cangrelor was no longer protective in rats treated with dimethylsphingosine to block sphingosine kinase. 133 Therefore, platelets in cangrelor-treated postischemic hearts enter the interstitial space early in reperfusion and release sphingosine 1 phosphate when they are in contact with collagen. The latter leads to heart postconditioning and protects against reperfusion injury (Figure 2). 134,135
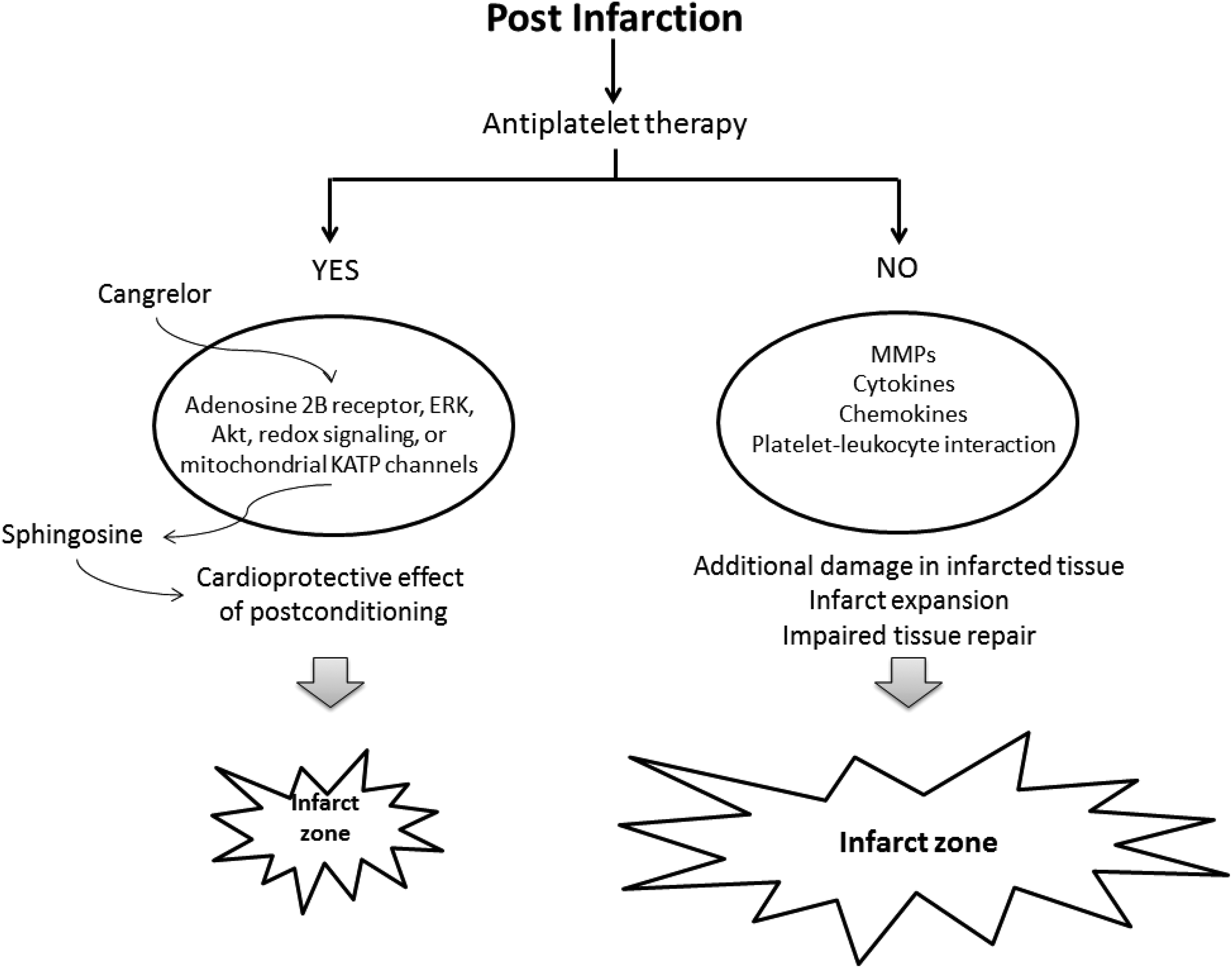
Platelet secretion of mediators that affect the recovery of the infarcted zone and the cardioprotective effect of postconditioning. MMP indicates matrix metalloproteinase.
Ticagrelor, a reversible binding oral P2Y12 receptor antagonist, has recently emerged as an antiplatelet drug with advantages over conventional clopidogrel and aspirin therapy. In a phase III clinical trial, ticagrelor provided better clinical outcomes than clopidogrel in patients with ACS. 136,137 Ticagrelor blocks adenosine diphosphate–induced platelet activation and aggregation, prevents platelet-mediated thrombosis, prolongs reperfusion time, and reduces reocclusion and cyclic flow variation. 138 Of relevance is that it significantly decreases infarct size and rapidly restores myocardial tissue perfusion. 138 In addition, the protective effect of ticagrelor was abolished by chelerythrine and wortmannin, supporting the participation of protein kinase C and PI3-kinase signaling pathways on ticagrelor protective effects. 139 The advantage of this drug, in addition to its effect on platelets, is that it prevents the resorption of adenosine in the infarcted tissue, exerting beneficial effects on patient recovery. In this sense, an increase in adenosine levels at the myocardial level has been shown, which is related to a protective effect on reperfusion injury and decreased infarct size. 140,141 Since adenosine is also capable of inhibiting platelet activation 142 and suppressing ROS generation at the environment of the reperfused tissue, 143 the search for drugs acting on the adenosine pathway would have benefits in patients with MI. The αIIbβ3 integrin inhibitor tirofiban has shown a myocardial improvement in hypoxia/reperfusion-induced injury in rats by decreasing myocardial myocyte apoptosis. This action is related to the reduction in oxidative stress markers in the affected area. 144 Since tirofiban can also decrease thrombin-induced ROS generation in platelets, 145 it could be an interesting drug to test on patients under MI and I/R injury.
Conclusion
A more complete understanding of the mechanisms that lead to deregulated platelet activity in disease states may provide additional therapeutic strategies. There is a close relationship between deregulated platelet activity and oxidative stress during the stages involving CVDs, in particular in the infarcted area, at the I/R stages, and during the expansion of the infarcted area. The persistent effect of platelet hyperreactivity following an acute coronary event and/or PCI provides the rationale for integrating antiplatelet strategies into standard medical management and substantially improving results in patients in this context. 146 Overall, therapies designed to interfere with oxidative stress might be beneficial in preventing myocardial failure.
Footnotes
Acknowledgments
Eduardo Fuentes thanks CONICYT/FONDECYT N° 1180427, CONICYT/REDES N° 170003, and Centro de Estudios en Alimentos Procesados (Chile, Talca). Andres Trostchansky was supported by CSIC-Grupos (N°536), Uruguay. Antonio Marcus de A. Paes is supported by Fundação de Amparo à Pesquisa e Desenvolvimento Científico do Maranhão—FAPEMA (BEPP-02511/18). The authors thank Cesar Sepulveda for his careful and critical reading of our article, especially his participation in the figures.
Author Contributions
Eduardo Fuentes contributed to conception and design and contributed to acquisition, analysis, and interpretation. Rodrigo Moore-Carrasco contributed to design and contributed to analysis. Antonio Marcus de Andrade Paes contributed to conception and contributed to analysis. Andres Trostchansky contributed to conception and design and contributed to analysis. All authors drafted the manuscript, critically revised the manuscript, gave final approval, and agrees to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.