
Abstract
Aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor that regulates multiple cellular processes. The anticancer drug doxorubicin (DOX) can activate AhR-mediated transcription of target genes. Because DOX in cells activates a DNA damage response involving ataxia telangiectasia-mutated (ATM)-mediated activation of p53, we investigated whether the activation of the p53 in cells by DNA-damaging agents such as DOX or bleomycin could regulate the AhR levels. Here we report that activation of p53 by DNA-damaging agents in human cells increased levels of AhR through a posttranscriptional mechanism. Accordingly, fibroblasts from ATM patients, which are defective in p53 activation, expressed reduced constitutive levels of AhR and treatment of cells with bleomycin did not appreciably increase the AhR levels. Further, activation of p53 in cells stimulated the expression of AhR target genes. In murine cells, activation of p53 reduced the levels of AhR messenger RNA and protein and reduced the expression of AhR target genes. Our observations revealed that activation of p53 in human and murine cells differentially regulates AhR levels.
Introduction
The aryl hydrocarbon receptor (AhR), a ligand-activated transcription factor, regulates signaling pathways that are critical for cellular homeostasis and the development of human diseases. 1 –4 These pathways regulate multiple cellular processes, including cell proliferation and differentiation, cell motility and migration, and inflammation. 2,3 Further, epidemiological and experimental animal studies have revealed an association between dysregulation of the AhR functions and the development of certain cancers. 3,4 Additionally, a study has implicated a role for AhR expression in cardioprotection against doxorubicin (DOX; a polyaromatic hydrocarbon and an anticancer drug) treatment-associated toxicity in mice. 5
The AhR mediates many of the responses induced by polyhalogenated and polycyclic hydrocarbons, which are ubiquitous environmental contaminants that cause toxic responses in humans. 1,6 –9 These responses depend on the type of toxin (ligand) and its dose. 1,7 –9 Upon binding to a variety of environmental toxins, including 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and DOX, the AhR and AhR nuclear translocator (ARNT), members of the basic helix–loop–helix family of proteins, form a heterodimeric transcription factor. 2,6 The activated AhR/ARNT heterodimer stimulates transcription of genes that contain a core nucleotide consensus sequence 5′-GCGTG-3′ (the sequence is termed dioxin responsive element or DRE) in the promoter region. 6
Treatment of cells with TCDD activates AhR-mediated transcription and also decreases steady-state levels of the AhR protein in a time-dependent manner. 10,11 The treatment induces ubiquitination of AhR, which is followed by its degradation by proteosomal degradation pathway. 11 Accordingly, inhibition of the proteosomal degradation by ubiquitin proteosomal pathway increases the levels of the AhR protein and the increased levels of AhR correlate well with increased expression of its target genes such as CYP1A1. 12 Inhibition of the activity of extracellular signal-regulated kinase (ERK) by its inhibitors (such as PD98059) increases steady-state levels of AhR by reducing TCDD-induced degradation. 13 Further, the ERK-dependent phosphorylation of AhR targets the carboxyl region of the receptor for its degradation by the proteosomal degradation pathway. 13
Ataxia telangiectasia-mutated (ATM) protein kinase (encoded by the ATM gene) is a key regulator of the cellular response to DNA double-strand breaks. 14 –16 Ataxia telangiectasia (AT) in individuals with ATM gene mutation is characterized by progressive cerebellar ataxia, immunodeficiency, premature aging, and increased cancer incidences. 15,16 Additionally, cells that are derived from individuals with AT are defective in activation of p53 upon treatment with ionizing radiations and certain DNA-damaging agents. 14 –18
The p53 tumor suppressor is either mutated or functionally inactivated in a variety of human cancers. 19,20 The p53 protein acts as a tumor suppressor through inhibiting cell cycle progression and inducing apoptosis in cells. Upon DNA damage, p53 is activated by the ATM protein kinase, which phosphorylates the p53 on Ser-15 residue. 15,18,21 The phosphorylation of p53 leads to its activation and stabilization, resulting in an increase in the p53 protein levels. 21,22 The activated p53 either stimulates (such as the gene that encodes for p21) or suppresses the transcription of its target genes. 19,22,23 Transcriptional regulation of its target genes by p53 is central to its ability to regulate cell cycle arrest, apoptosis, DNA repair, senescence, differentiation, and inhibition of angiogenesis. 20 –23
The lack of AhR expression in murine cardiomyocytes sensitizes them to DOX-induced cardiotoxicity. 5 Because DOX (also called adriyamycin) is a common anticancer drug, 24 which activates p53 through activation of ATM protein kinase, 25 we explored whether activation of p53 in cells by DOX or other DNA-damaging agents (such as bleomycin) could regulate the AhR expression levels. Here we demonstrate for the first time that activation of the p53 by DNA-damaging agents in human and murine cells differentially regulates the AhR levels and, thus, the expression of its target genes. Our observations implicate the ATM-p53-AhR axis in the regulation of DOX-mediated cellular toxicity and organ damage in patients with cancer.
Materials and Methods
Reagents
Protease inhibitors cocktail (complete mini EDTA-free) was purchased from Roche Applied Science (Indianapolis, Indiana). Universal recombinant interferon α (IFN-α) was purchased from R&D Systems (Minneapolis, Minnesota). TCDD and DOX hydrochloride were purchased from Sigma-Aldrich (St Louis, Missouri). 6-Formylindolo(3,2-b)carbazole (FICZ) was purchased from Enzo Life Sciences (Farmingdale, New York). Bleomycin (Zeocin) was purchased from (Life Technologies, Grand Island, New York).
Cell Lines, Culture Conditions, and Treatments
WI-38, human normal fetal lung fibroblasts, at lower population doubling (PD) were obtained from the National Institute of Aging Cell Culture Repository (Coriell Medical for Medical Research, Camden, New Jersey) and maintained in culture as described. 26 Additionally, human fibroblasts (GM00363 and GM00648) from individuals with AT at lower PDs were obtained from the Coriell Medical. All fibroblasts were maintained (5.5% CO2 and ∼21% O2) in DMEM cell culture medium with high glucose (4.5 g/L). The medium was supplemented with 10% fetal bovine serum and antibiotics (Invitrogen, Carlsbad, California). Murine macrophage cell lines (J774.A1 and RAW264.7) were purchased from the American Type Culture Collection (Manassas, Virginia), and these cell lines were maintained as suggested by the supplier. Subconfluent cultures of cells, when indicated, were treated with the indicated concentrations of IFN-α, DNA-damaging agents such as DOX or bleomycin (dissolved in phosphate-buffered saline [PBS]), or AhR ligands such as TCDD or FICZ (dissolved in PBS).
Saos2 (human osteosarcoma cells) that stably expressed a temperature-sensitive mutant (Val138) of the human p53 with Arg72 (cells indicated as SaosArg72) were generously provided by Dr Maureen Murphy (Fox Chase Cancer Center, Philadelphia, Pennsylvania). 27,28 For activation of the p53 in SaosArg72 cells, cells were incubated at 32°C (p53 favoring a wild-type conformation).
Antibodies
Antibodies specific for human AhR (sc-74572), IFI16 (sc-8023), p53 (sc-126), and p21 (sc-397) proteins were purchased from Santa Cruz Biotechnology (Santa Cruz, California). Antibodies to detect murine AhR (BML-SA550) and human CYP1A1 (BML-CR3120) were purchased from Enzo Life Science. Antibodies for β-actin (cat # 5125), STAT1 (#9172), p-p53 (Ser-15; #9284), ATM (#2873), and p-ATM (#4526) were purchased from Cell Signaling Technology (Danvers, Massachusetts). Horseradish peroxidase-conjugated secondary antimouse (NXA-931) and antirabbit (NA-934) antibodies were from GE Healthcare Biosciences (Piscataway, New Jersey).
Immunoblotting
Cells from subconfluent cultures were lysed with radioimmunoprecipitation assay buffer (50 mmol/L Tris-HCl (pH 7.4), 150 mmol/L NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate). The buffer was supplemented with complete mini EDTA-free protease inhibitor cocktail. Lysates were centrifuged at 12 000g for 5 minutes at 4°C. Cell lysates containing equal amounts of protein (˜50 μg) were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis, transferred to a polyvinylidene difluoride (from Millipore, Billerica, Massachusetts, USA) membrane, and immunoblotted according to the manufacturer’s guidelines.
RNA Isolation and Polymerase Chain Reaction
Total RNA was isolated from the cells with Trizol reagent (Invitrogen). Complementary DNA synthesis, reverse transcription-polymerase chain reaction (RT-PCR), and quantitative real-time TaqMan PCR were performed to detect the expression of the genes as described. 29 The following primers were used for RT-PCR: the human actin (forward: 5′-GCTCGTCGTCGACAACGGCTC-3′; reverse: 5′-CATGATCTGGGTCATCT TCTC-3′), human AhR (forward: 5′-CTGGCAATGAATTTCCA AGGGAGG-3′; reverse: 5′-CTTTCTCC AGTCTTAATCATGCG-3′), human CYP1A1 (forward: 5′-TTCATCCCTATTCTTC GCTAC-3′; reverse: 5′-TCCATCAGCATCTATGCC-3′), murine AhR (forward: 5′-CCGTCCATCCTG GAA ATT CGAACC-3′; reverse: 5′-CCTTCTTCAT CCGTTAGCGGT CTC-3′), and murine CyP1A1 (forward: 5′-TTGCCCTTCA TTGGTCACAT-3′; reverse: 5′-GAGCAGC TCTTGGTCATCAT-3′). Other RT-PCR primer pair sequences are available upon request.
Statistical Methods
When indicated, experiments involving immunoblotting and semiquantitative RT-PCR techniques were repeated at least 2 to 3 times. Fold changes in levels of certain proteins and messenger RNAs (mRNAs) are indicated based on a representative experiment (out of 2-3 repeats).
Results
Activation of p53 in Human Fibroblasts Increases AhR Levels
Treatment of rat cardiomyocytes with DOX activates AhR-mediated transcription of its target genes, including the CYP1A1. 5 Because DOX is a DNA-damaging agent and can activate the ATM-p53 pathway, 25 we investigated whether activation of p53 in human diploid fibroblasts (HDFs) could also regulate the expression of AhR (in addition to its activation). As shown in Figure 1A, treatment of WI-38 HDFs with a water-soluble ligand of AhR (FICZ) did not measurably affect constitutive levels of activated ATM (P-ATM), ATM, activated p53 (PS-p53), p53, and AHR (compare lane 2 with 1). However, treatment of HDFs with DOX activated increased levels of activated ATM (P-ATM), PS-p53, and p53. Notably, the activation of the ATM-p53 axis in cells by DOX increased steady-state levels of AhR ∼2- to 3-fold (compare lane 3 with 1). Expectedly, treatment of cells with FICZ reduced DOX-treatment induced levels of the AhR (compare lane 4 with lane 3). Significantly, treatment of cells with both DOX and FICZ did not appreciably decrease the AhR levels (compare lane 4 with lane 3). Interestingly, treatment of WI-38 HDFs with increasing concentrations of DOX did not change the steady-state levels of the AhR mRNA (Figure 1B, compare lanes 2 and 3 with lane 1). Similarly, treatment of cells with FICZ also did not change AhR mRNA levels (data not shown).
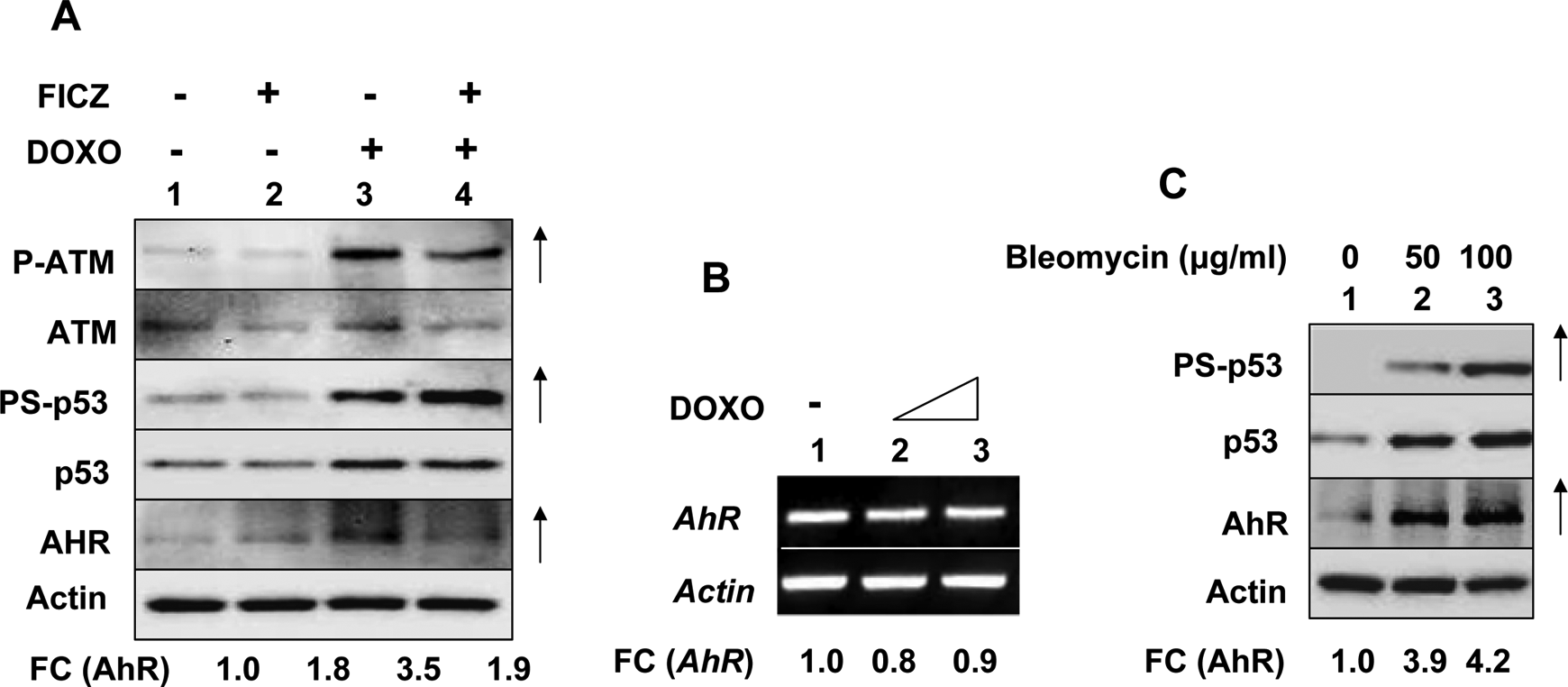
Activation of p53 in human fibroblasts increased AhR levels. A, Subconfluent cultures of human normal WI-38 fibroblasts at an early passage (passage 12) were either left untreated (lane 1), treated with doxorubicin (1 μmol/L, lane 2), 6-formylindolo(3,2-b)carbazole (FICZ; 10 nmol/L, lane 3), or both (lane 4) for 18 hours. After the treatments, total cell lysates containing equal amounts of proteins were analyzed by immunoblotting using antibodies specific to the indicated proteins. An upward arrow indicates appreciable increases in levels of the indicated protein. FC indicates the fold changes in the levels of the indicated proteins. B, Subconfluent cultures of human normal WI-38 fibroblasts were either left untreated (lane 1) or treated with increasing concentrations (1 or 2.5 μmol/L) of doxorubicin (lanes 2 and 3) for 18 hours. After the treatment, total RNA was isolated and analyzed by reverse transcription-polymerase chain reaction (RT-PCR) for steady-state levels of AhR mRNA. FC indicates fold changes in the levels of AhR mRNA. C, Subconfluent cultures of human normal WI-38 fibroblasts were either left untreated (lane 1) or treated with the indicated amounts of bleomycin (lanes 2 and 3). After the treatment, total cell lysates containing equal amounts of proteins were analyzed by immunoblotting. An upward arrow indicates appreciable increases in levels of the indicated protein. FC indicates the fold changes in the levels of AhR. These experiments were repeated 2 to 3 times with similar results. AhR indicates aryl hydrocarbon receptor; mRNA, messenger RNA.
We also tested whether treatment of WI-38 cells with bleomycin, an inducer of DNA double-strand breaks that mimics the ionizing radiation-induced DNA damage in cells, 30 could regulate levels of AhR. As shown in Figure 1C, treatment of cells with indicated concentrations of bleomycin, which activated p53 (as determine by Ser-15 phosphorylation of p53 and increased levels of p53), also increased steady-state levels of the AhR ∼4-fold in a dose-dependent manner (compare lanes 2 and 3 with lane 1). Together, these observations indicated that activation of p53 in human WI-38 fibroblasts by DNA-damaging agents such as DOX or bleomycin increased steady-state levels of AhR without increasing the AhR mRNA levels.
Defects in p53 Activation in AT Fibroblasts Reduced Constitutive and Bleomycin-Induced AhR Levels
Treatment of human WI-38 HDFs with DNA-damaging agents such as DOX is known to activate p53 through activation of ATM protein kinase, a key regulator of the cellular response to DNA double-strand breaks. 15,18,25 Therefore, our observation that treatment of WI-38 HDFs with bleomycin that activated p53 increased levels of AhR (Figure 1C) prompted us to investigate whether treatment of WI-38 HDFs with DOX activates the ATM-p53 axis. As shown in Figure 2A, treatment of cells with DOX resulted in activating phosphorylation of both ATM and p53 and increases in steady-state levels of AhR (compare lane 2 with 1). These observations encouraged us to test whether AT HDFs, which are defective in p53 activation by the ATM protein kinase, 14,31 are defective in the regulation of AhR levels following their treatment with bleomycin. As shown in Figure 2B, constitutive levels of the AhR were appreciably lower in extracts from AT HDFs than WI-38 HDFs (compare lanes 3 and 5 with lane 1). Further, treatment of HDFs with bleomycin, which robustly activated p53, increased levels of p21 and AHR in WI-38 HDFs, did not measurably increase the AhR levels in AT HDFs (compare lanes 4 and 6 with lane 2). These observations revealed that the ATM-p53 axis regulates the constitutive and bleomycin-induced levels of the AhR in human HDFs.
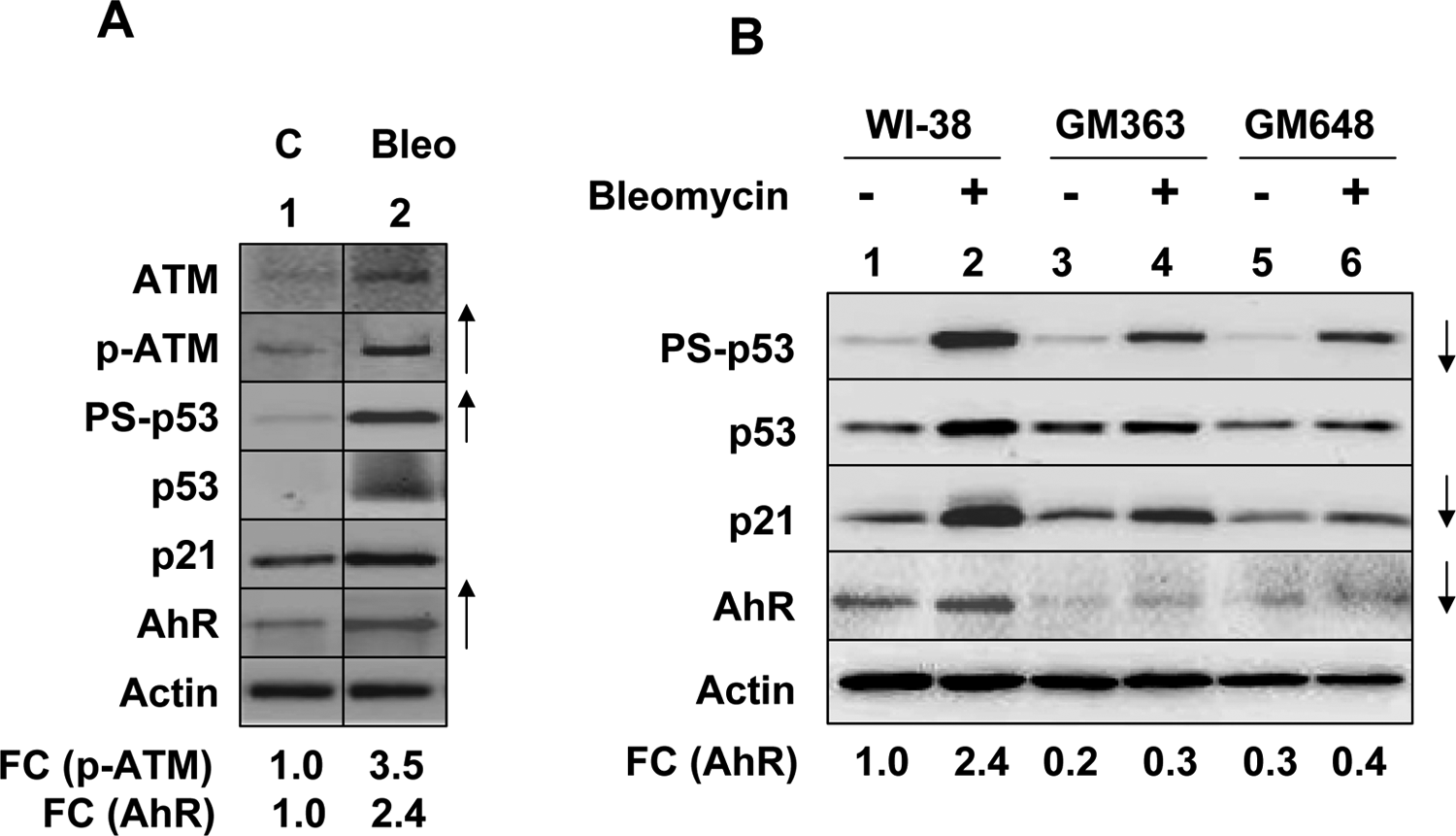
Defects in p53 activation in AT fibroblasts are associated with decreases in basal and bleomycin-induced AhR levels. A, Cultures of human normal WI-38 fibroblasts were either left untreated (lane 1) or treated with bleomycin (100 μg/mL; lane 2) for 18 hours. After the treatment, total cell lysates containing equal amounts of proteins were analyzed by immunoblotting using antibodies specific to the indicated proteins. An upward arrow indicates an appreciable increase in levels of the indicated protein. FC indicates the fold changes in the levels of the indicated proteins. B, Cultures of human normal WI-38 fibroblasts and fibroblasts derived from individuals with AT were either left untreated (lanes 1, 3, and 5) or treated with 50 μg/mL bleomycin (lanes 2, 4, and 6) for 18 hours. After the treatment, total cell lysates containing equal amounts of proteins were analyzed by immunoblotting. A downward arrow indicates an appreciable decrease in levels of the indicated protein. FC indicates the fold changes in the levels of AhR as compared to untreated WI-38 human diploid fibroblasts (HDFs). These experiments were repeated 2 times with similar results. AhR indicates aryl hydrocarbon receptor; AT, ataxia telangiectasia.
Restoration of p53 Function in Saos-2 Osteosarcoma Cells Increased AhR Levels
Expression of a temperature-sensitive mutant (Val138) of human p53 in Saos-2 (a human osteosarcoma cell line, which expresses a mutant p53) cells and incubation of cells at 32°C, but not 39°C, restores the p53 functions. 27,28 Therefore, to further investigate whether the restoration of p53 function in Saos-2 cells could regulate levels of AhR, we incubated Saos-2 cells that expressed the temperature-sensitive mutant of p53 at 32°C for increasing lengths of time. As shown in Figure 3A, the incubation restored p53 function in cells as indicated by p53-mediated increased levels of its target proteins p21 and IFI16. 28 Further, the incubation also increased levels of AhR protein (compare lanes 2 and 3 with lane 1). Encouraged by this observation, we tested whether treatment of Saos-2 cells, which expressed increased levels of AhR (after the restoration of p53 function), with TCDD could regulate levels of AhR. As shown in Figure 3B, treatment of Saos-2 cells with TCDD at 39°C (when p53 is not active) did not appreciably alter constitutive levels of AhR. Interestingly, the steady-state levels of AhR were ∼4- to 5-fold higher in Saos-2 cells at 32°C than 39°C (compare lane 3 with 1) and treatment of cells with TCDD at 32°C appreciably (˜50%) decreased the p53-mediated increased levels of AhR in cells (compare lane 4 with lane 3). These observations suggested that activation of p53 in Saos-2 cells increased the constitutive levels of AhR.
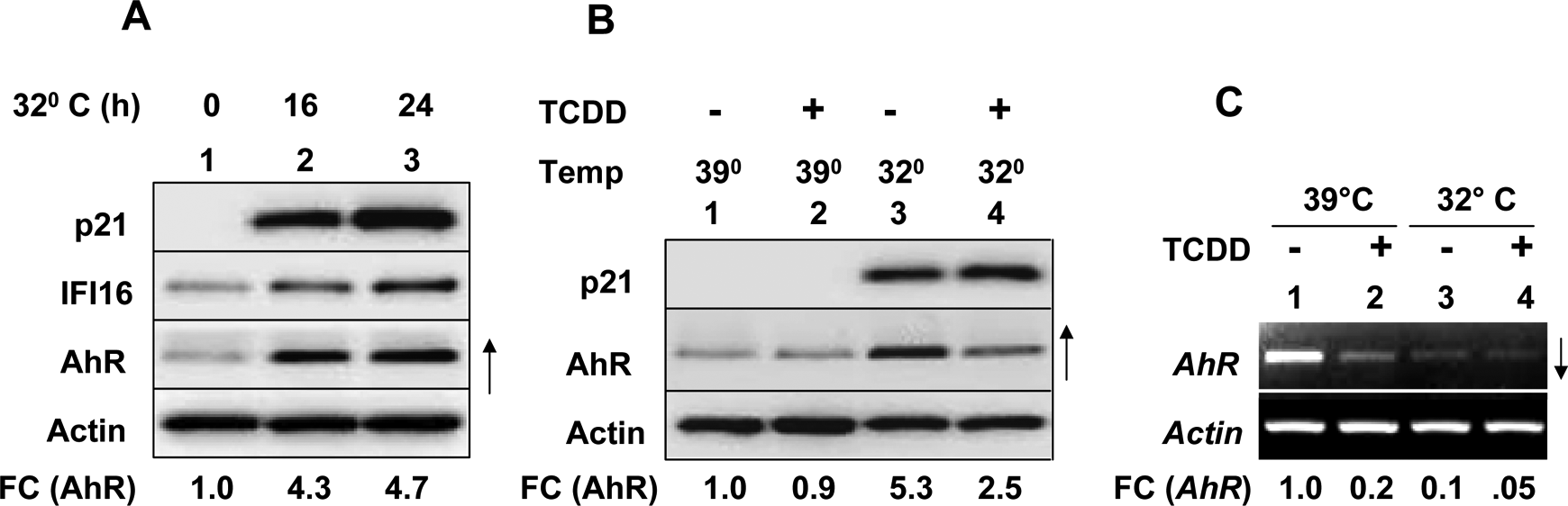
Restoration of p53 function in Saos-2 osteosarcoma cells increases AhR levels. A, Subconfluent cultures of human oseteosarcoma Saos-2 (SaosArg72) cells were either incubated at 39°C (lane 1) or 32°C (lanes 2 and 3) for the indicated time (hours). After the incubations, total cell lysates containing equal amounts of proteins were analyzed by immunoblotting. FC indicates fold changes in the levels of AhR. B, Cultures of SaosArg72 cells were incubated at 39°C (lanes 1 and 2) or 32°C (lanes 3 and 4) without any treatment (lanes 1 and 3) or with TCDD treatment for 24 hours. After the incubations, total cell lysates containing equal amounts of proteins were analyzed by immunoblotting. FC indicates fold changes in the levels of AhR. C, Cultures of SaosArg72 cells were incubated at 39°C (lanes 1 and 2) or 32°C (lanes 3 and 4) without any treatment (lanes 1 and 3) or with TCDD treatment for 24 hours. After the incubations, total RNA was isolated and analyzed by reverse transcription-polymerase chain reaction (RT-PCR). FC indicates the fold changes in the levels of the AhR messenger RNA (mRNA). AhR indicates aryl hydrocarbon receptor; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin.
Treatment of cells with TCDD, an AhR ligand, is known to activate the AhR and induce its degradation through the ubiquitin-proteasome degradation pathway. 10,11 Therefore, we explored whether activation of p53 in Saos-2 cells and their treatment with TCDD could regulate AhR mRNA levels. As shown in Figure 3C, we were able to detect constitutive levels of AhR mRNA in Saos-2 cells that were incubated at 39°C. However, treatment of cells with TCDD appreciably decreased the levels of AhR mRNA (compare lane 2 with 1). Further, incubation of Saos-2 cells at 32°C, which activated the p53 function and increased levels of AhR protein (Figure 3B), reduced steady-state levels of the AhR mRNA (compare lane 3 with 1). Treatment of cells with TCDD further decreased the levels of AhR mRNA (compare lane 4 with lane 3). Together, our observations indicated that restoration and activation of p53 function in Saos-2 cells increased steady-state levels of AhR by a posttranscriptional mechanism involving stabilization of the AhR.
Activation of p53 in Murine Cells Decreased AhR Levels
Our previous observations that activation of p53 in certain human cell types increased levels of AhR protein through a posttranscriptional mechanism prompted us to explore whether activation of p53 in murine cells could also regulate AhR levels. Because our preliminary experiments revealed that murine macrophage cell lines (such as RAW264.7 and J774.A1) express detectable levels of AhR and treatment of cells with IFN-α reduced AhR levels (data not shown), we decided to treat macrophage cell lines with either IFN-α (as a control) or DOX. As shown in Figure 4, treatment of RAW264.7 (Figure 4A) and J774.A1 (Figure 4B) cells with IFN-α, which activated an IFN-signaling (as revealed by an increased levels of the IFN-inducible STAT1 transcription factor), appreciably decreased levels of AhR. Similarly, treatment of RAW264.7 and J774.A1 cells with DOX, which increased levels of p53 and its target protein p21, measurably reduced levels of the AhR protein. Notably, DOX-mediated decrease in AhR levels was ∼2- to 3-fold greater than the decrease that was observed after the IFN-α treatment (Figure 4A and B, compare lane 3 with lane 2). Consistent with the previous observations, treatment of J774.A1 cells with DOX, which increased steady-state levels of p21 mRNA, decreased steady-state levels of AhR mRNA about 4-fold (Figure 4C, compare lane 3 with 1). Further, treatment of cells with DOX also decreased the basal levels of CyP1A1 mRNA (compare lane 3 with 1). These observations indicated that treatment of murine macrophage cell lines with DNA-damaging agents that activate p53 decreases levels of AhR mRNA and protein.
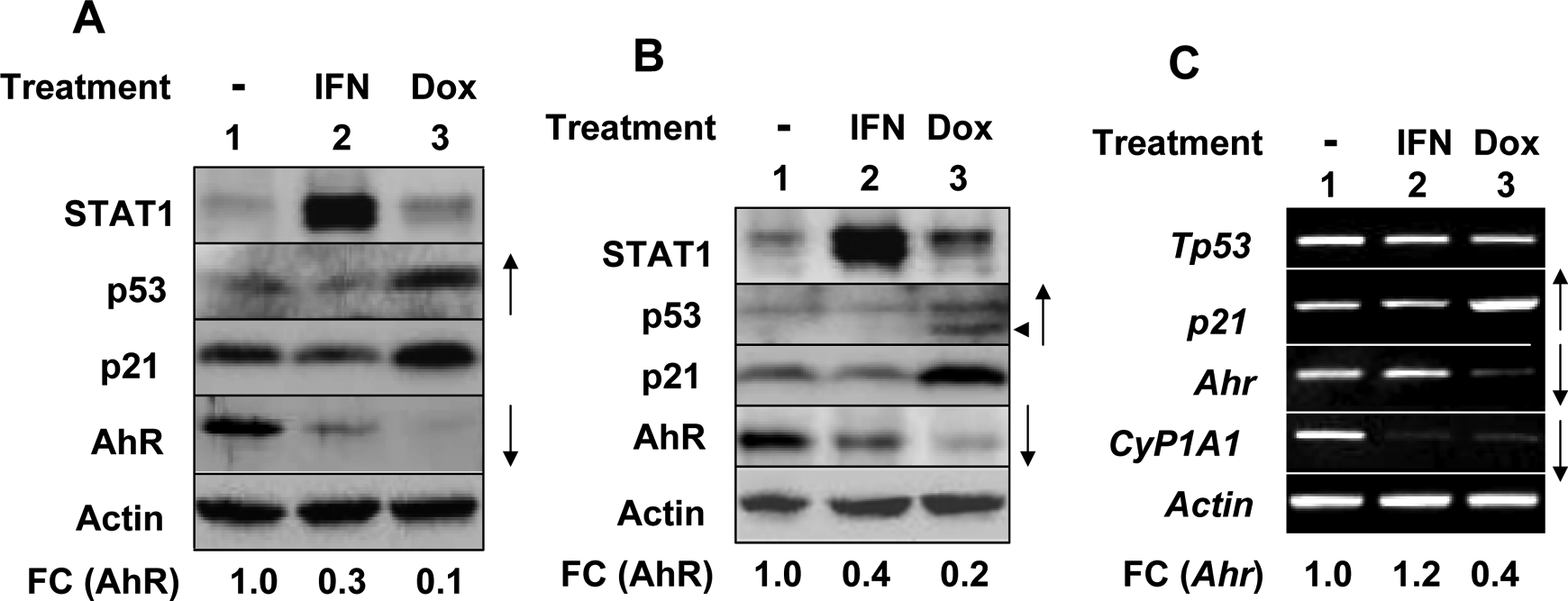
Activation of p53 in murine cells decreases AhR mRNA and protein levels. A, Subconfluent cultures of murine macrophage cell line RAW264.7 were either left untreated (lane 1), treated with universal IFN-α (1000 µg/mL; lane 2), or doxorubicin (10 ng/mL) for 18 hours. After the treatments, total cell lysates were analyzed by immunoblotting for the indicated proteins. FC indicates fold changes in AhR levels. B and C, Subconfluent cultures of murine macrophage cell line J774.A1 were either left untreated (lane 1), treated with universal IFN-α (1000 µg/mL; lane 2) or doxorubicin (10 ng/mL) for 18 hours. After the treatments, cells were harvested and total cell lysates (panel B) or total RNA (panel C) were prepared. Total cell lysates were analyzed by immunoblotting for the indicated proteins. FC indicates the fold changes in AhR levels. Total RNA was analyzed by reverse transcription-polymerase chain reaction (RT-PCR) for levels of the indicated mRNAs. AhR indicates aryl hydrocarbon receptor; IFN-α, interferon α; mRNA, messenger RNA.
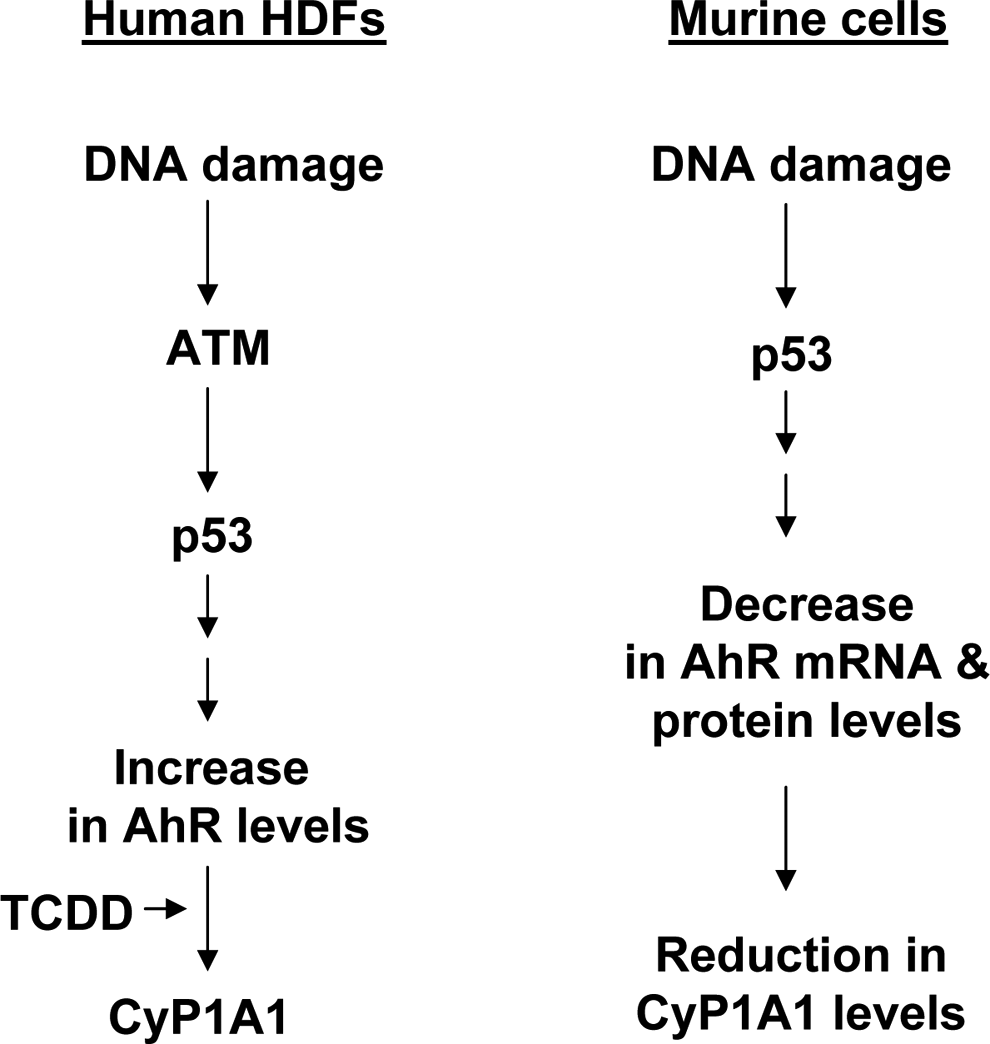
Proposed model for differential regulation of aryl hydrocarbon receptor (AhR) levels by activation of the ataxia telangiectasia-mutated (ATM)-p53 axis in human and murine cells.
Discussion
Signaling pathways that regulate levels of AhR in a variety of cell types remain largely unknown. Additionally, it is not known whether DNA damage-induced activation of p53 in human and murine cells could regulate the AhR levels. Therefore, our observations that activation of p53 by DNA-damaging agents such as DOX and bleomycin in certain human cell types increased levels of AhR are novel. Additionally, our observations that activation of p53 by DNA-damaging agents in certain murine macrophage cell lines downregulated the levels of AhR are also novel (Figure 4).
Although treatment of murine Swiss 3T3 fibroblasts that express AhR with benzo[a]pyrene, a DNA-damaging agent, resulted in p53-independent G1 arrest, 32 it remains unknown whether the treatment results in increases in levels of the AhR. Further, MEFs from the p53-deficient mice were found to express AhR levels that were comparable with the wild-type MEFs. 32 Therefore, it remains to be investigated whether the p53-mediated regulation of AhR levels depends upon the genetic background of mice.
Treatment of cardiomyocytes with DOX increases AhR migration to the nucleus and stimulates the expression of AhR target genes such as CyP1A1 and GSTA1. 5 Further, treatment of AhR-deficient mice with DOX increases activation of p53 and induces apoptosis of cardiomyocytes. These observations indicate that AhR expression in the murine cardiomyocytes protects against the DOX-induced toxicity. Therefore, our observations that HDFs from patients with AT, which are defective in activation of p53, 14 expressed reduced levels of AhR (Figure 2) predict that the patients with AT are likely to be more susceptible to cytotoxicity that is induced by certain anticancer agents, including the DOX. Accordingly, treatment of lymphoid malignancies in patients with AT having bleomycin or DOX has been associated with poor outcomes. 33 Further, our observations make it likely that tumor cells that express mutated p53 (or exhibit a defect in p53 function) are likely to be more susceptible to DOX therapy. Consistent with this prediction, the human ovarian cancer cell line that expressed wild-type p53 (as compared to a mutant p53) was found to be less susceptible to DOX treatment. 34 Therefore, our observations have important implications for cancer therapy.
Because activation of p53 by ATM protein kinase in response to DNA damage results in transcriptional regulation of the p53 target genes, 19,23 we tested whether treatment of WI-38 HDFs with actinomycin D, an inhibitor of transcription, 35 could inhibit the p53-mediated increases in AhR levels. Our preliminary experiments revealed that cotreatment of cells with actinomycin D under our experimental conditions inhibited bleomycin-induced increases in levels of p21 (a transcriptional target of p53). However, the treatment did not result in decreases in levels of AhR following treatment with bleomycin (data not shown). Although these observations are consistent with the transcriptional activity independent role of the p53 in the regulation of DNA damage-induced cellular stress responses, 36 further work will be needed to identify the molecular mechanisms through which p53 regulates the AhR levels in human HDFs.
Treatment of HepG2 cells with ERK inhibitors (such as PD98059) inhibits TCDD treatment-induced decreases in steady-state levels of AhR by reducing proteolytic degradation. 13 Therefore, we explored whether activation of the ATM-p53 axis in human WI-38 fibroblasts increases AhR levels through an inhibition of the ERK. Our preliminary experiments revealed that the activation of the axis in cells by bleomycin treatment also resulted in activation of ERK (data not shown). Therefore, it is conceivable that activation of the ATM-p53 axis in WI-38 cells increases levels of AhR independent of ERK inhibition. Further work is needed to identify the molecular mechanisms through which the axis increases levels of AhR in certain human cell types by posttranscriptional mechanisms.
Doxorubicin is known to activate the ATM-p53 axis through the generation of reactive oxygen species. 25 Therefore, our observations that treatment of human fibroblasts with DOX activated the axis and increased steady-state levels of AhR raised the possibility that the generation of reactive oxygen species in cells could regulate the levels of AhR. Consequently, we tested whether treatment of human WI-38 HDFs with hydrogen peroxide could regulate levels of AhR. Our experiments revealed that the treatment under our experimental conditions only moderately (∼2-fold) increased steady-state levels of AhR (data not shown). Therefore, it is possible that DOX-induced signaling in human HDFs increases levels of AhR through additional mechanisms.
A previous study involving analysis of AhR expression in murine cell lines that were derived from different tissues had indicated that alterations (increase or decrease) in the expression of the AhR gene in response to treatment with dioxin, retinoic acid, cyclic adenosine monophosphate, or 12-O-tetradecanoylphorbol-13-acetate depends on tissue origin and cell types. 37 Accordingly, a study has noted that treatment of human lung carcinoma cells (A549) and hepatoma (HepG2) cells with transforming growth factor β1 regulated the AhR expression in cell-type-dependent manner. 38 Given that AhR regulates cell proliferation, apoptosis, differentiation, and inflammation, 1 –4 our observations that activation of the ATM-p53 axis in certain human and murine cell types differentially regulated the levels of AhR have implications for cancer therapy in patients. These observations are likely serve as a basis to further investigate the role of AhR in the regulation of cell cycle, cell survival, and differentiation. An improved understanding of the AhR functions is needed to identify new approaches to effectively treat cancers in human.
Footnotes
Acknowledgments
We thank Dr M. Murphy for generously providing p53 expression plasmids and human cell lines. We also thank Dr Alvaro Puga in our Department for thoughtful suggestions with respect to the detection of aryl hydrocarbon receptor levels in various human and murine cell types.
Author Contribution
Ravichandran Panchanathan: contributed to conception or design; contributed to acquisition, analysis, or interpretation; drafted the manuscript; critically revised the manuscript gave final approval; Agrees to be accountable for all aspects of work ensuring integrity and accuracy. Hongzhu Liu: contributed to acquisition, analysis, or interpretation; critically revised the manuscript; gave final approval; Agrees to be accountable for all aspects of work ensuring integrity and accuracy. Divaker Choubey: contributed to acquisition, analysis, or interpretation; drafted the manuscript; critically revised the manuscript; gave final approval; Agrees to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors declared the following financial support for the research, authorship, and/or publication of this article: This research was supported in part by a Merit Award (I01 BX001133) to DC from the Department of Veteran Affairs, Medical Research and Development Service.