
Abstract
Decitabine (5-aza-2′-deoxycytidine; DAC) in combination with tetrahydrouridine (THU) is a potential oral therapy for sickle cell disease and β-thalassemia. A study was conducted in mice to assess safety of this combination therapy using oral gavage of DAC and THU administered 1 hour prior to DAC on 2 consecutive days/week for up to 9 weeks followed by a 28-day recovery to support its clinical trials up to 9-week duration. Tetrahydrouridine, a competitive inhibitor of cytidine deaminase, was used in the combination to improve oral bioavailability of DAC. Doses were 167 mg/kg THU followed by 0, 0.2, 0.4, or 1.0 mg/kg DAC; THU vehicle followed by 1.0 mg/kg DAC; or vehicle alone. End points evaluated were clinical observations, body weights, food consumption, clinical pathology, gross/histopathology, bone marrow micronuclei, and toxicokinetics. There were no treatment-related effects noticed on body weight, food consumption, serum chemistry, or urinalysis parameters. Dose- and gender-dependent changes in plasma DAC levels were observed with a Cmax within 1 hour. At the 1 mg/kg dose tested, THU increased DAC plasma concentration (∼10-fold) as compared to DAC alone. Severe toxicity occurred in females receiving high-dose 1 mg/kg DAC + THU, requiring treatment discontinuation at week 5. Severity and incidence of microscopic findings increased in a dose-dependent fashion; findings included bone marrow hypocellularity (with corresponding hematologic changes and decreases in white blood cells, red blood cells, hemoglobin, hematocrit, reticulocytes, neutrophils, and lymphocytes), thymic/lymphoid depletion, intestinal epithelial apoptosis, and testicular degeneration. Bone marrow micronucleus analysis confirmed bone marrow cytotoxicity, suppression of erythropoiesis, and genotoxicity. Following the recovery period, a complete or trend toward resolution of these effects was observed. In conclusion, the combination therapy resulted in an increased sensitivity to DAC toxicity correlating with DAC plasma levels, and females are more sensitive compared to their male counterparts.
Introduction
Decitabine (5-aza-2′-deoxycytidine; DAC) modifies cellular epigenetics, gene expression, and differentiation by depleting DNA methyltransferase 1 (DNMT1), a key member of the network of chromatin-modifying enzymes that mediate transcription repression. This property of DAC is being explored as a potential remedy for hemoglobinopathies such as sickle cell disease and β-thalassemia, via a DNMT1 depletion-induced shift in hematopoietic differentiation toward erythropoiesis and an increase in fetal hemoglobin (HbF) expression. 1 –3 Low doses of DAC do not terminate DNA chain elongation during S-phase and can deplete DNMT1 without causing significant DNA damage or cytotoxicity in-vitro and in-vivo. 4 –8 This DNMT1-depleting action is S-phase dependent; hence, long exposures of progenitor cells to DAC (a broad-shaped area under the curve [AUC]) are critical determinants of the therapeutic epigenetic effect. For these reasons, a pharmacologic goal is to achieve drug levels that are above the minimum concentrations required to deplete DNMT1, but below thresholds associated with off-target effects and cytotoxicity, and can be sustained with practical dosing routes/schedules so that appropriate hematopoietic precursors are affected for an in vivo therapeutic effect.
Decitabine reactivates HbF expression in baboons following intravenous, subcutaneous, and oral administration 6,9 –11 and in patients with sickle cell disease following intravenous and subcutaneous administration. 2,12 However, oral administration of DAC has not been examined in patients with sickle cell disease or β-thalassemia. Oral administration is more likely to achieve low levels of exposure for prolonged time periods than its parenteral counterpart, while avoiding high peak DAC levels that cause DNA damage/cytotoxicity. However, oral bioavailability of DAC (and cytidine analogs in general) is severely curtailed because of rapid metabolism in gut and liver by the enzyme cytidine deaminase (CDA), which converts cytidines and analogs thereof to uridine counterparts. 13,14 Furthermore, gender differences in CDA expression and nonsynonymous single-nucleotide polymorphisms in CDA 15,16 cause clinically significant variation in pharmacokinetics, efficacy, and toxicity parameters. 17 To overcome these aforementioned barriers to oral administration, an inhibitor of CDA can be used in combination with DAC. The uridine analog tetrahydrouridine (THU), a competitive inhibitor of CDA, has been widely used in combination with cytosine analogs in preclinical and clinical settings. 13,14 Thus, an oral combination therapy of DAC with THU is being explored as a potential mean to surmount barriers to oral DAC single-agent therapy. In brief, the current good laboratory practice-compliant toxicity study was conducted to evaluate safety of the combination therapy in mice and to help determine a safe clinical starting dose for DAC in combination with a fixed dose of THU.
Materials and Methods
Test Article and Formulation Preparation
All formulations were prepared prior to each dosing, maintained on wet ice, and used within 7 hours following preparation.
Tetrahydrouridine (Purity 94.9%)
Sodium phosphate buffer (THU vehicle) was prepared by adding the appropriate amounts of sodium phosphate dibasic (2.50 mg/mL) and sodium phosphate monobasic (0.67 mg/mL) to sterile water for injection (SWFI). The THU vehicle was used without further formulation for groups 1 and 5, and for groups 2 to 4, a 16.7 mg/mL solution of THU was prepared in THU vehicle.
Decitabine (Purity 98.6%)
Potassium phosphate buffer was prepared by adding the appropriate amounts of potassium phosphate monobasic (2.72 mg/mL) and sodium chloride (5.40 mg/mL) to SWFI; the pH of the solution was adjusted to 6.90 (±2.90%). For group 1 (DAC vehicle), a 5% solution of potassium phosphate buffer in sodium chloride for injection (SCFI) was prepared; for groups 2 to 5, a 2-mg/mL stock solution of DAC was prepared by adding the appropriate amount of DAC to the potassium phosphate buffer; the pH was adjusted to 6.90% ± 2.90%. The dosing formulations of DAC (0.02, 0.04, and 0.1 mg/mL) were prepared by diluting the 2 mg/mL stock solution with SCFI.
Animals
CD-1 mice (male 30-38 g and female 24-31 g) from Charles River Laboratories (Raleigh, North Carolina) were individually housed in polycarbonate cages suspended on stainless steel racks with SaniChip-certified hardwood bedding. The mice were provided with Certified Global Harlan Teklad Laboratory Diet 2018 (pellets). Water was provided with automatic watering system and supplemented with water bottles as necessary. The Institutional Animal Care and Use Committee of AVANZA approved this protocol in accordance with provisions of the US Department of Agriculture Animal Welfare Act, and the Public Health Service Policy on Humane Care and Use of Laboratory Animals.
Experimental Design
Mice were assigned to 4 dose groups and a vehicle control group as shown in Table 1. Animals were gavaged with DAC or its vehicle 1 hour ± 5 minutes after administration of THU or its vehicle at a dose volume of 10 mL/kg. The DAC doses were selected based on the range finding study in which the mice tolerated 6 oral doses (2×/week) of 0.1, 0.2, and 0.4 mg/kg DAC in combination with a fixed dose of 167 mg/kg THU. A fixed THU dose (500 mg/m2) and the optimal timing between THU and DAC administration (60 minutes) were selected based on the previous studies. 11 Conversion of milligrams per body surface area dose in mice into milligrams per kilogram body weight dose estimation was based on conversion factor (km) for mice obtained from US Food and Drug Administration published guidelines. In brief, the mouse dose in milligrams per body surface area (500 mg/m2) was divided by the km of 3 to convert the dose to milligrams per kilogram body weight (167 mg/kg). The working body weight range of mice in the guideline is 11 to 34 g; the body weight range of mice used in this study was 24 to 38 g.
Study Design.a
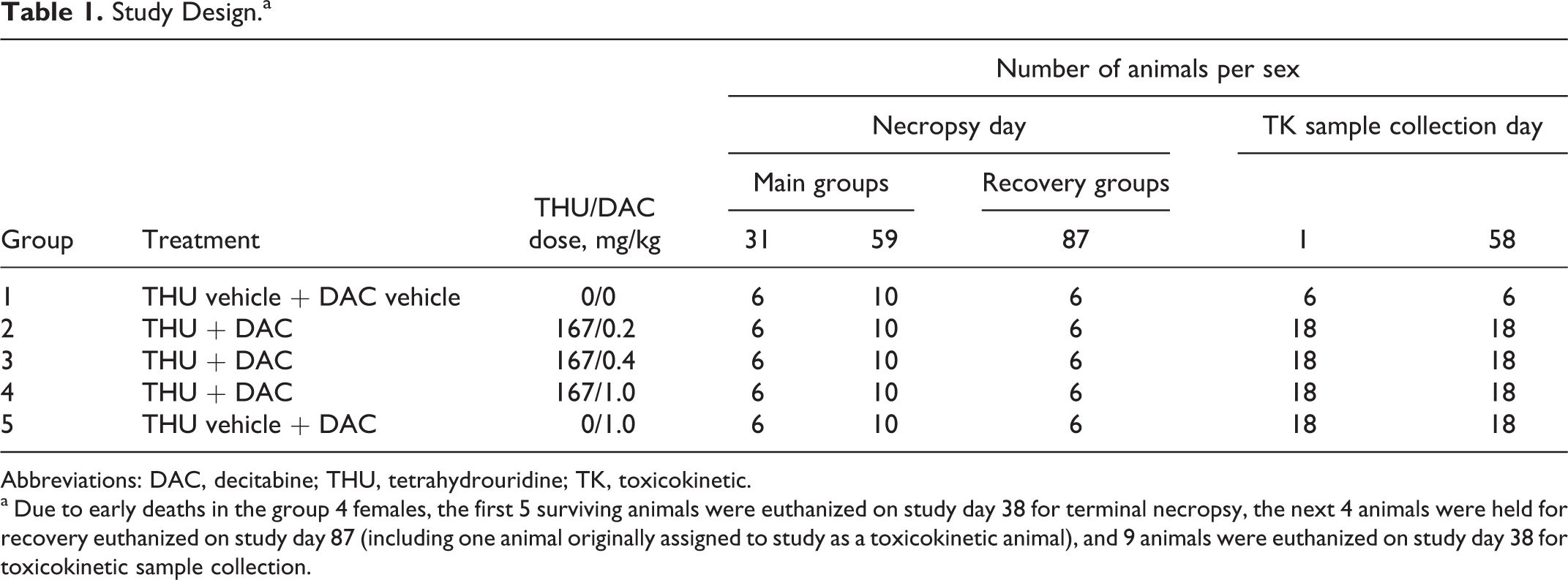
Abbreviations: DAC, decitabine; THU, tetrahydrouridine; TK, toxicokinetic.
a Due to early deaths in the group 4 females, the first 5 surviving animals were euthanized on study day 38 for terminal necropsy, the next 4 animals were held for recovery euthanized on study day 87 (including one animal originally assigned to study as a toxicokinetic animal), and 9 animals were euthanized on study day 38 for toxicokinetic sample collection.
All animals, with the exception of group 4 females, were dosed 2 times per week for a total of 18 doses (final dose was administered on study day 58) followed by a 4-week recovery period. Due to early deaths, group 4 recovery females were dosed on 2 consecutive days/week for 5 weeks (total of 10 doses; final dose administered on study day 30), and the remaining surviving group 4 main study and toxicokinetic females received an 11th dose in the sixth week of study prior to terminal euthanasia (administered on study days 37 and 38, respectively).
Parameters Evaluated
Clinical observations
Cageside observations for morbidity and death were made twice daily throughout the study. Starting on day 1, clinical observations were conducted at least once each day.
Body weights/food consumption
Body weights were recorded for all animals on day 1 and weekly thereafter throughout the study and prior to each scheduled necropsy. All body weights collected on dosing days were collected prior to dosing and used for dose volume determinations. Food consumption was recorded weekly.
Toxicokinetics
Sample collection tubes were prepared prior to each collection day by adding 10 μL/tube of a 10-mg/mL THU solution. Blood samples (∼0.5 mL) were collected via intracardiac puncture from nonfasted, anesthetized toxicokinetic animals on study day 1 (groups 2-5) and study day 58 (groups 2-5 with the exception of group 4 females) at 15, 30, 60, 90, 120, and 180 minutes after administration of DAC from 3 animals/sex/group at each time point. Due to mortality in the group 4 females, the first 5 surviving animals were necropsied on study day 38, and blood samples were collected from 3 females per time point at 15, 30, and 120 minutes following administration of DAC. All samples were collected within 5 minutes of the target time.
Plasma samples were stored at −75°C ± 15°C until shipped on dry ice to The Ohio State University for DAC analysis using liquid chromatography-tandem mass spectrometry method. 18
Concentrations versus time data were analyzed by compartmental and noncompartmental methods. WinNonlin was used to fit the data to a 1-compartment oral absorption model (first-order input) with an appropriate weighting factor. Cmax was obtained from the observed values. Area under curves were calculated using the linear trapezoidal rule to the last observed time point (noncompartment) or with extrapolation to time infinity (compartment). In the case of gender comparisons, the data were truncated at the same time point.
Clinical pathology
Clinical pathology samples were collected from fasted animals (3 animals/sex/group) via intracardiac puncture on study days 31, 59, and 87. Additional samples were collected from nonfasted animals prior to moribund euthanasia, when possible. Terminal euthanasia for all group 4 females was performed on study day 38; therefore, no samples were collected for this group on study day 59. Hematology and urinalysis samples were collected from all group 4 females (nonfasted) euthanized on study day 38. Chemistry samples were not collected for group 4 females at terminal euthanasia. Hematology analysis included white blood cell (WBC), red blood cell (RBC) counts, hemoglobin (HGB), hematocrit (HCT), mean corpuscular volume, mean corpuscular hemoglobin (MCH), MCH concentration, platelets, differential leukocyte counts (neutrophils, monocytes, eosinophils, basophils, and lymphocytes), reticulocytes, and nucleated RBCs. Clinical chemistry analysis included alkaline phosphatase, aspartate aminotransferase, alanine aminotransferase, γ glutamyl transferase, total protein, albumin, glucose (GLU), blood urea nitrogen, creatinine, phosphates, creatine kinase, lactose dehydrogenase, sodium, potassium, chloride, globulin, albumin/globulin ratio, total bilirubin, direct bilirubin, and indirect bilirubin. Coagulation parameters included prothrombin time, activated partial thromboplastin time, and fibrinogen concentration.
Urinalysis
Urine specimens were collected on study days 31 and 59 and examined for bilirubin, GLU, ketones, leukocytes, nitrites, occult blood, protein, urobilinogen, color, pH, volume, and specific gravity.
Bone marrow micronucleus analysis
The micronucleus analysis was performed by BioReliance, Inc (Rockville, Maryland) to evaluate the genotoxic potential of the combination treatment. At the scheduled terminal necropsies on study days 38 and 59, 2 femoral bone marrow smear slides were prepared for each main study animal for bone marrow micronucleus analysis. Smears were stained with May-Grunwald/Giemsa stain and, if possible, 2000 polychromatic erythrocytes (PCEs) per animal were evaluated microscopically for the presence of micronucleated PCEs. In addition, the number of PCEs per 1000 bone marrow erythrocytes per animal and treatment group was also determined to evaluate the effects on bone marrow erythropoiesis.
Termination, necropsy, and histopathology
All animals were euthanized by carbon dioxide inhalation followed by exsanguination. Toxicokinetic animals were euthanized and discarded without necropsy following their scheduled sample collection. Group 4 toxicokinetic females euthanized on study day 38 were subjected to gross necropsy and tissue preservation as described subsequently.
Scheduled necropsies for main study animals were conducted on study day 31 (6 animals/sex/group), study day 38 (5 group 4 females, euthanized 24 ± 1 hours following the final dose), study day 59 (10 animals/sex/group for all other groups), and study day 87 (all surviving animals).
All main study animals (including scheduled euthanasia animals and those found dead or euthanized moribund) and toxicokinetic animals that were found dead were necropsied as soon as possible after the time of death or discovery. The following tissues were collected for microscopic examination: adrenal glands, aorta, femur, bone marrow (from sternum and costochondral junction), brain, cecum, colon, duodenum, epididymides, esophagus, eyes, heart, ileum, jejunum, kidneys, liver, lungs, lymph nodes (mandibular and mesenteric), mammary gland, ovaries, pancreas, parathyroid gland, pituitary gland, prostate gland, salivary gland (mandibular), sciatic nerve, seminal vesicle, skeletal muscle (thigh), skin (ventral abdomen), spinal cord (thoracolumbar), spleen, stomach (forestomach, glandular), testes, thymus, thyroid glands, trachea, urinary bladder, and uterus.
The tissues were grossly examined and fixed in 10% neutral-buffered formalin (NBF) with exception of the eyes (and associated ocular tissue) and testes (with epididymides), which were preserved in modified Davidson fixative and transferred to 10% NBF within 24 to 72 hours. All tissues for main study animals in groups 1 and 4 from the study days 31, 38, and 59 necropsies were processed to approximately 5-μm sections, stained with hematoxylin and eosin, and examined microscopically by a board certified pathologist. Target tissues that were examined in all remaining groups and intervals (including those found dead or euthanized moribund) included brain, bone marrow, duodenum, epididymides, ileum, jejunum, kidneys, liver, lungs, mandibular lymph nodes, ovaries, Peyer's patches, spleen, testes, thymus, uterus, and any gross lesions.
Statistical analyses
Quantitative data were analyzed using the Kolmogorov-Smirnov test for normality, the Levene Median test for equal variance, and 1-way analysis of variance (ANOVA). If normality or equal variance test failed, then the Kruskal-Wallis ANOVA was performed on rank-transformed data. For parametric data, the Dunnett t test was used to delineate which groups (if any) differed from the control. For nonparametric data, the Dunn test was used to delineate which groups (if any) differed from the control. The P value of less than 0.05 (2 tailed) was used as the critical level of significance for all tests.
Results
Overview
Dose formulation analysis results indicated that all dosing concentrations were within ±10% of the target values (except low-dose formulation samples on study day 44/43 were approximately 15% lower than the target). Administration of 167 mg/kg THU followed by 1.0 mg/kg DAC (group 4) resulted in death in 1 male and 8 females. Animals surviving to scheduled termination were generally asymptomatic with no treatment-related effects observed in body weights, food consumption, clinical chemistry, and urinalysis for a treatment up to 1.0 mg/kg DAC in combination with 167 mg/kg THU in mice.
Toxicokinetics
At all dose levels and measured intervals, Cmax for plasma DAC was attained from 15 to 60 minutes postdose (Table 2, panels A and B; Figures 1 and 2) and was in a range (>0.5 µmol/L) expected to cause DNA damage and cytotoxicity. The DAC was detectable through 180 minutes postdose in all treated groups with the exception of group 5 males (DAC alone) on study day 58 where DAC was detected through 120 minutes postdose. Plasma DAC levels increased in a dose-dependent manner in groups 2 to 4 (combined treatment of DAC with THU), and there was an overall trend for higher plasma levels in female mice when compared to male mice in all treatment groups. For animals receiving a combined treatment of DAC with THU (groups 2-4), AUC values were higher on study day 1 when compared to study day 58 (all groups except group 4 females), and t1/2 was generally longer on study day 58. For DAC alone animals (group 5), AUC and t1/2 values were generally comparable between the sampling days.
Toxicokinetics of DAC Following Oral Treatment of DAC Alone or in Combination With THU in Mice.a
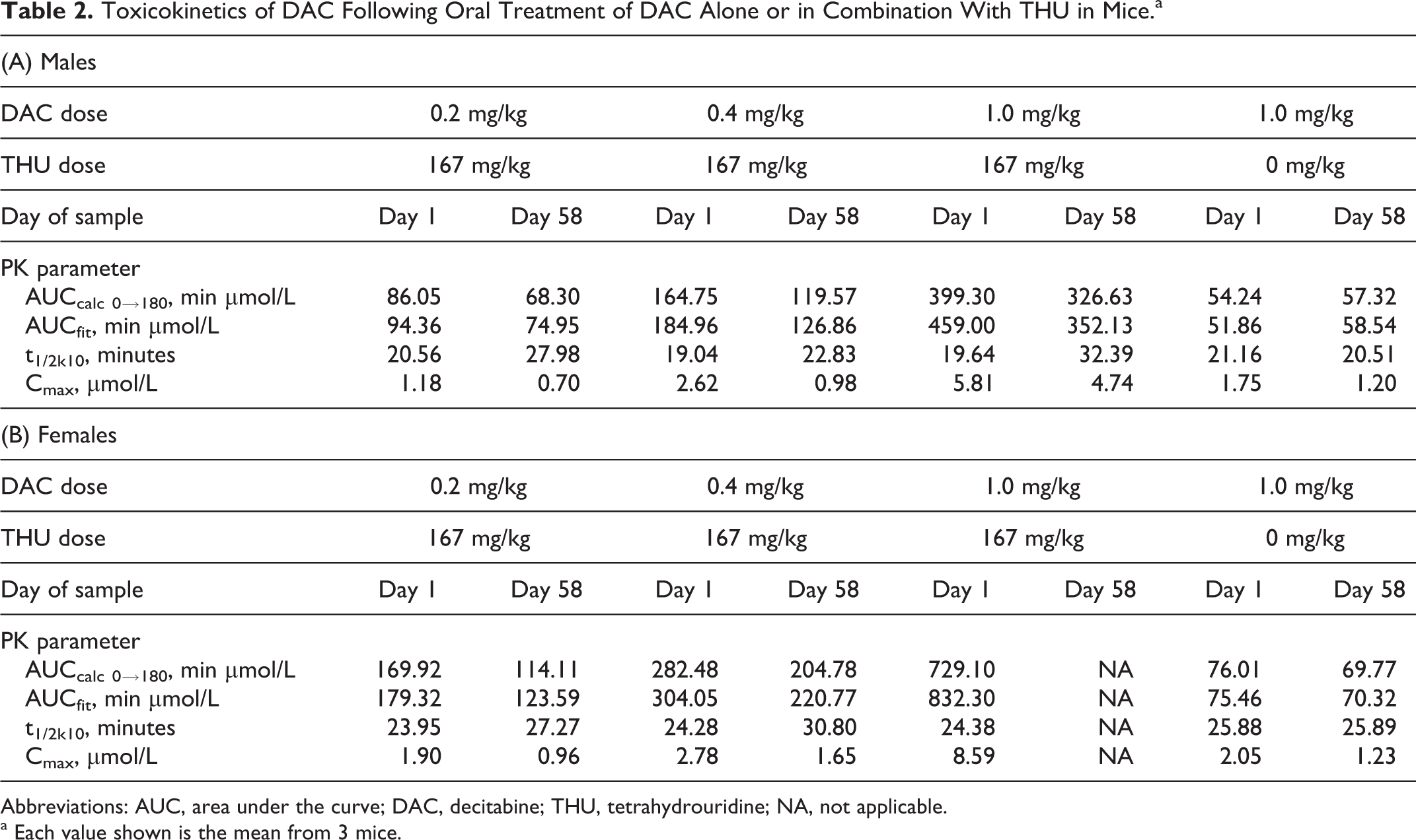
Abbreviations: AUC, area under the curve; DAC, decitabine; THU, tetrahydrouridine; NA, not applicable.
a Each value shown is the mean from 3 mice.

Plasma decitabine (DAC) levels in group 2 (A), group 3 (B), group 4 (C), and group 5 (D) mice with genders separated for day 1 ( male;  female).
female).

Plasma decitabine (DAC) levels in group 2 (A), group 3 (B), group 4 (C), and group 5 (D) mice on day 38/58 ( male;  female).
female).
The fold increase in DAC exposure (AUCfit) produced by preadministration of THU before DAC 1.0 mg/kg compared to vehicle followed by DAC 1.0 mg/kg was 8.8 in males and 11.8 in females (study day 1).
Hematology
On study day 31 (Table 3), decreases in RBCs, HGB, HCT, and absolute reticulocytes (ABRETis) were noted in males and females receiving THU and DAC. Females were affected more severely and also showed decreases in WBCs and absolute lymphocyte (ABLYMP) counts in groups 3 and 4. Animals receiving DAC alone (group 5) had decreases in ABRETis and females had decreases in WBCs, ABLYMPs, RBCs, and HGB.
Effect of DAC Alone or in Combination With THU on Hematologic Parameters on Day 31.a
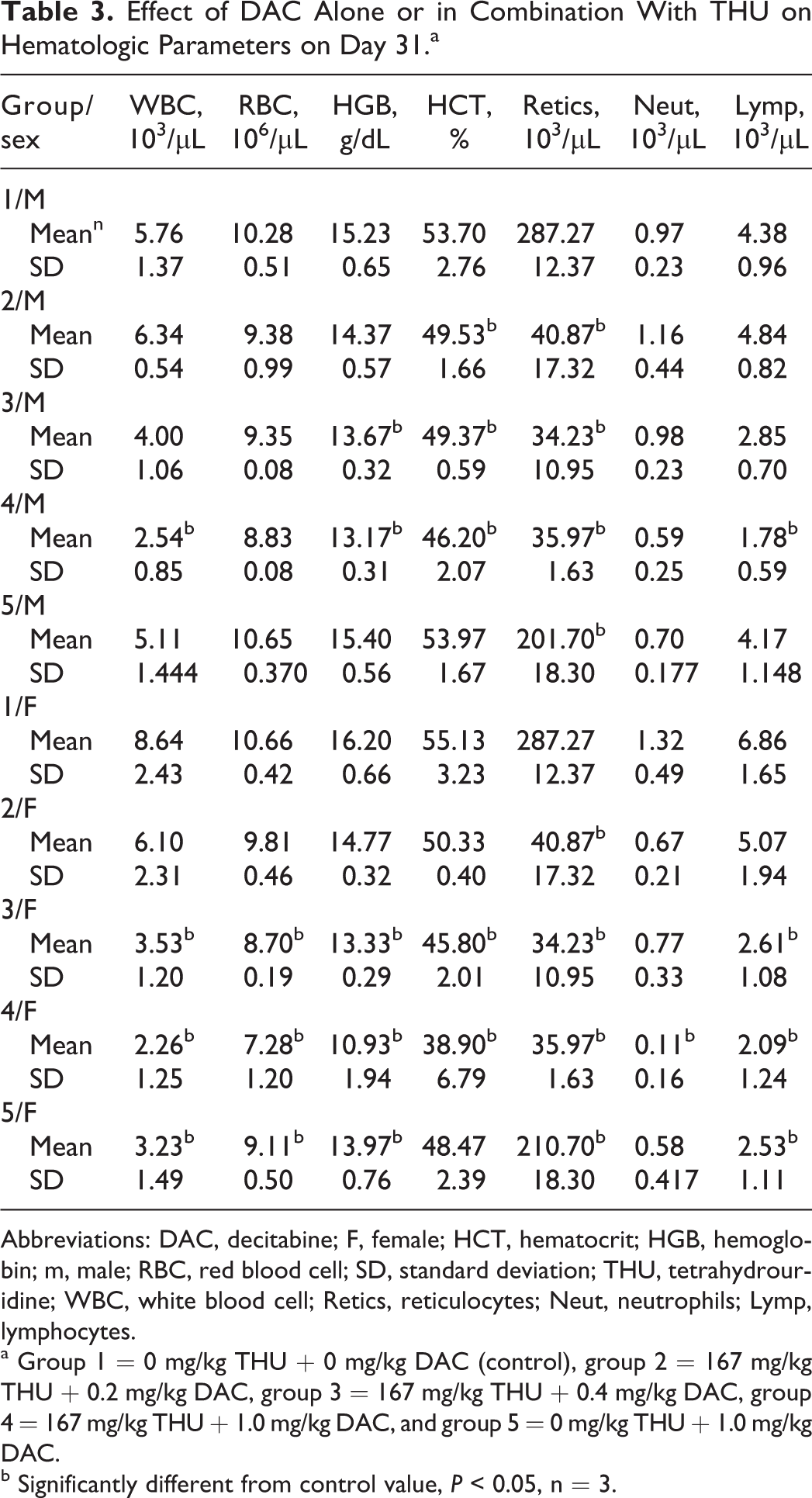
Abbreviations: DAC, decitabine; F, female; HCT, hematocrit; HGB, hemoglobin; m, male; RBC, red blood cell; SD, standard deviation; THU, tetrahydrouridine; WBC, white blood cell; Retics, reticulocytes; Neut, neutrophils; Lymp, lymphocytes.
a Group 1 = 0 mg/kg THU + 0 mg/kg DAC (control), group 2 = 167 mg/kg THU + 0.2 mg/kg DAC, group 3 = 167 mg/kg THU + 0.4 mg/kg DAC, group 4 = 167 mg/kg THU + 1.0 mg/kg DAC, and group 5 = 0 mg/kg THU + 1.0 mg/kg DAC.
b Significantly different from control value, P < 0.05, n = 3.
On study day 38 (Table 4), decreases in RBCs, HGB, absolute neutrophils (ABNEUTs), absolute monocytes (ABMONOs), and absolute eosinophils (ABEOSs) were noted in group 4 females. On study day 59 (Table 4), decreases in WBCs, RBCs, HGB, HCT, and ABRETis were noted in males and females receiving THU and DAC or DAC alone. In addition, ABNEUTs and ABLYMPs were also reduced. These changes were corroborated with effects on the bone marrow and lymphoid tissues including microscopic effects on the bone marrow.
Effect of DAC Alone or in Combination With THU on Hematologic Parameters on Day 38/59.a
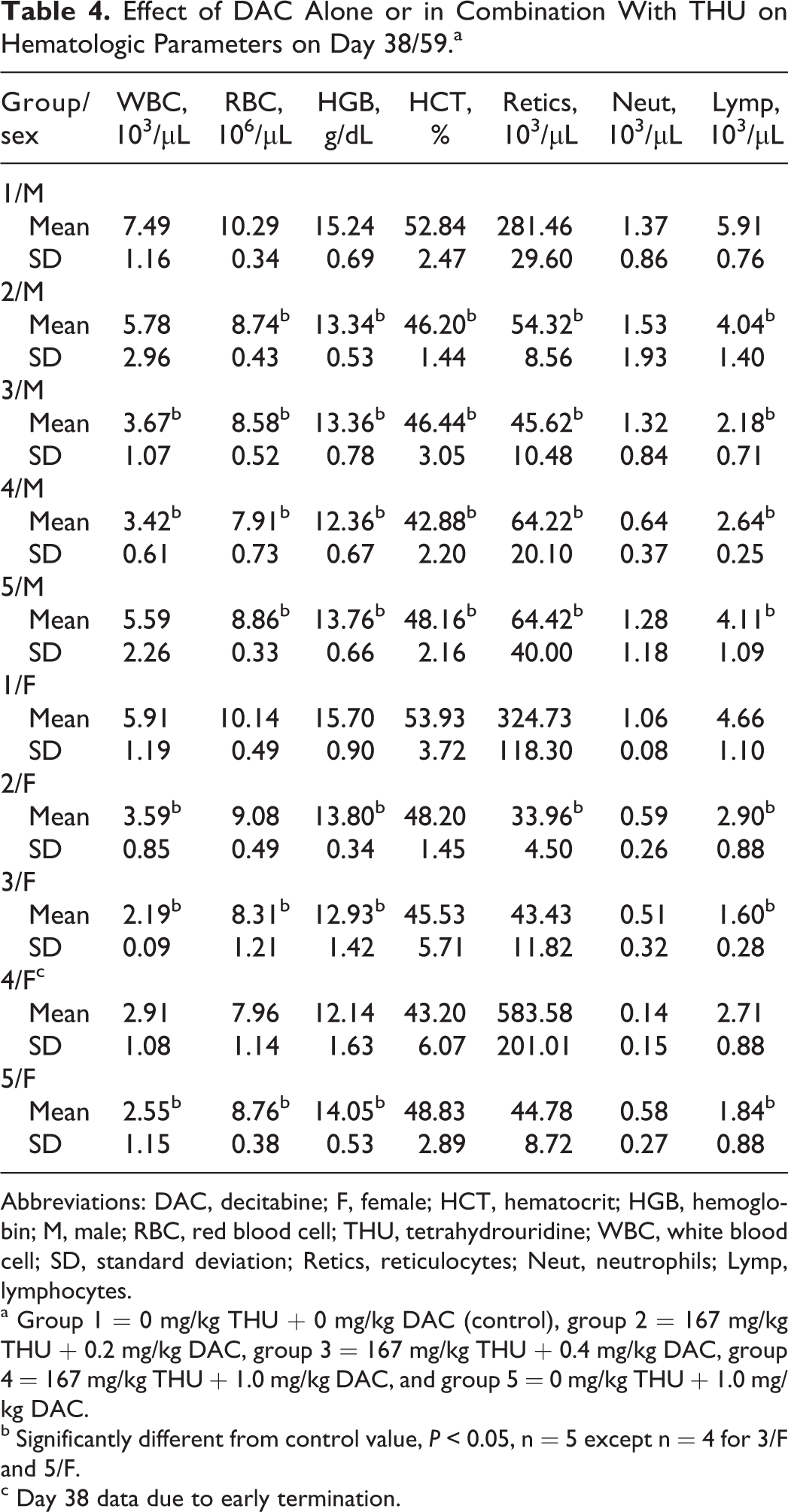
Abbreviations: DAC, decitabine; F, female; HCT, hematocrit; HGB, hemoglobin; M, male; RBC, red blood cell; THU, tetrahydrouridine; WBC, white blood cell; SD, standard deviation; Retics, reticulocytes; Neut, neutrophils; Lymp, lymphocytes.
a Group 1 = 0 mg/kg THU + 0 mg/kg DAC (control), group 2 = 167 mg/kg THU + 0.2 mg/kg DAC, group 3 = 167 mg/kg THU + 0.4 mg/kg DAC, group 4 = 167 mg/kg THU + 1.0 mg/kg DAC, and group 5 = 0 mg/kg THU + 1.0 mg/kg DAC.
b Significantly different from control value, P < 0.05, n = 5 except n = 4 for 3/F and 5/F.
c Day 38 data due to early termination.
On study day 87 following a 28-day recovery period, there were no residual or delayed-onset effects on hematology parameters.
Microscopic Findings
As shown in Tables 5 and 6, in the interim and terminal euthanasia animals (study days 31, 38, and 59), microscopic findings related to treatment were observed in bone marrow (hypocellularity, proliferation of megakaryocytes, and cytoplasmic alteration [emperipolesis] of megakaryocytes), lymphoid tissues (lymphoid depletion of the thymic cortex and proliferation of megakaryocytes in the splenic red pulp), small intestine (epithelial apoptosis with occasional regeneration in glands), and reproductive tissues (seminiferous tubule degeneration in the testes, hypospermia and germ cell exfoliation in the epididymides, and atrophy of the uterus). These findings were generally observed in increasing incidence and severity as the dose of DAC increased, with the incidence/severity of most of the findings higher in animals receiving 1.0 mg/kg DAC with 167 mg/kg THU (group 4) than in animals receiving 1.0 mg/kg DAC alone (group 5). Hypocellularity was characterized by a generalized loss of hematopoietic cells, leaving primarily adipose tissue. When the finding was less severe, the erythropoietic lineage seemed to be lost, leaving a higher density of myeloid cells. However, when at higher severity, all cell types were affected. In addition, many of the findings were slightly higher in severity in females than in males (except for the reproductive organs). All findings were observed at interim euthanasia (study day 31), and they were slightly increased in incidence/severity at terminal euthanasia (study day 38 for group 4 females and study day 59 for all others). A small testis in a group 5 male and soft bilateral testes in a group 4 male were correlated microscopically with seminiferous tubule degeneration. All findings resolved by the recovery euthanasia (study day 87), with the exception of findings in the male reproductive tissues.
Effect of DAC Alone or in Combination with THU on Microscopic Findings on Day 31.
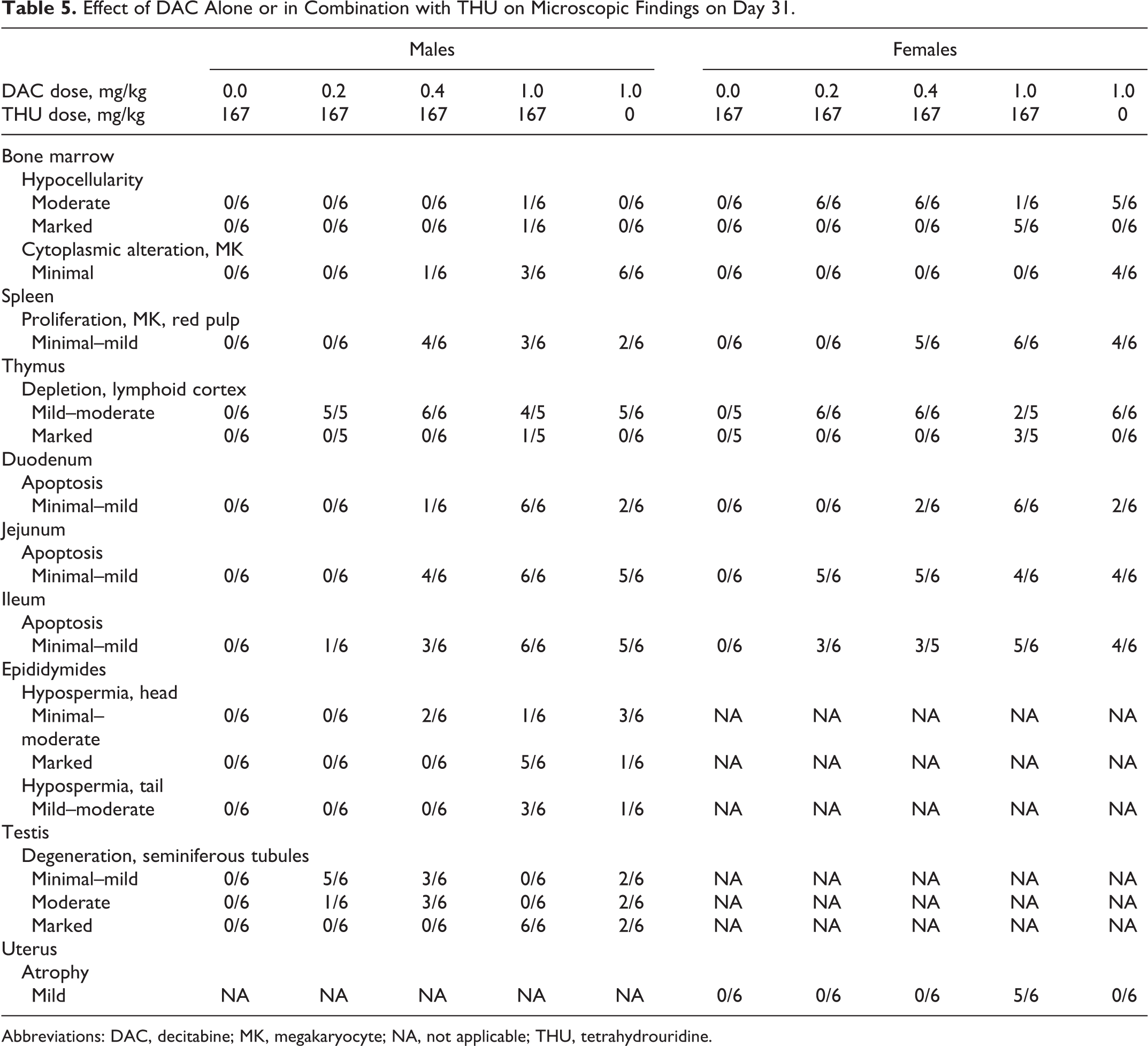
Abbreviations: DAC, decitabine; MK, megakaryocyte; NA, not applicable; THU, tetrahydrouridine.
Effect of DAC Alone or in Combination on Microscopic Findings on Day 38/59.
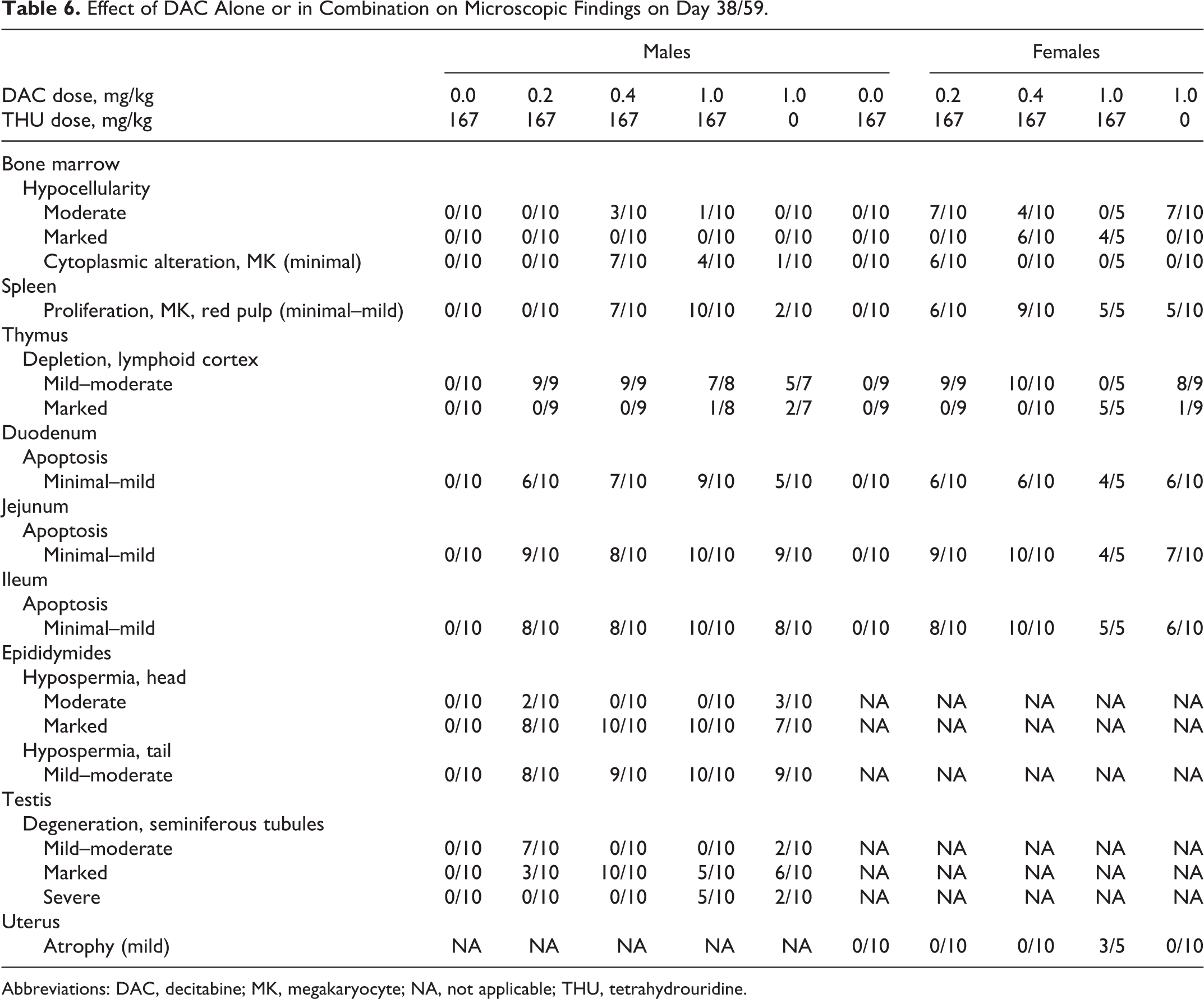
Abbreviations: DAC, decitabine; MK, megakaryocyte; NA, not applicable; THU, tetrahydrouridine.
Bone Marrow Micronucleus Analysis
Administration of up to 167 mg/kg THU followed by 1.0 mg/kg DAC caused bone marrow cytotoxicity and suppression of erythropoiesis in all treated groups (Table 7). This was indicated by marked decreases in the incidence of PCEs and a resultant reduction of up to 73% in the ratio of PCEs to erythrocytes when compared to controls. Genotoxicity was apparent in the treated groups by the statistically significant higher incidence of micronucleated PCEs when compared to controls.
Summary of Bone Marrow Micronucleus Analysis.a
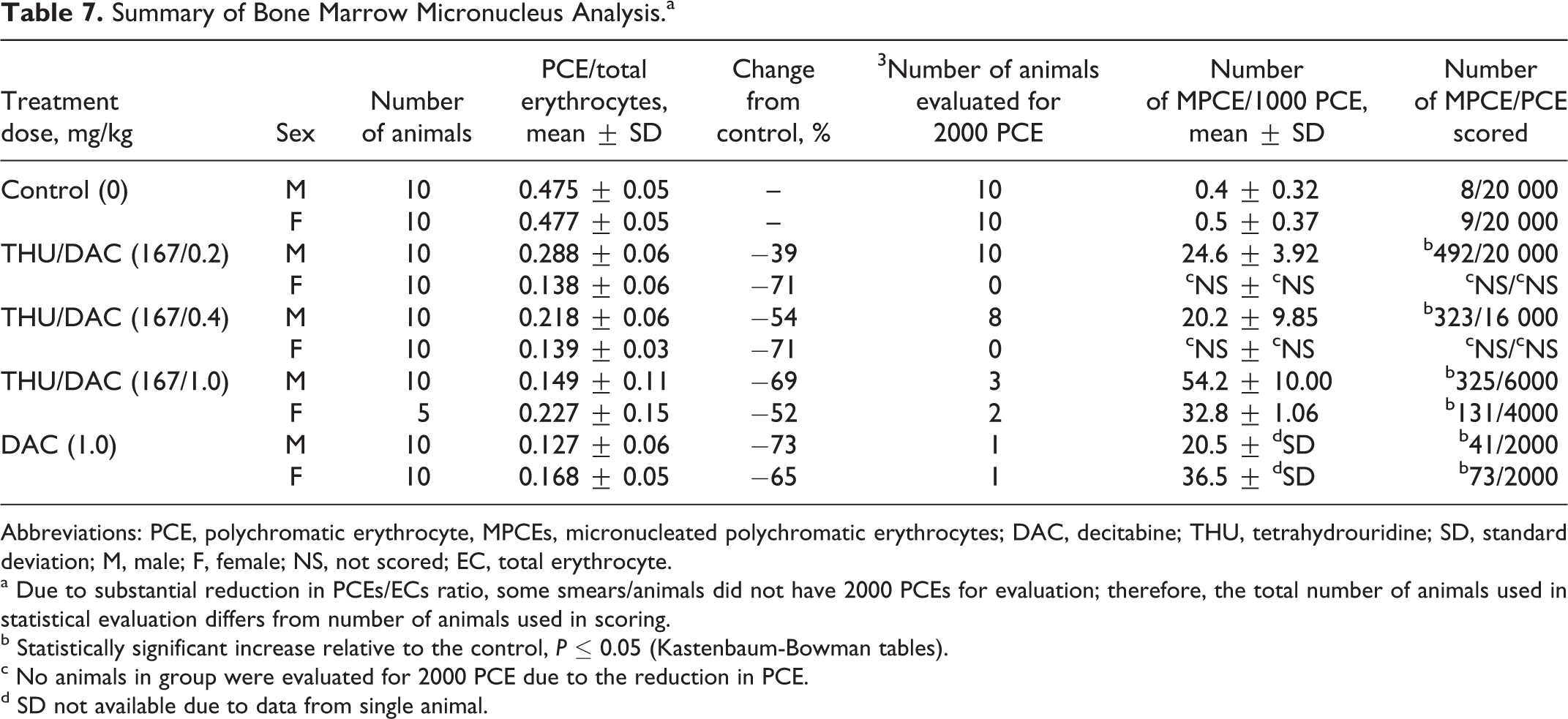
Abbreviations: PCE, polychromatic erythrocyte, MPCEs, micronucleated polychromatic erythrocytes; DAC, decitabine; THU, tetrahydrouridine; SD, standard deviation; M, male; F, female; NS, not scored; EC, total erythrocyte.
a Due to substantial reduction in PCEs/ECs ratio, some smears/animals did not have 2000 PCEs for evaluation; therefore, the total number of animals used in statistical evaluation differs from number of animals used in scoring.
b Statistically significant increase relative to the control, P ≤ 0.05 (Kastenbaum-Bowman tables).
c No animals in group were evaluated for 2000 PCE due to the reduction in PCE.
d SD not available due to data from single animal.
Discussion
Decitabine at low doses (≤0.5 µmol/L plasma concentrations) is a potential therapeutic agent for sickle cell disease and β-thalassemia. Low concentrations but extended time above a threshold concentration are required to deplete DNMT1 without causing significant cytotoxicity. To achieve these lower systemic levels of DAC for extended period of time, an oral route of administration should be more promising than parenteral alternatives for logistic and accessibility reasons. However, oral administration of cytidine analogs such as DAC is impeded by high expression of the enzyme CDA in the gut and liver, an enzyme that rapidly metabolizes and inactivates this class of drugs. Therefore, inhibiting CDA by pretreatment with THU was used to increase oral bioavailability of DAC and to facilitate production of the desired wide (low Cmax with extended duration) pharmacologic profile. Pharmacokinetic properties of THU in mice have been evaluated previously demonstrating 100 mg/kg oral dose of THU is 20% bioavailable (Cmax—10 µg/mL and half-life 85 minutes) and is sufficient to produce and sustain plasma concentrations (>1 µg/mL) for several hours that inhibit CDA. 14
Toxicity of DAC as a single agent has been evaluated extensively in mice, rats, dogs, and monkeys to support oncology clinical trials. Toxicological effects of DAC are mainly characterized as hematological (leukopenia, anemia, and thrombocytopenia), myeloid (bone marrow hypoplasia), gut associated (necrosis of intestinal mucosa), lymphoid (thymic/testicular atrophy), and developmental (teratogenicity). 6,8 These effects are very much consistent with its cytotoxic biologic action at high (generally >0.5 µmol/L) drug concentrations. Toxicity of THU has been evaluated in beagle dogs and monkeys with single intravenous injections demonstrating no notable toxic effects up to 1 g/kg doses. 19 Toxicological evaluation of orally administered DAC in combination with THU has not been reported in the literature. Therefore, the present study was conducted to support evaluation of the combination therapy in the clinic.
Treatment-related moribundity and death occurred in 9 high-dose group (167 mg/kg THU + 1.0 mg/kg DAC) animals. In 8 of these 9 animals, septicemia (as evident in microscopic evaluation) was secondary to bone marrow hypocellularity and appeared to be the cause of moribundity/death. In addition to bone marrow hypocellularity, dead animals had marked lymphoid depletion of the thymus and/or bacteria in the mandibular lymph nodes, spleen, liver, and/or adrenal cortex.
The THU pretreatment increased plasma concentration (∼10-fold) of DAC compared to the same dosage of DAC without THU. In all DAC treatment groups, DAC Cmax exceeded thresholds (>0.5 µmol/L) expected to cause cytotoxicity. In addition, female mice appear to have higher exposure (∼2-fold) to DAC compared to males, which can be explained by lower CDA expression in female mice. 17 Furthermore, THU pretreatment appeared to increase sex difference in DAC exposure (Figures 1 and 2), which could be explained by gender differences in CDA expression and/or its affinity to THU. The increased toxicity of the combination therapy correlated with increased plasma DAC exposure. The DAC treatment caused marked myelosuppression as indicated by decreases in erythroid mass parameters, panleukopenia, and bone marrow hypocellularity. The decreases in erythroid mass parameters were marked in extent and lacked a regenerative response. The leucopenia was caused by decreases in ABNEUT, ABMONO, ABEOS, and ABLYMP values, that is, panleukopenia. Genotoxicity was also observed in all treatment groups other than control mice as indicated by increased incidence of micronucleated PCEs.
Microscopic findings related to the treatment with DAC + THU and/or DAC alone were observed in the bone marrow, lymphoid tissues, small intestine, and male and female reproductive tissues. Dose-dependent mild to marked hypocellularity of the femoral bone marrow was noticed in all test article-treated animals with increased severity in females than in males. Hypocellularity was more severe in the combined dose of 167 mg/kg THU + 1.0 mg/kg DAC than in animals treated with 1.0 mg/kg DAC alone. Hypocellularity correlated with hematology changes in RBC mass parameters and decreased ABLYMP and WBC counts. Bone marrow findings of low incidence were proliferation of megakaryocytes and cytoplasmic alteration in megakaryocytes. Megakaryocyte proliferation, observed in 1 interim euthanasia male treated with 167 mg/kg THU + 1.0 mg/kg DAC, was characterized by increased density of megakaryocytes within the hypocellular bone marrow. When the bone marrow hypocellularity was most severe, megakaryocyte proliferation was not observed (ie, more low- and mid-dose animals were observed to have megakaryocyte proliferation, but most high-dose animals were not). It may have been the normal population of megakaryocytes that was more resistant than other hematopoietic cell types in low- and mid-dose animals compared with the high-dose group; however, the possibility that the mid-dose group had a stimulatory effect on the megakaryocyte population could not be ruled out and is consistent with previous clinical observations. 1 Cytoplasmic alteration was an increase in emperipolesis within megakaryocytes (ie, intact cells, typically neutrophils, or other hematopoietic cells within the megakaryocyte cytoplasm). This was observed with much lower frequency in control bone marrow megakaryocytes compared with treated groups. Emperipolesis has been associated with increased demand for hematopoietic cell delivery. 20
Lymphoid changes were DAC-dose responsive. Mild to marked lymphoid depletion in the cortex of the thymus in most treated animals and minimal or mild proliferation of megakaryocytes in the red pulp of the spleen of animals given ≥0.4 mg/kg DAC with or without THU were evident. Lymphoid depletion was characterized by loss of (primarily) cortical thymic lymphocytes. As with bone marrow hypocellularity, the highest severity of thymic lymphoid depletion was primarily observed in animals administered 167 mg/kg THU + 1.0 mg/kg DAC. Thymic lymphoid depletion probably contributed to decreased ABLYMP counts observed among hematology changes. Proliferation of megakaryocytes in the spleen was characterized by increased numbers of megakaryocytes, mostly along trabeculae in the red pulp, in the absence of a multilineage increase in extramedullary hematopoeisis.
Conclusion
In summary, the combination therapy resulted in increased sensitivity to DAC-induced toxicity as compared to the DAC treatment alone, correlating with DAC plasma levels. The DAC Cmax was in a range expected to cause cytotoxicity (>0.5 µmol/L) in all DAC treatment groups. Female mice had higher Cmax and are more sensitive than males. Furthermore, hematologic effects appeared to be the most sensitive safety biomarkers, suitable for monitoring in clinical trials. Treatment-related effects resolved fully or showed a trend toward resolution by the end of the recovery period. An oral route of administration for this combination therapy appears to be promising for clinical evaluation in sickle cell disease and β-thalassemia. Thus, the current study in CD-1 mice yielded important toxicity parameters pertinent to potential therapeutic application of THU and DAC combination oral therapy in human settings.
Footnotes
Acknowledgments
The authors acknowledge BioReliance, Rockville, Maryland, for conducting the micronucleus analysis and also thank Pramod Joshi for editorial assistance.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article. Yogen Saunthararajah possesses a patent application for combination of oral THU and DAC.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This study was supported by NCI contract N01-CM-42204, N01-CM-52205, and NHLBI and NIDDK under NIH-BrIDGs Program. YS is supported by National Institutes of Health (1R01CA138858) and Department of Defense (PR081404).