
Abstract
Liver disease is a major health issue characterized by several pathological changes, with steatosis (fatty liver) representing a common initial step in its pathogenesis. Steatosis is of critical importance because prevention of fatty liver can obviate downstream pathologies of liver disease (eg, fibrosis). Recent studies have shown a strong correlation between chemical exposure and steatosis. The work described here identifies chemicals on the US Environmental Protection Agency’s Integrated Risk Information System (IRIS) that induce steatosis and investigates putative mechanisms by which these chemicals may contribute to this pathological condition. Mitochondrial impairment, insulin resistance, impaired hepatic lipid secretion, and enhanced cytokine production were identified as potential mechanisms that could contribute to steatosis. Taken together, this work is significant because it identifies multiple mechanisms by which environmental chemicals may cause fatty liver and expands our knowledge of the possible role of environmental chemical exposure in the induction and progression of liver disease.
Introduction
Liver disease is a serious health concern for the world’s population. Nonalcoholic fatty liver disease is the most prevalent form of liver disease in the United States, affecting over 20% of the population. 1 Of those people afflicted with nonalcoholic fatty liver disease, 20% to 25% will succumb to a liver-associated death within 10 years. The most common early pathological change associated with liver disease is steatosis (accumulation and retention of fat in liver cells) followed by steatohepatitis (inflammation of the liver with concurrent fat accumulation). If the insult responsible for steatosis persists, more severe disease pathologies such as fibrosis and cirrhosis can develop (Figure 1, panel A is a flow chart of liver disease progression). Steatosis (also known as fatty liver) is characterized by fat infiltration in the hepatocytes of the liver. These increased lipids account for over 5% of hepatic wet weight in a steatotic liver (Figure 1, panels B and C are a pathological representation). 2 Steatosis is a critical step in the progression of liver disease as its attenuation has been suggested to prevent more severe hepatic damage. Previous studies in both rodents and humans have also shown that the presence of fat in the liver induces metabolic changes that render the liver susceptible to more severe pathologies (eg, fibrosis) caused by exposure to hepatotoxicants. 3 Therefore, a thorough understanding of how a toxicant causes fatty liver could be crucial in determining that toxicant’s role in the progression of liver disease.
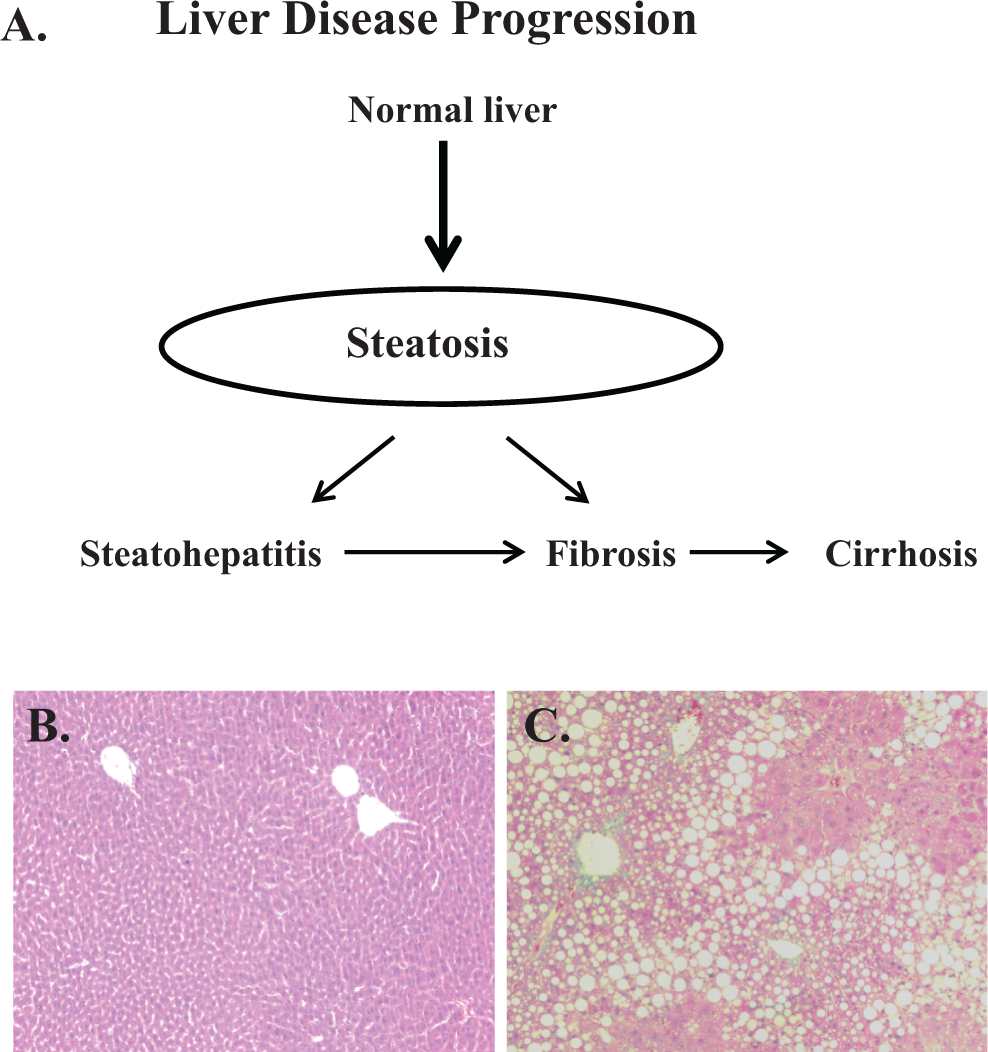
The progression of liver disease and histopathology of steatosis. A flow chart for liver disease progression is depicted in A, and representative photomicrographs are shown depicting steatosis in C57BL/6 mice following 4 weeks of an isocaloric maltose-dextrin (B) diet or ethanol-containing (C) diet.
There are multiple, diverse biochemical perturbations that could be responsible for the development of steatosis. Hepatic lipid accumulation generally occurs when the influx of lipids via increased import and synthesis exceeds the ability of hepatic fat clearance by lipid breakdown and export. 4,5 For example, antiarrhythmic and antiretroviral drugs induce hepatic fat accumulation by impairing mitochondrial function leading to attenuated hepatic lipid breakdown. 6 Furthermore, antibiotics and antidepressants cause steatosis by preventing the formation of very-low-density lipoproteins (VLDLs) leading to decreased hepatic lipid clearance. 7 Increased peripheral lipolysis and subsequent export of free fatty acids from adipose tissue into the liver due to insulin resistance has also been shown to be a mechanism for steatosis. 8,9 Finally, a causal role of increased cytokine production (eg, tumor necrosis factor-alpha [TNF-α]) has been determined in alcohol-induced steatosis. 10 It is possible that steatosis-inducing chemicals could be causing this pathological condition via one or more of these biochemical perturbations.
Although alcohol consumption, 11 the metabolic syndrome, 12 and hepatitis 13 are all common causes of liver disease, studies in humans have demonstrated a strong correlation between chronic toxicant exposure and liver disease seemingly unrelated to other confounding factors (eg, alcohol consumption). For example, Cave et al 14 reported that industrial workers exposed to vinyl chloride for an average of nearly 19 years had increased pathological indices of liver disease. Specifically, over 80% of liver biopsies sampled from 25 industrial workers chronically exposed to vinyl chloride displayed steatosis and steatohepatitis. Of the workers with steatohepatitis, 55% also presented with fibrosis 14 ; similar results were mirrored in other human studies. 15 –18 However, these studies do not fully characterize the biochemical mechanisms by which chemicals could contribute to the development of liver disease.
The objective of this study was to identify putative biochemical mechanisms by which well-studied chemicals cause the accumulation of fat in the liver. Integrated Risk Information System (IRIS) contains a rich database of environmental chemicals with substantial repeated-dose toxicity information. Human health assessments provided by the IRIS program are based on high-quality science and are highly accessible; therefore, the IRIS database was mined to identify steatosis-inducing chemicals.
Materials and Methods
The IRIS database was searched (October 2011) for chemicals that resulted in fatty liver effects. The detailed selection criteria are provided in Figure 2. The selection of chemicals was not limited to those in which steatosis was the critical effect used to derive an oral or inhalation reference value by IRIS. Briefly, the IRIS database was searched using the term fatty in the search engine (criterion 1 in Figure 2) to ensure that steatosis was observed and to discern this pathology from other lesions (eg, vacuolization). This search revealed a select number of chemicals that were associated with the induction of fatty liver in experimental studies (criterion 2 in Figure 2). Literature searches were performed on these chemicals using PubMed to examine their link to several potential mechanisms known to cause steatosis. For example, mitochondrial impairment is a known contributory factor in fatty liver disease. By reviewing the published literature, the role that a given chemical may play in mitochondrial impairment was determined. A lack of supporting studies about a chemical’s mechanistic association with steatosis excluded it from further investigation in this particular analysis (criterion 3.1 in Figure 2). For some chemicals, mechanistic support could only be provided from a nonrodent model (eg, bacteria), and although information of this nature is not definitive, it can still provide insight into how a given chemical may induce steatosis. Other criteria evaluated the quality of the experimental study in which a chemical was shown to cause steatosis (criterion 3.2 in Figure 2). For example, a chemical was excluded if studies relating to its potential to cause steatosis had an inadequate number of animals (less than 4, recognized as the minimum number of animals per dose group for toxicity testing by the Organization for Environmental Cooperation and Development) 19 treated per dose group. A high rate of mortality in steatosis-related studies unrelated to chemical exposure (ie, due to infection) also resulted in a chemical being excluded from this investigation. The lack of a control group in a study led to a chemical being removed from consideration or if a comparison of fatty liver incidence could not be made between control and treated groups. Also, a chemical was removed from the analysis if the reported incidence of steatosis was not statistically significantly (P > .05) increased as determined by Fisher's exact test. Furthermore, if a chemical was shown to cause fatty liver in a single study only written in a foreign language, the chemical was deemed invalid because the quality of the study could not be verified. It was also required that a chemical be shown to cause steatosis in an experimental study of at least subchronic duration (ie, more than 30 days) and repeated exposure by the oral or inhalation route. A chemical with evidence for causing steatosis via an acute-duration exposure only (ie, a single or short-term exposure ≤24 hours) was excluded for further analysis. Finally, chemicals exposed via inhalation were withdrawn if no physiologically based pharmacokinetic (PBPK) models were available to accurately convert inhalation concentrations to equivalent oral doses (criterion 3.3 in Figure 2). Criteria 3.2 and 3.3 were not applied when assessing supportive mechanistic studies performed in other models (eg, in vitro).
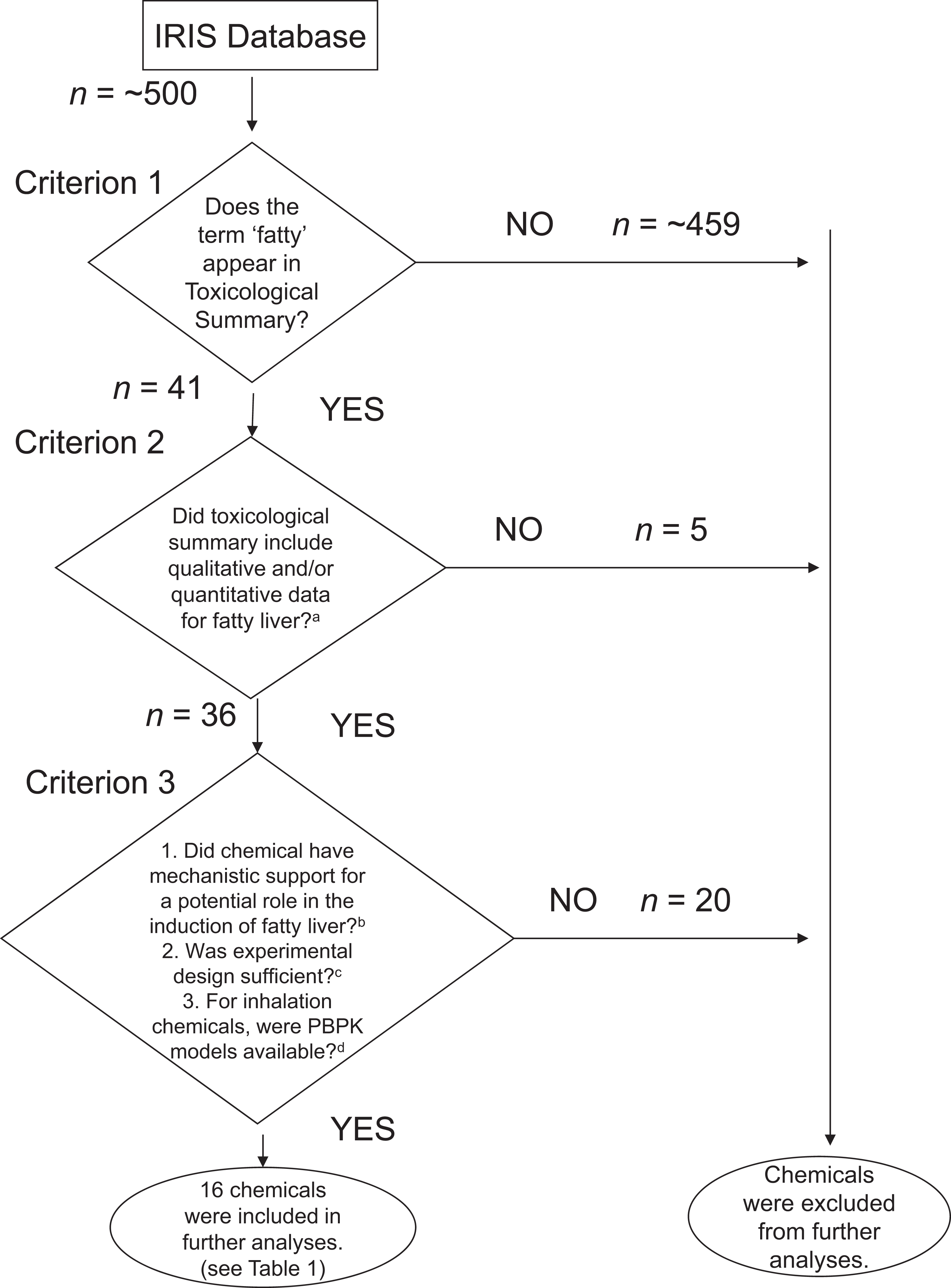
Flow chart for selection of chemicals. The selection of chemicals to include in our analyses was based on various criteria as depicted in this flow chart. a Chemicals excluded based on criterion 2: cerium oxide, naphthalene, nitrobenzene, acetone, and chromium (III). b Chemicals excluded based on criterion 3.1: Bromoform, dibromochloromethane, disulfoton, isoxaben, londax, merphos oxide, metalaxyl, nustar, 1,1,1,2-trichloropropane, and trans-1,2-dichloroethylene. c Chemicals excluded based on criterion 3.2: 1,2-dichloropropane, dichlorvos, N, N-dimethylformamide, ethyl chloride, phosphine, 1,2,3 trichloropropane, 1,1,2,2-tetrachloroethane, bentazon, and 1,1,1-trichloroethane. d Chemicals excluded based on criterion 3.3: antimony trioxide.
After identifying chemicals that met all of the criteria, relationships were examined between dose-response and various mechanisms (eg, mitochondrial impairment) hypothesized to be involved in the induction of steatosis. No observed adverse effect levels (NOAELs) and lowest observed adverse effect levels (LOAELs) for fatty liver induction were identified for each chemical from available experimental studies for a given toxicant (Table 1). To provide an accurate assessment of NOAEL/LOAEL values, all doses were duration adjusted and inhalation NOAELs and LOAELs were converted to equivalent oral doses in mg/kg-day. For vinyl chloride and carbon tetrachloride, only inhalation points of departure were available. Because PBPK models are available for these chemicals, they were used to develop estimates of equivalent oral doses on the basis of concentrations (area under the concentration [AUC] versus time) for parent chemical in the liver, the site of anticipated toxicity. To ensure consistency with hepatic toxicity values for other chemicals in this analysis, the PBPK methodologies utilized in the IRIS Toxicological Reviews for vinyl chloride 35 and carbon tetrachloride 36 were used. These include PBPK models for vinyl chloride by Clewell et al 37 and for carbon tetrachloride by Thrall et al. 38 For both chemicals, inhalation toxicity was observed and PBPK modeling was conducted in rats. Partition coefficients and metabolic parameters from these publications were included in the respective PBPK models. Simulations for inhalation and oral exposures were performed until AUC values changed less than 0.01% per hour. Eight-hour inhalation simulations and single oral bolus dose scenarios were conducted for both chemicals, with simulation times being roughly 6 hours for vinyl chloride and 13 hours for carbon tetrachloride. The AUC values for parent chemical in the liver were determined for each inhalation point of departure, and iterative simulations of oral exposures were conducted to determine oral doses resulting in the same AUC value for each inhalation point of departure.
Summary of IRIS Chemicals Included in the Analyses of Mechanisms of Steatosis.a
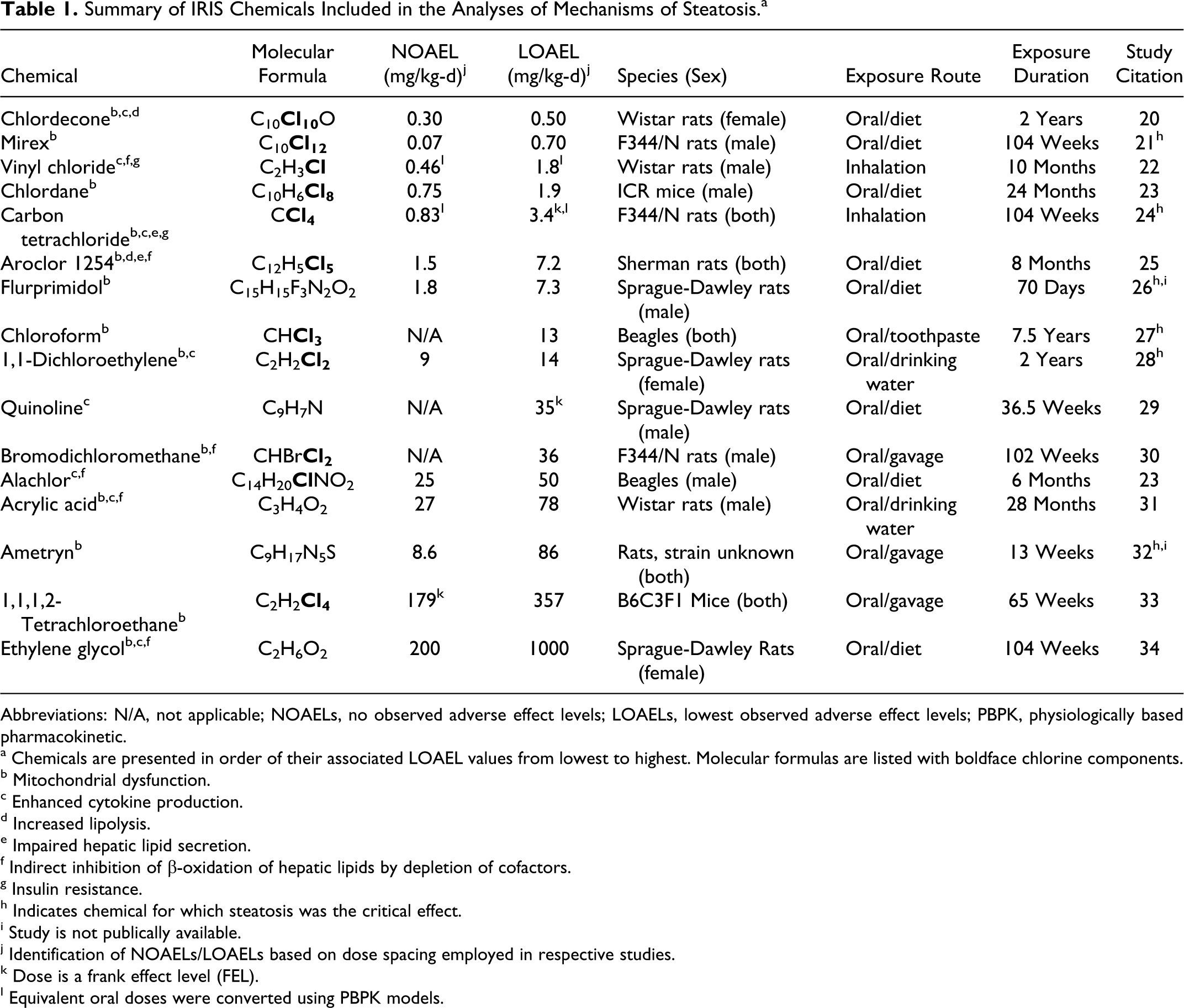
Abbreviations: N/A, not applicable; NOAELs, no observed adverse effect levels; LOAELs, lowest observed adverse effect levels; PBPK, physiologically based pharmacokinetic.
a Chemicals are presented in order of their associated LOAEL values from lowest to highest. Molecular formulas are listed with boldface chlorine components.
b Mitochondrial dysfunction.
c Enhanced cytokine production.
d Increased lipolysis.
e Impaired hepatic lipid secretion.
f Indirect inhibition of β-oxidation of hepatic lipids by depletion of cofactors.
g Insulin resistance.
h Indicates chemical for which steatosis was the critical effect.
i Study is not publically available.
j Identification of NOAELs/LOAELs based on dose spacing employed in respective studies.
k Dose is a frank effect level (FEL).
l Equivalent oral doses were converted using PBPK models.
After ensuring that doses were duration and dose (mg/kg-day) adjusted, the chemicals were organized by their LOAEL values to compare their respective potencies in the development of steatosis. The possible biochemical mechanisms by which the chemicals could induce fatty liver were also identified. Each chemical included in the analysis was evaluated for all known biochemical mechanisms of fatty liver induction. The ranking of chemicals by potency was then analyzed to examine possible trends. Chemicals that were determined to be the most potent in the development of fatty liver were investigated for comparisons of both structure and their respective hypothesized mechanisms for steatosis to determine whether commonalities existed among the chemicals examined that could explain the observed differences in the potencies.
Results and Discussion
Results
The number of chemicals eliminated from the study and by what criteria is presented in Figure 2. Specifically, criterion 1 resulted in 41 chemicals being included, of which 36 met the requirements for criterion 2. Ten chemicals were excluded based on criterion 3.1, 9 according to criterion 3.2, and 1 according to criterion 3.3; this elimination process resulted in 16 chemicals for inclusion in the study. Table 1 shows the 16 chemicals in this investigation of mechanisms of hepatic fat accumulation organized by LOAEL values. Of the 16 chemicals investigated in the current study, six list steatosis as the critical effect. For the remaining 10 chemicals, other nonliver effects were identified as more sensitive. With regard to any structural similarities between the chemicals, it was determined that the majority of the investigated chemicals (11 of 16) were chlorinated (≥1 chlorine molecule in the chemical structure; Table 1). The PBPK models were used to estimate oral equivalent doses for inhalation point of departure values. For each chemical, simulations of inhalation exposures were uncomplicated—both blood and liver AUC values increased with exposure time and reached a plateau (data not presented); oral simulations demonstrated a rapid increase and biphasic decline in tissue concentrations following oral exposures. Differences in blood and liver concentrations reflected chemical-specific liver–blood partition coefficient values. Oral equivalents to inhalation points of departure are presented in Table 1. Because of the role that hepatic metabolism plays in the clearance of vinyl chloride and carbon tetrachloride (parent) compounds and the portal circulation from the gastrointestinal tract to the liver, it is not surprising that the ratios of NOAEL-LOAEL values were not completely consistent between routes of administration. Relative to inhalation point of departure values, a disproportionate difference in oral equivalents was observed for vinyl chloride, which may have its basis on high rates of metabolism of the parent compound in liver, especially at low exposure concentrations. Given the effective metabolism of vinyl chloride, higher doses are needed to reach the level of parent chemical in the liver. Once oxidative metabolism is saturated, an increased fraction of the parent chemical remains available for interaction with tissues or for glutathione S-transferase–mediated metabolism, which is consistent with what is known about vinyl chloride’s mode of action. In contrast, a nearly proportionate relationship was seen in oral equivalents to inhalation point of departure values for the more slowly metabolized carbon tetrachloride.
Among the chemicals with authentic oral dose-response information, chlordecone, mirex, and chlordane were the most potent with respect to fatty liver induction (Figure 3). Once adjusted from inhalation to oral exposures, the potencies of vinyl chloride and carbon tetrachloride were similar to these chemicals. After reviewing the possible mechanisms by which various chemicals could induce steatosis, it was found that impaired mitochondrial function, insulin resistance, decreased VLDL synthesis, and increased cytokine production (eg, TNF-α) were all possible biochemical perturbations that could lead to hepatic fat accumulation due to chemical exposure and are discussed in further detail below. It was also determined that a single chemical could be acting via multiple hypothesized mechanisms to induce steatosis.
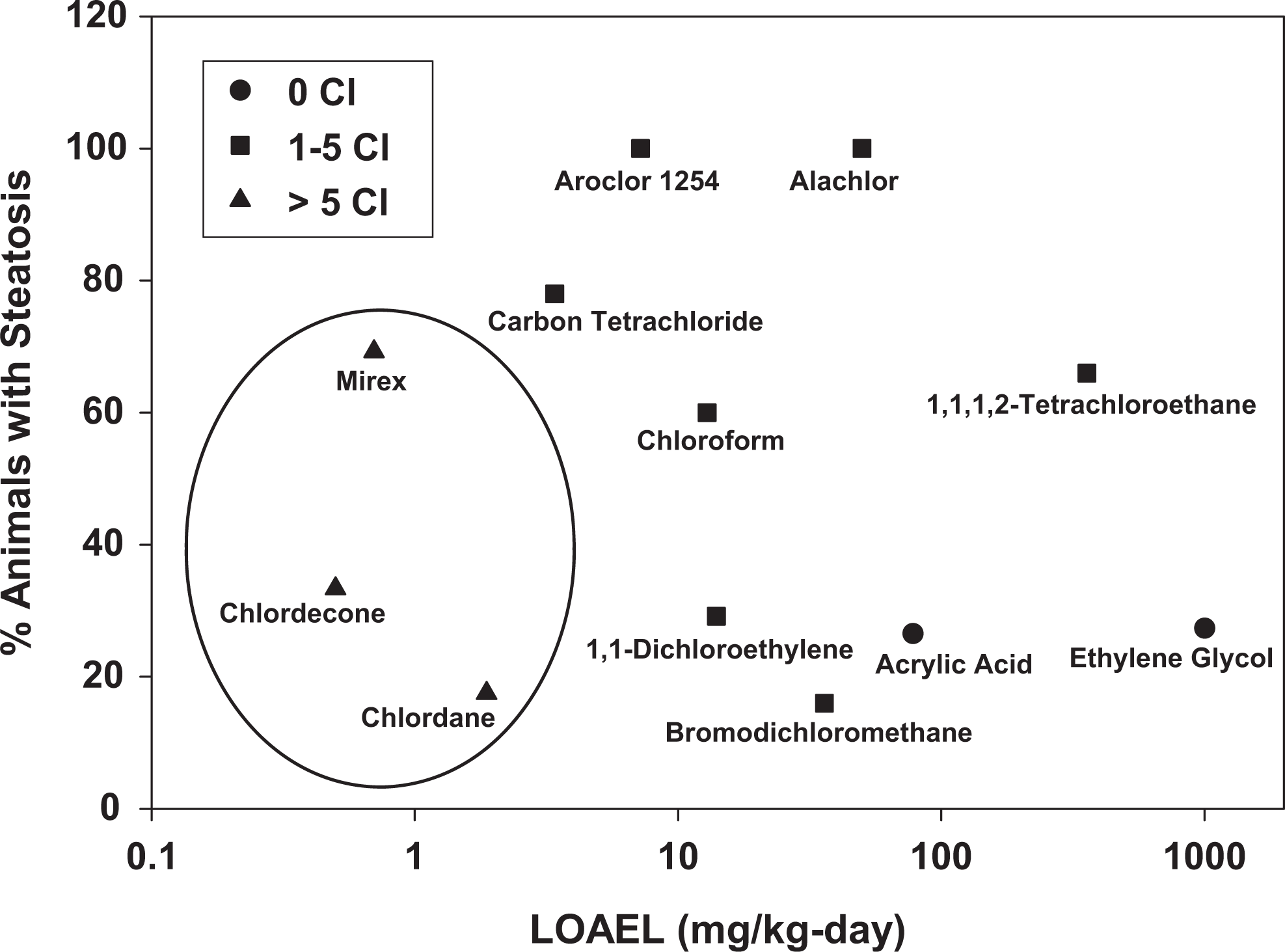
Dose-responses for chemical-induced steatosis. The 3 circled chemicals (chlordecone, mirex, and chlordane) were determined to be the most potent with respect to the induction of fatty liver following oral (dietary) treatment. These chemicals not only share a common mechanism of mitochondrial dysfunction, but they also have similar molecular structures with a high degree of chlorination (≥8 chlorine [Cl] molecules). Incidence data for vinyl chloride, quinoline, ametryn, and flurprimidol were not available.
Impaired Mitochondrial Function
One of the main processes responsible for hepatic lipid breakdown is mitochondrial β-oxidation. 39 Inhibition of mitochondrial function, via decreased supply of necessary oxidized cofactors (eg, NAD+), is a known mechanism for impaired mitochondrial β-oxidation. Therefore, compounds that hinder mitochondrial function may be potential mediators of steatosis (Figure 4). Of the mechanisms potentially associated with the induction of steatosis, mitochondrial impairment (eg, induction of mitochondrial permeability transition, decreased mitochondrial respiration, impaired mitochondrial electron transport, attenuated mitochondrial transmembrane potential, mitochondrial structural damage, etc) was the most common with 13 of 16 chemicals shown to impair mitochondrial function across multiple studies. For example, chlordecone, mirex, chlordane, 1,1,1,2-tetrachloroethane, chloroform, and Aroclor 1254 disrupt mitochondria in in vitro experiments utilizing primary rat liver cells. 40 –44 Similarly, intraperitoneal injections of carbon tetrachloride and 1,1-dichloroethylene cause mitochondrial impairment in the livers of various mouse strains. 45,46 Because these chemicals were explicitly tested to determine how they alter mitochondrial function in rodent livers, these studies provide strong support that these chemicals may be causing steatosis via impaired hepatic mitochondrial function.
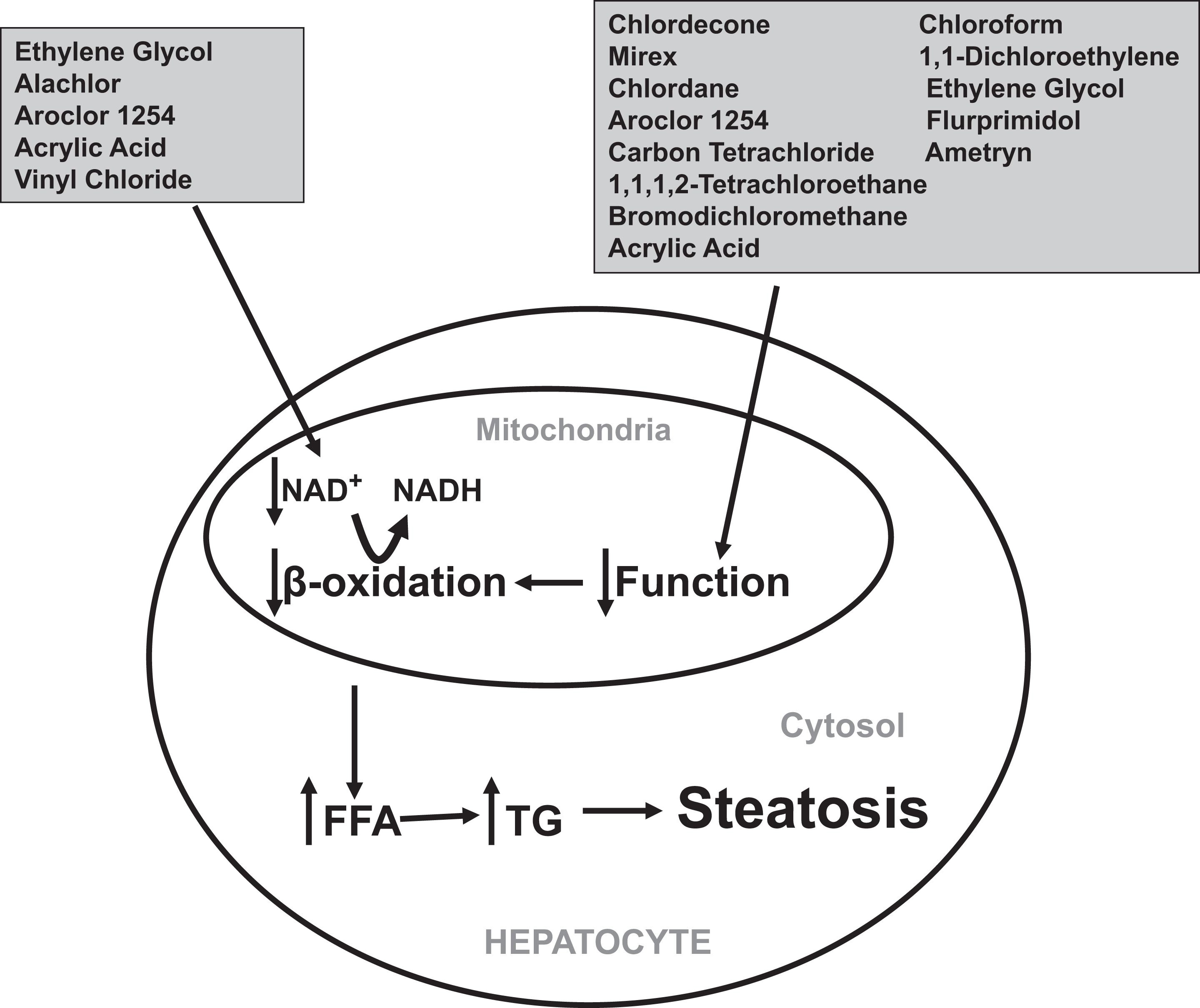
The role of mitochondrial dysfunction and inhibition of β-oxidation in the induction of steatosis. Chemicals can impair hepatic β-oxidation of lipids by preventing the regeneration of the required cofactor NAD+ via mitochondrial dysfunction or metabolic-induced depletion. FFA indicates free fatty acids; TG, triglycerides.
For some chemicals, evidence for a metabolite was found in the impairment of mitochondrial function. For example, bromodichloromethane is metabolized to formic acid in rats, and this metabolite disrupts mitochondrial function in Drosophila melanogaster. 47,48 Calcium oxalate monohydrate, a metabolite of ethylene glycol, impairs mitochondrial function in in vitro experiments using primary rat kidney cells. 49 Whereas the effects of the respective metabolites of these chemicals have not been directly tested on hepatic mitochondria in vivo or in vitro, it is plausible that similar mitochondrial impairment would be observed in the liver, possibly leading to the onset of steatosis.
Although mitochondrial impairment has not been evaluated for some chemicals in rodent studies, a role in disrupting mitochondrial function has been investigated in other experimental models. For example, ametryn attenuates electron transport in bacteria (ie, Rhodopseudomonas capsulata and Rhodopseudomonas sphoeroides). 50 Similarly, flurprimidol decreases mitochondrial functioning in European black alder leaves. 51 Due to a dearth of experimental studies in rodents for these chemicals, it is not possible to ascertain with certainty the mechanisms by which these toxicants may cause steatosis.
Of the 13 chemicals hypothesized to cause fatty liver via mitochondrial impairment, 9 were chlorinated. Furthermore, the 3 most potent chemicals with respect to fatty liver induction following oral (dietary) treatment (ie, chlordecone, mirex, and chlordane) were chemicals with a high degree of chlorination (≥8 chlorine molecules). The potentially enhanced ability of chlorinated chemicals to cause mitochondrial impairment is supported by previous laboratory studies which determined that mitochondrial impairment is directly related to the degree of chlorination of a respective molecule. 52,53 Furthermore, exposure to polychlorinated biphenyls (a class of organic compounds including Aroclor 1254 and made up of 2-10 chlorine atoms) has been correlated with steatosis and increased liver disease in American adults. 54 Although mitochondrial impairment could possibly explain the increased potency of the polychlorinated chemicals causing fatty liver following oral treatment, it is not the only mechanism by which chlorinated chemicals could induce steatosis (Table 1). Furthermore, vinyl chloride and alachlor, both chlorinated, were not found to impair mitochondrial functioning. 55 –57 The lack of mitochondrial impairment by these chemicals could be because they are not polychlorinated. Taken together, these findings suggest that the increased potency for causing fatty liver of polychlorinated chemicals following oral exposure observed in our analysis could possibly be explained by their enhanced ability to impair mitochondrial function.
Although mitochondrial impairment plays a major role in decreased β-oxidation, it is not the sole cause. A separate mechanism that could be responsible for decreased β-oxidation of hepatic lipids is a metabolic-induced depletion of cofactors (eg, NAD+) that are needed for lipid breakdown. A similar mechanism is proposed for steatosis due to ethanol exposure. Specifically, ethanol is metabolized to acetaldehyde via alcohol dehydrogenase and then further oxidized to acetate by aldehyde dehydrogenase; both enzymes require the cofactor NAD+. This oxidative metabolism of ethanol increases the ratio of NADH to NAD+ which subsequently attenuates lipid metabolism in the liver. 58 Therefore, chemicals that are metabolized similarly to ethanol may share a common mechanism of steatosis induction. Ethylene glycol and vinyl chloride are both metabolized via alcohol dehydrogenase and aldehyde dehydrogenase therefore decreasing the levels of NAD+ and potentially inhibiting β-oxidation, leading to the accumulation of hepatic lipids. 59,60 Similarly, Aroclor 1254 increases the activity of aldehyde dehydrogenase in the liver of rats. 61 β-oxidation also requires a series of related enzymes (eg, acyl CoA dehydrogenase, β-ketoacyl CoA thiolase, etc) to breakdown lipids. The blunting of one or more of these enzymes could compromise the entire lipid oxidation process. Acrylic acid is a known inhibitor of β-oxidation and prevents lipid breakdown by inactivating β-ketoacyl CoA thiolase, the enzyme that catalyzes the final step in the process. 62 Therefore, it is likely that acrylic acid may cause steatosis by blocking the activity of this key enzyme in the β-oxidation pathway.
Insulin Resistance
Insulin resistance refers to the biochemical scenario in which normal amounts of the hormone are no longer sufficient to facilitate the transport of glucose into a cell due to disrupted insulin signaling. Impaired insulin signaling can cause tissue-specific insulin resistance. 63 The major target tissues of insulin resistance are muscle, adipose, and liver. 64 Adipocyte and hepatic insulin resistance can have important roles in the development of steatosis. A decrease in insulin sensitivity can cause biochemical changes in the liver (ie, impaired VLDL synthesis and decreased lipid oxidation) 65,66 and in the adipose (ie, increased peripheral lipolysis) 65,67 that lead to hepatic lipid accumulation and subsequently steatosis (Figure 5). Although insulin resistance is commonly associated with obesity and the metabolic syndrome, recent studies have also shown a role for chemical-induced insulin resistance. Of the 16 chemicals investigated, it was hypothesized that carbon tetrachloride and vinyl chloride may induce fatty liver via insulin resistance, as both compounds have been shown to cause insulin resistance in the published literature. Carbon tetrachloride impairs insulin signaling in rats following chronic exposure as indicated by decreased insulin-stimulated total body glucose disposal and increased steady-state serum insulin levels. 68 Vinyl chloride caused insulin resistance in factory workers occupationally exposed to the compound at a chemical plant in Louisville, Kentucky, indicated by increased concentrations of insulin in serum samples. 14 Based on these findings, a causal role of carbon tetrachloride and vinyl chloride in the development of steatosis may be explained, at least in part, by the their respective abilities to cause insulin resistance. It is feasible that other chemicals included in this work may also cause insulin resistance leading to steatosis, but their exposure effects on insulin sensitivity parameters have yet to be determined experimentally.
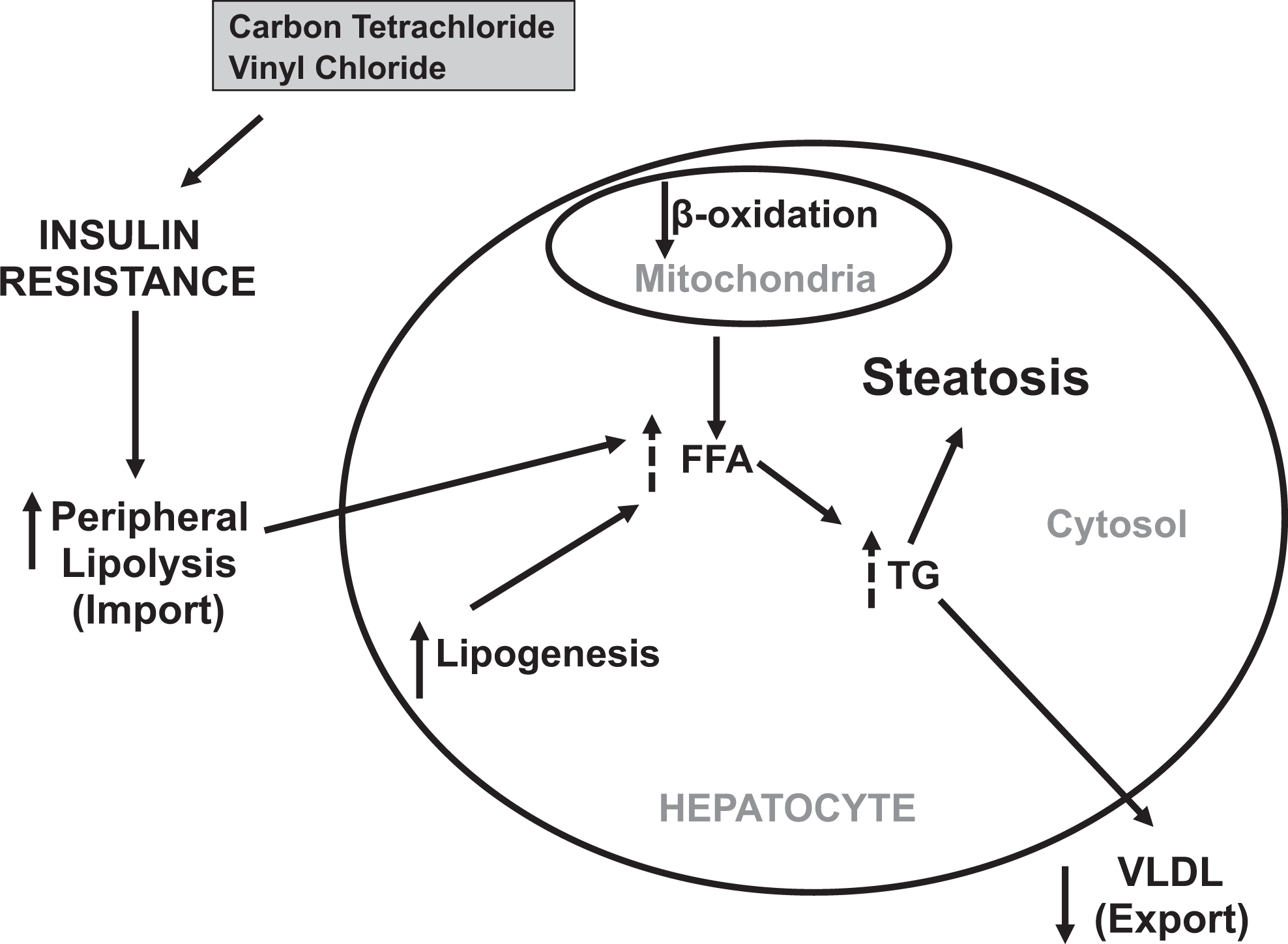
The role of insulin resistance in the development of steatosis. Insulin resistance can cause fat accumulation in the liver by multiple biochemical pathways. Carbon tetrachloride and vinyl chloride are 2 chemicals that are hypothesized to induce fatty liver via insulin resistance. FFA indicates free fatty acids; TG, triglycerides; VLDL, very-low-density lipoprotein. Dotted lines indicate indirect effects of insulin resistance.
Impaired Hepatic VLDL Synthesis and Secretion
In normal liver function, excess free fatty acids are esterified to triglycerides through a series of biochemical processes, these triglycerides are ultimately secreted in the plasma as VLDLs. 69 Impairment of any step within this lipid export pathway could result in the development of steatosis. For example, alcohol exposure, a known causal agent of steatosis, decreases VLDL secretion in rats. 70 Aroclor 1254 is hypothesized to induce fatty liver by a similar mechanism supported by the in vitro observation that fat accumulation in rat hepatocytes correlated with decreased VLDL secretion following exposure. 71 Furthermore, carbon tetrachloride administered to rats via a stomach tube was shown to cause fatty liver in a study in which the authors observed decreased VLDL synthesis and hypothesized that this was the cause of the fat accumulation. 72 Therefore, both Aroclor 1254 and carbon tetrachloride could contribute to steatosis by impairing different steps in hepatic lipid secretion; Aroclor 1254 prevents the secretion of triglycerides as VLDLs, whereas carbon tetrachloride attenuates the formation of VLDLs (Figure 6). A role for other chemicals examined in the present study in preventing hepatic lipid secretion has not been determined.
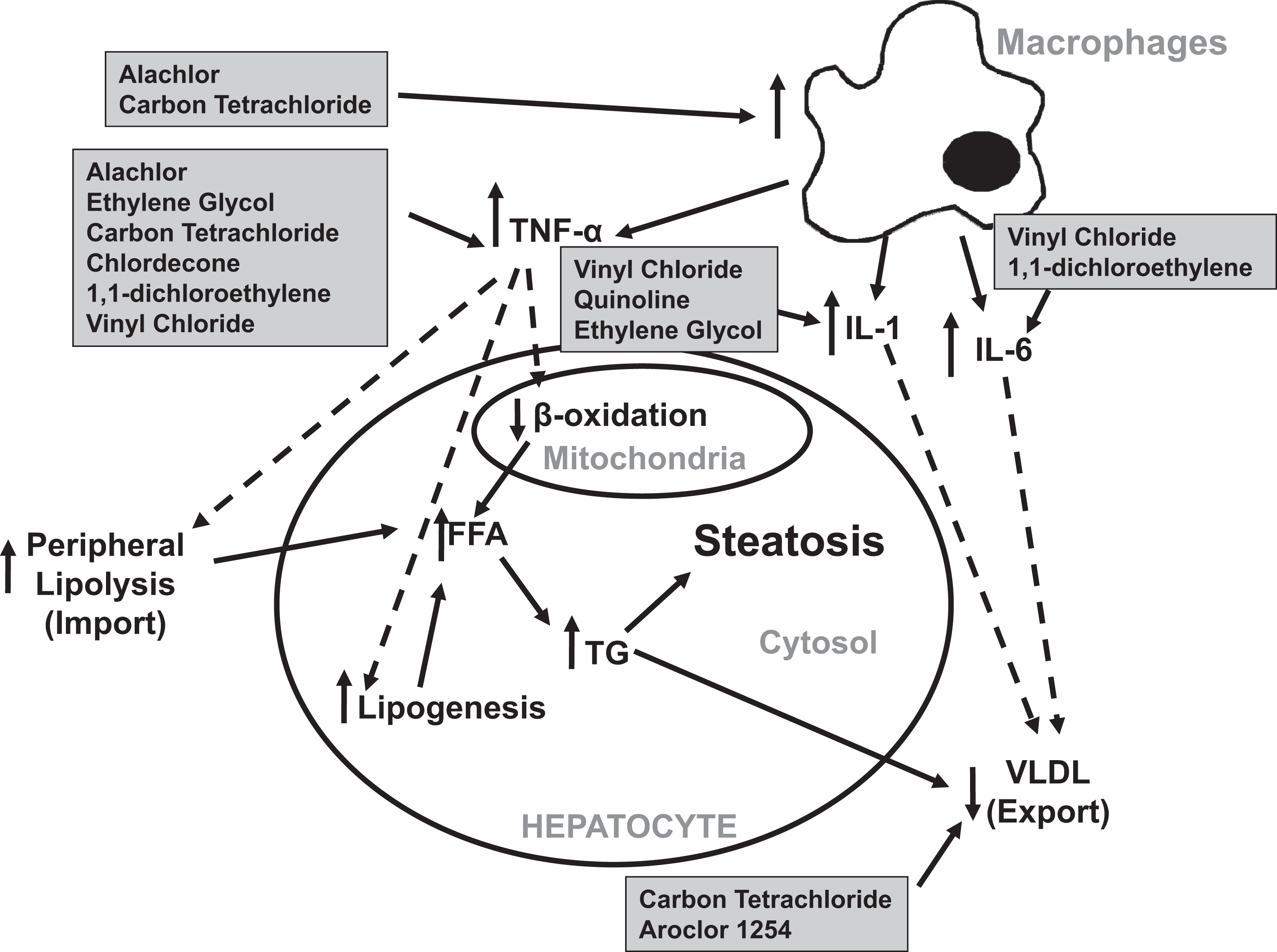
The role of impaired hepatic lipid secretion and increased cytokine production in the induction of steatosis. Macrophage activation and cytokine release can induce fatty liver via numerous biochemical manipulations. FFA indicates free fatty acids; TG, triglycerides; VLDL, very-low-density lipoprotein. Dotted lines indicate indirect effects of chemicals via cytokines and transcription factors while full lines indicate direct actions of chemicals.
Increased Cytokine Production
The role of cytokines in the development of steatosis is multifaceted (Figure 6). For example, TNF-α promotes lipolysis in 3T3-F442A adipocytes in vitro, increases the production of hepatic lipids in rats, and impairs the β-oxidation of fatty acids in isolated rat hepatocytes. 73 –75 Further support for a role of cytokines in fatty liver is provided by a mouse study in which the deletion of the tumor necrosis factor receptor-1 gene completely blunted steatosis following alcohol administration. 76 Of the 16 chemicals examined in the present study, 8 have been shown to increase cytokines known to play a role in causing hepatic lipid accumulation. The major producer of cytokines in the liver is a specialized cell type referred to as Kupffer cells, the resident macrophages of the liver. 77 Although the effects of alachlor and ethylene glycol have not been directly tested in Kupffer cells, alachlor enhanced the production of TNF-α from mouse peritoneal macrophages treated with lipopolysaccharides under ex vivo conditions. 78 Similarly, ethylene glycol (in the form of a hydrogel) upregulates the messenger RNA expression of TNF-α and interleukin (IL)-1 in bone marrow-derived primary macrophages in vitro. 79 Both IL-1 and IL-6 inhibit the lipid export process in HepG2 cells in vitro. 80 Carbon tetrachloride increases TNF-α synthesis via stimulation of Kupffer cells in rats administered the compound via intraperitoneal injection. 81 Although there is a strong evidence for a role of Kupffer cells in mediating cytokine release due to carbon tetrachloride exposure, a similar mechanism could be likely for alachlor- and ethylene glycol-induced cytokine production.
Other chemicals have been determined to upregulate cytokines without evidence of macrophage involvement. Chlordecone increases splenic T-cell TNF-α secretion in mice implanted subcutaneously with pellets containing the chemical. 82 Similarly, gavage treatment of 1,1-dichloroethylene induces TNF-α and IL-6 secretion in mice. 83 In an in vitro study, it was determined that quinoline facilitated the release of IL-1 from human monocytes. 84 Vinyl chloride increased the levels of TNF-α, IL-6, and IL-1 in the serum of vinyl chloride–exposed factory workers. 14 Because the effects of these chemicals on Kupffer cells have not been directly tested, a causal role for these hepatic macrophages in the development of hepatic fat accumulation cannot be fully determined. However, these toxicants have been shown to cause both steatosis and cytokine release from various cell lines. Given the abundant ability of activated Kupffer cells to indirectly lead to steatosis, it could be hypothesized that Kupffer cells are causally involved in the biochemical pathway by which chlordecone, quinoline, 1,1-dichloroethylene, and vinyl chloride are causing this hepatic pathological condition.
The Induction of Steatosis via Multiple Mechanisms
It is apparent that some chemicals have multiple hypothesized mechanisms that may be responsible for their causal role in fatty liver. It is possible that multiple mechanisms for an individual chemical’s involvement in the development of steatosis may not be mutually exclusive but instead entail a series of connected pathways that eventually lead to the accumulation of fat in the liver. For example, Table 1 illustrates that mitochondrial impairment, increased cytokine production, decreased hepatic lipid secretion, and insulin resistance are all probable biochemical disruptions by which carbon tetrachloride could cause steatosis; these hypothesized mechanisms could potentially have synergistic or additive roles in hepatic fat accumulation due to the exposure of this chemical as suggested by Weber et al. 85 Figures 5 and 6 illustrate the pathways by which both insulin resistance and increased cytokine production may contribute to steatosis caused by exposure to carbon tetrachloride. Both pathways share common biological perturbations (ie, increased lipolysis, lipogenesis, and impaired β-oxidation) resulting in hepatic fat accumulation. It is possible that these two pathways could be connected as TNF-α has been linked to the development of insulin resistance in humans and has also been shown to induce mitochondrial impairment in mouse fibrosarcoma cells. 86,87 For example, one possible scenario could be that carbon tetrachloride upregulates TNF-α which then attenuates insulin signaling and leads to increased lipid import and production as well as decreased hepatic fat clearance. Concomitantly, TNF-α causes mitochondrial impairment and subsequent inhibits the β-oxidation of hepatic lipids. The net effect of TNF-α and insulin resistance is increased supply of lipids to the liver combined with a decrease in fat breakdown and export, culminating in carbon tetrachloride-induced steatosis. Similar scenarios involving numerous biochemical disturbances could be occurring for chemicals that have multiple hypothesized mechanisms for the induction of fatty liver. Ultimately, it is critical to identify the key steps among mechanisms of steatosis to provide the best understanding of how a chemical could cause fatty liver.
The Sensitization Effect of Steatosis
A predominant theory in liver research is the “two-hit” hypothesis also known as sensitization which proposes that hepatic steatosis is not an inert pathology but may actually sensitize the liver to a second insult. 3 The sensitization effect of steatosis has been observed in multiple experimental studies. For example, Yang and colleagues determined that obese mice with fatty livers were more sensitive to lipopolysaccharide-induced steatohepatitis compared to their lean counterparts that did not have hepatic fat accumulation. 88 The two-hit hypothesis could have major implications on chemical-induced liver disease, as possible cumulative hepatotoxic effects could occur from the combination of chemical exposure and a separate potential stressor (eg, alcohol, obesity, etc). The presence of steatosis due to chemical exposure could sensitize the liver to a second “hit” and greatly increase the risk for liver disease.
The Role of Chemicals in Later Stages of Liver Disease
As described above, steatosis is a common early pathological change associated with liver disease. Although the determination of the mechanisms by which a chemical contributes to the development of hepatic lipid accumulation is critical, how a compound causes later stages of liver disease (eg, fibrosis) is of equal importance. Mirex, chlordecone, and Aroclor 1254 cause steatohepatitis in various experimental animals. 21,40,89 Furthermore, carbon tetrachloride is a commonly used toxicant to induce fibrosis in rodent experimental models. 85 Although exposure to these chemicals may result in later stages of liver disease via the natural progression from steatosis to steatohepatitis to fibrosis, it is possible that these toxicants could be causing more advanced stages/pathologies of liver disease unrelated to and in the absence of steatosis.
In a methionine choline-deficient mouse model of liver disease, Yamaguchi and colleagues determined that the therapeutic inhibition of diacylglycerol acyltransferase 2 prevented steatosis but provided no protection against steatohepatitis or hepatic fibrosis. 90 In a separate study, mice pretreated with ethanol and then lipopolysaccharide develop liver inflammation and necrosis with no steatosis. 91 Given the possible disconnect between steatosis and subsequent liver injury, there is a need to determine the role of toxicants in the development of later stages of liver disease which could potentially result in mortality.
Current Research Needs and Limitations
This study employs a novel method to identify possible mechanisms of chemical-induced steatosis, but it is limited to the current research on hepatotoxicity. As mentioned above, although 36 chemicals on Environmental Protection Agency’s IRIS database caused fatty liver, 20 of these chemicals were withdrawn from further analysis in this study. The chemicals not included could be potent inducers of steatosis but require further investigation. This gap in the scientific literature highlights the need for additional studies of high quality that examine the ability of a chemical to cause fatty liver or that fully investigate the biochemical pathways by which these 16 chemicals may specifically induce steatosis. For example, one possible mechanistic approach would be to test the exposure of one of these chemicals on mice that are genetically deficient for a cytokine known to cause fatty liver (eg, TNF-α). Also, some chemicals included in the study like mirex and quinoline have only a single hypothesized mechanism based on available data. Because chemicals such as these have not been thoroughly researched on a molecular biology/mechanistic level, their potential mechanisms for fatty liver cannot be fully identified. Despite this work relying heavily on the current available literature, it still provides an in-depth analysis into how environmental chemicals could cause steatosis.
Conclusions
This work shows that mitochondrial impairment, insulin resistance, attenuated lipid secretion, and increased cytokine production are potential mechanisms by which environmental chemicals on the IRIS database may cause fatty liver. A significant finding of the study is that the most potent IRIS chemicals with respect to the induction of fatty liver following oral exposure were polychlorinated, and this potency could possibly be due to increased mitochondrial impairment. This work could help provide a framework for the identification of key events in mechanisms of steatosis that could serve as sensitive endpoints for risk assessment. Furthermore, by adopting the data mining techniques applied in this article, one could identify important mechanistic processes for a given critical effect in other target organs. This work also demonstrates the value of the IRIS database for further data mining exercises similar to that described in this article. These findings could also be useful for mixtures/cumulative risk assessment, aiding to categorize chemicals into common liver outcome groupings. The work described here is further important because the results could elucidate mechanisms by which environmental chemicals could be significant inducers of not only steatosis but ultimately advanced liver disease as well.
Footnotes
Acknowledgments
The authors thank Dr Gavin Arteel (University of Louisville) for providing the liver pathology photomicrographs and Dr Kannan Krishnan (Université de Montréal) for providing expert assistance with PBPK modeling. The views expressed in this article are those of the authors and do not necessarily reflect the views and policies of the US Environmental Protection Agency.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.