
Abstract
Pharmaceutical therapies for non-insulin-dependent diabetes mellitus (NIDDM) include plasma glucose lowering by enhancing glucose utilization. The mitochondrial pyruvate dehydrogenase (PDH) complex is important in controlling the balance between glucose and fatty acid substrate oxidation. Administration of pyruvate dehydrogenase kinase inhibitors (PDHKIs) to rats effectively lowers plasma glucose but results in myocardial steatosis that in some instances is associated primarily with atrial and to a lesser degree with ventricular pathology. Induction of myocardial steatosis is not dose-dependent, varies from minimal to moderate severity, and is either of multifocal or diffuse distribution. Ventricular histopathology was restricted to few myocardial degenerative fibers, while that in the atrium/atria was of either acute or chronic appearance with the former showing myocardial degeneration/necrosis, acute myocarditis, edema, endothelial activation (rounding up), endocarditis, and thrombosis associated with moderate myocardial steatosis and the latter with myocardial loss, replacement fibrosis, and no apparent or minimal association with steatosis. The evidence from these evaluations indicate that excessive intramyocardial accumulation of lipid may be either primarily adverse or represents an indicator of other adversely affected cellular processes.
Introduction
Non-insulin-dependent diabetes mellitus (NIDDM) is a disease of rapidly increasing prevalence internationally, and numerous strategies for its treatment are currently being investigated and evaluated. The key requirement of such treatments is adequate control of plasma glucose toward achievement of a more stable glycemic condition and avoidance of the often substantial daily swings in plasma glucose concentration in the untreated diabetic state. Numerous therapeutic strategies target the β-cell of islets of Langerhans in order to enhance insulin secretion, but numerous approaches regarding control of plasma glucose without involvement of hormonal manipulation by either its excretion, for example, through inhibition of renal sodium-glucose co-transporters (SGLTs), or enhanced abstraction and metabolism are being investigated (Bakris et al. 2009; Chao and Henry 2010; Wright, Hirayama, and Loo 2007).
The mitochondrial pyruvate dehydrogenase (PDH) complex is key to the regulation of the balance between glucose and fatty acid oxidation (Mayers et al. 2003; Mayers, Leighton, and Kilgour 2005), as it catalyzes the irreversible decarboxylation of pyruvate to acetyl CoA. Inactivation of PDH occurs by reversible phosphorylation through the PDH kinases (PDHKs) and the PDH phosphatases. Four PDHK isozymes occur, which express distinctive regulatory activities and tissue distributions. Tissue distributions of the different isozymes are distinct: PDHK1, heart; PDHK2, ubiquitous; PDHK3, testis; PDHK4, cardiac and skeletal muscle, the latter tissues being of particular interest for diabetes therapy as they are major utilizers of glucose. Further attraction to PDHK4 as a target of small molecule manipulation lies in its regulation through metabolic condition (Roche et al. 2001; Sugden and Holness 2003), its expression being downregulated by insulin and peroxisome proliferator–activated receptor γ agonists and upregulated in response to high-fat feeding, in starvation and in Zucker Diabetic Fatty (ZDF) rat skeletal muscle. Sustained activation of PDH would be expected to benefit diabetic patients with their associated, multisystemic pathological and functional impairments, by controlling hyperglycemic episodes through stimulating glucose disposal in peripheral tissues and by inhibition of gluconeogenesis. Such sustained activation is induced by inhibition of PDHK, illustrated by stimulation of pyruvate oxidation in rat ex vivo primary hepatocyte cultures, elevated PDH activity in vivo, and lower plasma glucose concentration in obese Zucker rats (Mayers et al. 2003).
Dichloroacetate (DCA) nonspecifically inhibits PDHK and increases liver and skeletal muscle PDH activity in several, experimentally initiated rat diabetes models, namely, dexamethasone-, streptozotocin-, and alloxan-induced diabetes and in the ZDF rat model (Eichner, Stacpoole, and Forsham 1974; Mann et al. 1998; Liu et al. 1998). With the exception of the alloxan-induced diabetic model, DCA administration results in decreased plasma glucose. In hyperglycemic humans, but not in normally fed or nondiabetic individuals, DCA is efficacious in glucose lowering, consistent with PDH activation decreasing the availability of gluconeogenic substrates (Stacpoole, Moore, and Kornhauser 1978). While this glucose-lowering capability of DCA was encouraging as a potential therapy in diabetes, its further development was regarded as unsupportable due to low potency, metabolism, and toxicity (Stacpoole et al. 1990), although the target of specific PDHK inhibition employing specific molecules (PDHKIs) remained attractive.
Xenobiotic-induced shifts in metabolism such as those generated by drugs designed to specifically and potently inhibit PDHKs may have unexpected consequences on tissue function such as hepatocellular, cardiac, and skeletal myocyte excessive lipid accumulation. In studies of up to 28 days duration, conducted during evaluation of the safety of several potential PDHKIs, most dosed animals showed increases in cardiomyocyte lipid of variable degree and some animals were terminated prematurely due to clinical condition. Several animals showed cardiac pathology, primarily involving the atria with or without markedly elevated myocardial steatosis. This report communicates our investigations directed at understanding whether or not the cardiac pathology observed in drug-administered animals was associated with induction of myocardial steatosis and employed two PDHKIs, namely AZD7545 and AZ10219759 (Figure 1).
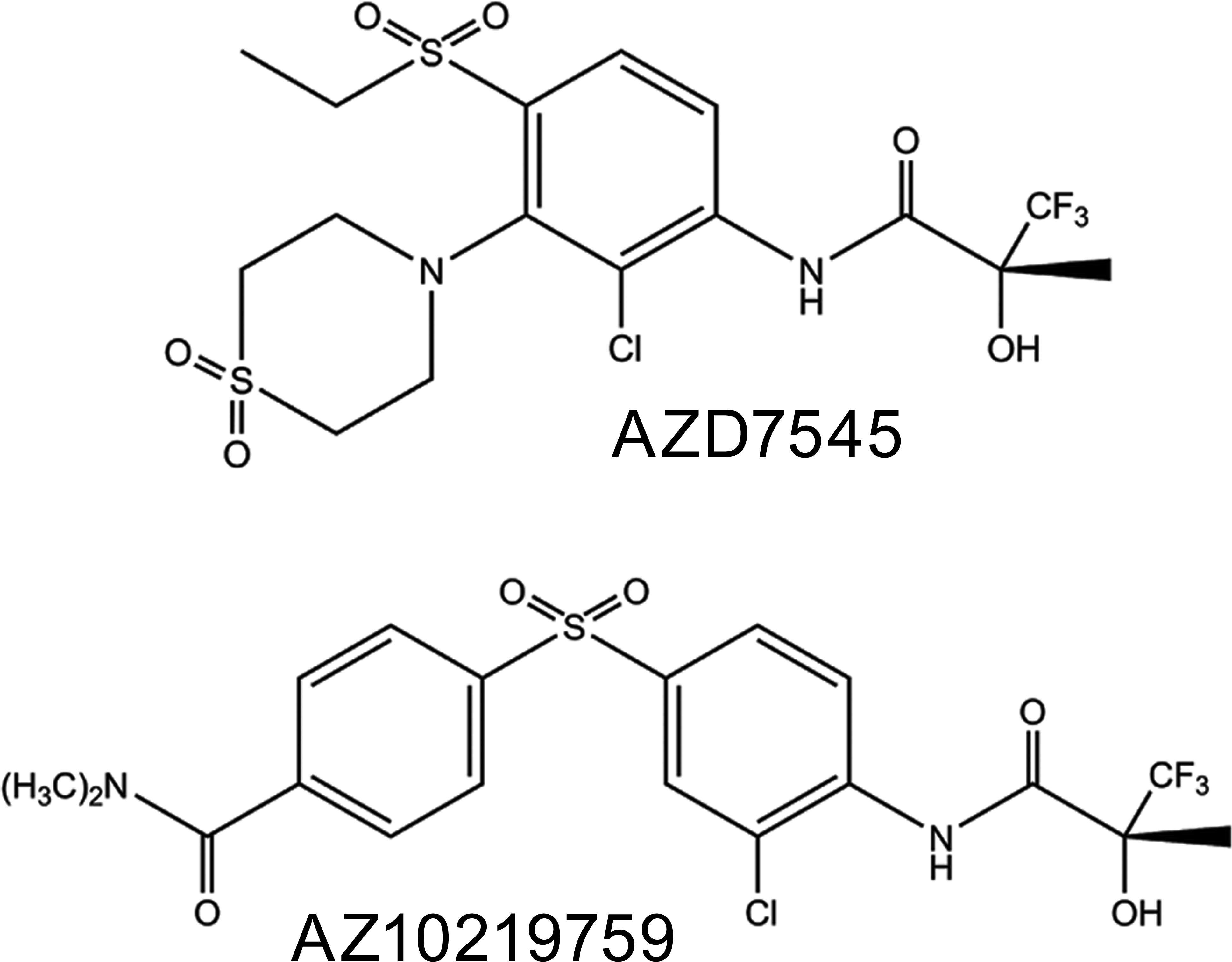
Molecular structures of AZD7545 (4-[3-Chloro-4-[3,3,3-trifluoro-2(R)-hydroxy-2-methyl propionamido] phenylsulfonyl]-N,N-dimethyl benzamide) and AZ10219759 (N-[2-Chloro-3-(1,1-dioxido thiomorpholin-4-yl)-4-(ethylsulfonyl) phenyl]-3,3,3-trifluoro-2 (R)-hydroxy-2- methylpropionamide).
Materials and Methods
Hannover Wistar rats aged 7 to 9 weeks (Charles River, Margate, Kent, UK) were maintained on sawdust in smooth-bottomed, plastic cages, and permitted free access to diet (SDS modified R&M No 1, Special Diet Services, Witham, Essex, UK) and potable drinking water in a climate-controlled room (maintained at a mean 20°C temperature, 12-hr dark/light cycle with approximately 70% relative humidity). Three animals/sex/group were used in the initial 7 day AZ10219759 toxicity study (study 1) and 5 males per group employed in the 7-day investigative AZ10219759 study (study 2). The 28 days drug development study with AZD7545 used 10 males and 10 females per group (study 3). Study 4 was designed to seek a no-effect dose for cardiotoxicity with daily dosing for 28 days and a 28 days recovery (drug-free) period of a cohort of high-dose animals and used 10 males/group. Appropriate controls that received the vehicle only, but otherwise were treated in the same way, were employed in all studies. In studies 1 and 3 and 4, both sexes were evaluated, and numbers of animals were in accordance with those prescribed by regulatory authorities. Study 2 employed males only, as no distinct sex-related differences in cardiac pathology were observed in study 1. Conventional fixation of tissue and processing into paraffin wax was performed in all studies with immunohistochemical and ultrastructural pathological assessments performed in the 7 days investigative study with AZ10219759 (study 2) only.
Clinical biochemistry analyses were performed in each study on plasma samples obtained at necropsy immediately after termination of the animals as indicated subsequently. The studies were conducted in strict adherence to the U.K. Home Office regulations for animal welfare (1986 Animal Scientific Procedures Act).
Study 1: AZ10219759: 7-Day Toxicity Study
The objective of this study was the assessment of systemic toxicity in the rat as a component of the drug development process for this drug candidate. For this study and the investigative study (study 2), AZ10219759 was formulated as a 45-mg AZ10219759/ml suspension in 0.5% w/v hydroxypropyl methyl cellulose (HPMC) in 0.1% w/v aqueous polysorbate 80 and administered by oral gavage at doses of 30-, 150-, or 450-mg AZ10219759/kg, once daily. Controls received the vehicle only.
At necropsy, multiple tissues including the heart were taken for pathological evaluation and lithium heparin (LH)-anticoagulated plasma sampled for markers of multiple organ system toxicity, including creatine kinase and troponin T.
Study 2: AZ10219759: Investigative, 7-Day Study
This study investigated pathological alterations in cardiac samples that were first observed in study 1 and therefore attempted to replicate those findings by employment of the high doses utilized in that study. AZ10219759 was formulated as described previously and administered by oral gavage at doses of either 150- or 450-mg AZ10219759/kg, once daily for 7 days.
At necropsy, only the heart was taken for histopathological, ultrastructural pathological, and immunohistochemical evaluation, and LH-anticoagulated plasma sampled for markers of multiple organ system toxicity, including creatine kinase and troponin I. Note that different troponins were evaluated in studies 1 and 2 in order to assess merits of each in relation to their usage as potential indicators of myocardial damage.
Studies 3 and 4: AZD7545: 28-Day Studies
These studies followed several acute/subacute toxicity studies in which neither cardiac pathology nor altered biochemical indicators of cardiac toxicity were observed in conventionally prepared H&E-stained tissue sections or in plasma samples. For both studies, AZD7545 was formulated as a 30-mg AZD7545/ml suspension in 0.5% w/v HPMC in 0.1% w/v aqueous polysorbate 80 and animals dosed once daily by oral gavage. Controls received the vehicle only. Both of these studies assessed multiple tissues for histopathology and plasma clinical biochemistry.
Study 3
A standard design, pivotal drug development study to investigate systemic toxicity using 10 animals/sex/group, given 0-, 30-, 100-, or 600-mg AZD7545/kg/day for 28 days. On day 16, due to adverse clinical observations that required premature termination of 1 animal given the highest dose, this dose was reduced from 600- to 300-mg AZD7545/kg/day, until scheduled termination on day 29. An extensive tissue list was sampled at necropsy for histopathogical diagnoses and specific cardiotoxicity clinical biochemistry assessments were made using creatine kinase only.
Study 4
A study undertaken to evaluate the dose response of drug-induced, myocardial steatosis, and cardiac pathology in 10 animals/sex/group, given 0-, 5-, 15-, or 300-mg AZD7545/kg/day for 28 days and recovery of satellite groups of high-dose animals for the same drug-free period. An extensive tissue list was sampled at necropsy for histopathogical diagnoses and multiple clinical biochemistry assessments were made including creatine kinase.
Pathology
Animals were killed by administration of halothane 1 day after the final dose. In the 7-day investigative study with AZ10219759 (study 2), the heart was removed first at necropsy to permit rapid weighing and fixation. In the other studies, the heart was removed within 10 to 15 min during postmortem sampling of multiple tissues. At necropsy, in order to optimize fixation for transmission electron microscopy, following removal of the heart, a very thin sliver (2–4 mm long by less than 1 mm thick, orientated transversely) from the left ventricle, midway between apex and base, was taken and fixed in 2.5% glutaraldehyde in 0.1M phosphate buffer (pH 7.2–7.4) prior to standard processing into epoxy resin for examination of toluidine blue–stained sections by light microscopy and ultrathin sections (70–90 nm thick) were stained using uranyl acetate and lead citrate with a JEOL 1400 transmission electron microscope at 80 KV (JEOL UK Ltd., Welwyn Garden City, UK). The remainder of the heart was fixed in 10% neutral buffered formalin for 24 to 48 hr and a 4- to 5-mm longitudinal section taken to sample the 4 chambers, before processing routinely into paraffin wax and 3- to 4-μm thick sections stained with H&E examined by light microscopy. Oil Red O–stained cryostat sections of formalin-fixed, right ventricular tissue were prepared from all animals in all studies.
Immunohistochemistry
Additional staining of wax-embedded sections from all animals in the 7-day investigative study (study 2) only was performed using the following histochemical and immunohistochemical procedures: Masson Trichrome for collagen, ED1 for monocytes/macrophages, and both human fatty acid binding protein (FABP) and troponin I for cardiac myocytes.
For all immunohistochemical techniques, 4-µm thick heart sections were dewaxed in xylene and rehydrated through graded alcohols to water. Sections were placed onto a Labvision Immunostainer (Fisher Scientific UK Ltd., Loughborough, UK) where the remainder of the staining was performed. Sections were first washed in Tris–buffered saline with 0.1% Tween (TBST) followed by blockade of endogenous peroxidase with 3% hydrogen peroxide in TBST for 10 min. After a buffer wash, nonspecific Ig-binding sites were blocked for 20 min using a background blocker with casein (MP-966-P500, A. Menarini Diagnostics, Winnersh–Wokingham, Berkshire, UK). This was followed by application of primary antibody, performed with a technique appropriate for each antibody, followed by another TBST wash and visualized with 3,3′-diaminobenzidine (DAB; A. Menarini Diagnostics, Winnersh–Wokingham, Berkshire, UK) for 10 min. After a water wash, sections were counterstained in Carazzi’s hematoxylin for 1 min and mounted nonaqueously. Appropriate negative and positive controls were utilized. All dilutions were in TBST and procedures were performed at room temperature.
Cardiac Troponin I immunohistochemistry was performed using a monoclonal mouse anticardiac troponin-I antibody (ab19615, 1:400, Abcam, Cambridge, UK) for 60 min incubation and detected with Mouse EnVision-HRP system (K4007, Dako, Ely, UK) for 30 min.
Cardiac FABP immunohistochemistry was performed using a monoclonal mouse anticardiac FABP antibody (ab16916, 1:10, Abcam, Cambridge, UK) for 60-min incubation, followed by a biotinylated rabbit antimouse IgG secondary antibody (E0464, 1:200, Dako, Ely, UK) for 30-min incubation and then detected with X-Cell plus polymer-HRP system (A. Menarini Diagnostics, Winnersh–Wokingham, Berkshire, UK) for 15 min.
ED1 immunohistochemistry required heat-induced epitope retrieval with 0.01 M Citrate (pH 6.0) at 110°C for 2 min prior to staining. A monoclonal mouse anti-rat ED1 (MCA341R, 1:100, Serotec, Kidlington, UK) was applied for 60 min and detected with Mouse EnVision-HRP system (K4007, Dako, Ely, UK) for 30 min.
Clinical Biochemistry
At necropsy, prior to tissue sampling, blood was collected from the vena cava immediately postmortem by venipuncture into tubes containing LH or ethylenediaminetetraacetic acid (EDTA). Following centrifugation at 3,000 rpm (1,940 g) for 10 min, plasma samples were frozen and stored at −20°C prior to thawing for analyses. LH plasma was used for measurements of troponin I and creatine kinase, while EDTA plasma was used for myoglobin and nonesterified fatty acids (NEFAs) in accordance with the manufacturer’s instructions. Please note that one or more of the following specific cardiac markers were assessed in different studies, in addition to the standard hematological and clinical biochemical parameters employed in drug development studies. We will report only those parameters that were altered in individual studies.
Troponin I was assessed in a minimum volume of 0.3-ml LH plasma using a Siemens Human Ultrasensitive TI assay (Siemens Healthcare Diagnostics, Tarrytown, NY) validated for use in the rat, on an Advia Centaur CP immunoassay system (Siemens Healthcare, UK).
Troponin T was assessed using 50-μl EDTA plasma using the Roche Troponin T high sensitivity assay (Ref: 05092744) on the Roche Elecsys 2010 Immunoassay analyzer (Roche, Welwyn Garden City, UK).
Creatine kinase was analyzed in a minimum of 0.2-ml LH plasma using a human Creatine Kinase liquid assay kit (Roche) validated for use in the dog, rat, and mouse, on an automated clinical chemistry analyzer (Modular P800, Roche) in accordance with the manufacturer’s instructions.
Myoglobin was assessed using 40-μl EDTA plasma with a rat myoglobin ELISA kit (Life Diagnostics Inc., Stoke on Trent, UK) on a Grifols Triturus Immunoassay plate analyzer (Grifols UK Ltd., Cambridge, UK).
NEFAs were analyzed in 0.2-ml EDTA plasma using a human NEFA (Free Fatty Acid) assay kit (WAKO Diagnostics, Richmond, VA) validated for use in the dog, rat, and mouse on the Modular P800 automated analyzer (Roche, Welwyn Garden City, UK).
Results
Study 1: AZ10219759: 7-Day Toxicity Study
There were no premature decedents in this study and the drug was well tolerated. Several control animals displayed minimal, multifocal cardiomyocyte steatosis but neither these nor those that showed none were associated with any other associated pathological features. One animal (designated 1 in Table 1) showed bilateral, atrial chronic inflammation with fibrosis of moderate severity associated with minimal, multifocal, myocardial steatosis (representative illustrations in Figure 2B and F) while two others (animals 2 and 3 in Table 1) exhibited mild left atrial myocardial degeneration/necrosis with minimal diffuse or multifocal myocardial steatosis, respectively. Several animals showed equivalent or greater degrees of myocardial steatosis with either, other associated pathologies primarily, mononuclear cell infiltration or, none. No clinical biochemistry parameters were altered due to dosing with AZ10219759.
Study 1: AZ10219759: 7-day drug development, toxicity study.
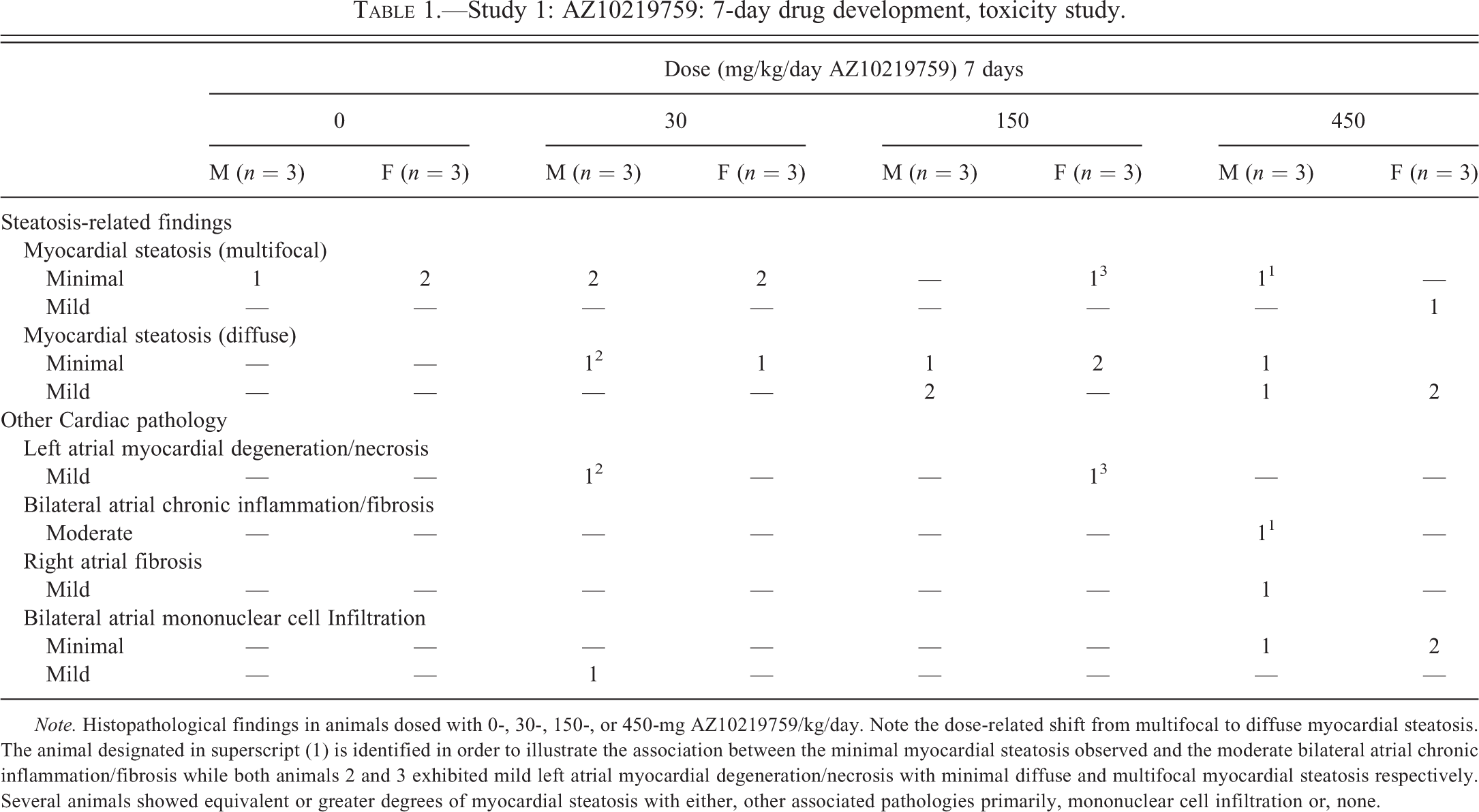
Note. Histopathological findings in animals dosed with 0-, 30-, 150-, or 450-mg AZ10219759/kg/day. Note the dose-related shift from multifocal to diffuse myocardial steatosis. The animal designated in superscript (1) is identified in order to illustrate the association between the minimal myocardial steatosis observed and the moderate bilateral atrial chronic inflammation/fibrosis while both animals 2 and 3 exhibited mild left atrial myocardial degeneration/necrosis with minimal diffuse and multifocal myocardial steatosis respectively. Several animals showed equivalent or greater degrees of myocardial steatosis with either, other associated pathologies primarily, mononuclear cell infiltration or, none.
(A) and (B) Hematoxylin and eosin–stained heart tissue. (A) Left atrium of animal given 600-mg AZD7545/kg/day for 28 days showing substantial myocardial myocyte loss with replacement fibrosis and activated endocardial endothelial cells. The subacute inflammation seen in Figure 2B has been replaced with a chronic inflammatory profile of few monocyte/macrophages with no polymorphonuclear leucocytes and minimal diffuse myocyte vacuolation. (B) Left atrial myocardial degeneration/necrosis (arrows) with mononuclear cell infiltration and interstitial fibrosis in a fascicle of myofibers from an animal following 7 days dosing with 150-mg AZ10219759 /kg/day. An adjacent fascicle shows no such features illustrating the differences between different locations of affected myofibers within the heart and their possible anatomical predilection sites (arrowheads define fascicle boundary). (C–F) Micrographs of Oil Red O–stained/hematoxylin counterstained, right ventricular cryosections. (C) Vehicle-dosed control animal showing absence of any red-stained myocardial, cytoplasmic droplets characteristic of normal appearance. (D) Moderate, generally diffuse myocardial steatosis in most of the tissue shown in this image from a deceased animal (designated 2 in Table 2) given 450-mg AZ10219759/kg/day for 3 days, although a myofiber fascicle displays significantly less stain, illustrative of different degrees of staining in distinct cardiac regions. (E) Increased magnification of tissue from D showing moderate, generally diffuse myofiber Oil Red O staining with linear arrangements of numerous, rounded fat droplets in most myofibers although a few exhibit no staining. (F) Minimal diffuse myocardial steatosis from an animal given 450-mg AZ10219759/kg/day for 7 days illustrative of substantial interanimal differences. (See [D] for comparison of tissue from animals dosed for similar durations. Original objective lens magnifications—A, 10×; B, C, D, 20×; E, F, 40×).
Study 2: AZ10219759: Investigative 7-Day Study
One animal (designated animal 2) given 450-mg AZ10219759/kg/day was deceased on day 3. The heart was taken from this animal and processed for light and electron microscopy, although no plasma sample was obtained.
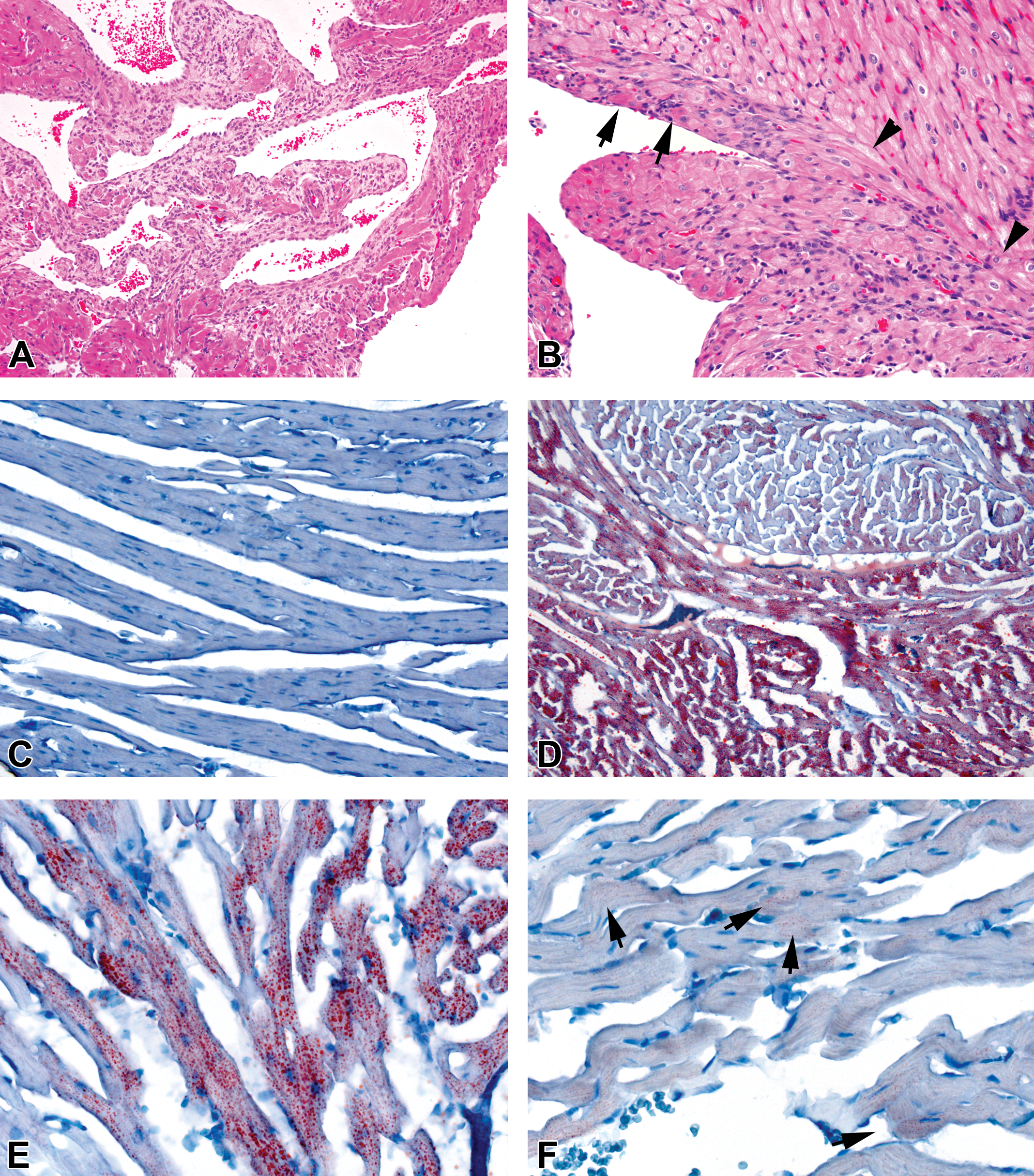
Vehicle-treated controls showed no significant histological changes and clinical biochemical values were within normal ranges for animals of this age (Figures 2C and 3A). In animals given either 150- or 450-mg AZ10219759/kg/day, respectively, significant alterations in cardiac pathology and myocardial steatosis were observed (Figures 2B–F and 3C, D, F) with clear, dose-related separation of the latter on the basis of either, distribution, severity or, incidence (Table 2). Group incidences of pathological findings are shown in Table 2 with individual animals identified by superscript, in order to illustrate first, the dose-related nature of the pathological findings and second, the association together of several features. Ventricular myocardial degeneration with associated inflammation and fibrosis was observed at minimal severity in 1 animal per dose group, but at greater incidence (3 and 2, respectively, in animals given 150- or 450-mg AZ10219759/kg/day) and minimal or moderate severity in either the right atrium only or in both atria. Other histopathological changes, namely atrial endothelial activation (rounding up) and a resolving thrombus in the left atrium of 1 animal given 150-mg AZ10219759/kg/day, were also observed (Figure 3C). Light microscopically visible (i.e., moderate) myocardial vacuolation in H&E-stained sections was present in 1 animal only namely, the decedent, high-dose male (animal 2, Figure 3F). Histochemical staining with Oil Red O (Figure 2C–F) revealed no detectable myocardial lipid staining in control animals indicative of the very low levels present normally in cardiomyocytes. By contrast, minimal or mild Oil Red O staining was seen in 5 animals in both drug-dosed groups, and only in the decedent animal was this change observed at marked severity: the remainder of the animals in these groups showed no increase in myocardial lipid. Myocardial steatosis graded as of minimal or mild severity was normally distributed multifocally, that is, individual myocytes showed this change, and these were separated from others showing similar staining, by unstained cells, while severe steatosis was diffusely distributed. Importantly, in those animals showing atrial pathology, the severity of myocardial steatosis while present in all such affected animals showed no correlation (Table 2).
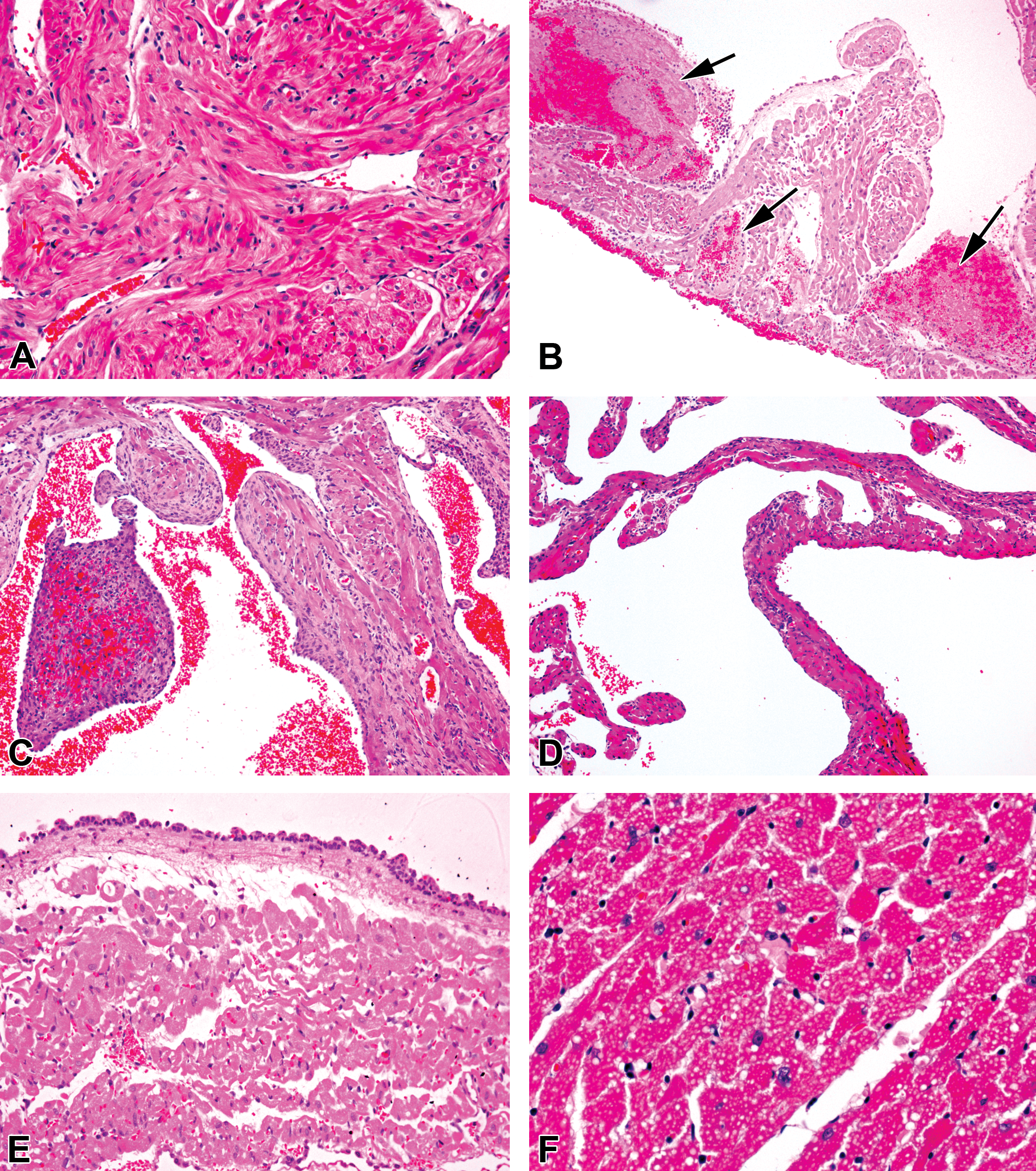
Histopathological appearances of heart from animals given pyruvate dehydrogenase kinase inhibitors. (A) Control animal—normal atrial histology with multi-orientated myofibers showing no vacuolation. (B) Left atrium of prematurely terminated animal (number 1 in Table 3), following 16 days of daily 600-mg AZD7545/kg/day dosing showing multiple, premortem thrombi (arrows) with substantial left atrial edema and myocardial and endocardial subacute inflammation and moderate, diffuse myocyte vacuolation, shown clearly in (F) and (C). Diffuse right atrial myocardial loss with inflammation and interstitial fibrosis and some endocardial endothelial activation in an animal (designated 3 in Table 2) following 7 days dosing with 150-mg AZ10219759 /kg/day. A resolving thrombus is prominent. (D) The same animal as in C displaying contralateral left atrial myocardial loss with associated inflammation. (E) Left atrium of prematurely terminated animal (described in [B]) after 16 days of daily dosing of 600-mg AZD7545/kg/day showing substantial left atrial edema, endocardial acute inflammatory cell attachment with no migration into myocardium, and mild diffuse myocyte vacuolation. (F) Right ventricle of prematurely deceased animal, following 3 days of daily 450-mg AZ/10219759 kg/day dosing (designated as animal 2 in Table 2), showing moderate, diffuse myocyte vacuolation with no evidence of degeneration/necrosis. (Hematoxylin and eosin. Original objective lens magnifications—A, E 20×; B, C, D 10×; F 40×).
Study 2: AZ10219759: Investigative, 7-day study.
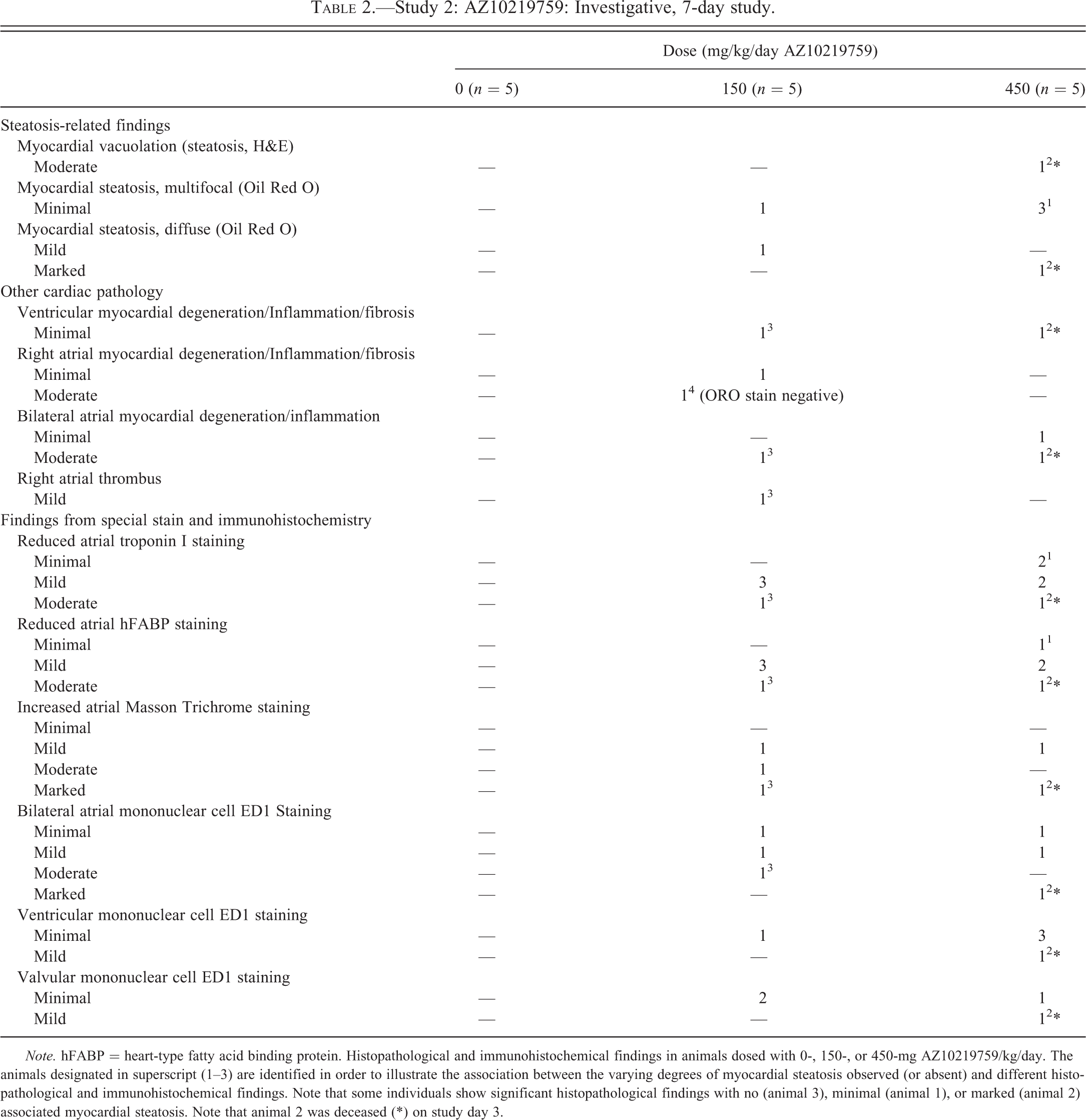
Note. hFABP = heart-type fatty acid binding protein. Histopathological and immunohistochemical findings in animals dosed with 0-, 150-, or 450-mg AZ10219759/kg/day. The animals designated in superscript (1–3) are identified in order to illustrate the association between the varying degrees of myocardial steatosis observed (or absent) and different histopathological and immunohistochemical findings. Note that some individuals show significant histopathological findings with no (animal 3), minimal (animal 1), or marked (animal 2) associated myocardial steatosis. Note that animal 2 was deceased (*) on study day 3.
Myocardial histological alterations were most clearly observed in specifically stained tissue. Troponin I and FABP are markers of functionally intact cardiac myocytes. The intensity and ubiquitous distribution of FABP in control animals was identical to that of troponin I (Figure 4A). Multifocal or, in some areas, diffuse loss of myocardial troponin I staining was seen in atria only of 9/10 dosed animals (Figure 4A–C). Further corroborative indication of individual myofiber sarcoplasmic disruption and myocardial degeneration or loss in dosed animals was provided by multifocal or locally diffuse reduced FABP staining that was seen in the atria only of 8/10 animals with no effects seen in ventricular myocardium (Figure 4D–F). Similarly, Masson Trichrome–stained tissue collagen, which in controls was largely absent, was increased in 5/10 drug-dosed animals, its extent and degree ranging from mild to marked (Figure 5A–C). Immunohistochemical staining of tissue monocytes/macrophages (ED1) showed no significant staining in controls indicative of the absence of these cells in uninjured cardiac tissue (Figure 5D). By contrast, a majority of drug-dosed animals showed bilateral atrial ED1 staining at minimal to marked severity with greater numbers of these cells than were present in controls (Figure 5E and F). Left ventricular ED1 staining at generally minimal levels only was also increased above control levels in about half of the drug-dosed animals. By comparison with controls, in which no monocyte/macrophage infiltration into valves was observed, several AZ10219759-dosed animals showed increased, valvular ED1 positivity to minimal or, mild degree, indicative of increased infiltration of these cells into this tissue and alluding to valvular injury. Whether or not such valvular monocytic infiltrations were primarily responsible for associated atrial pathology or occurred as a component feature of such damage cannot be determined from the data presented here, although their absence in animals showing other indications of drug-induced cardiac pathology (atrial cardiomyocyte degeneration/loss) suggests that they are secondary to initial tissue damage. No clinical biochemical parameters were adversely altered in this study.
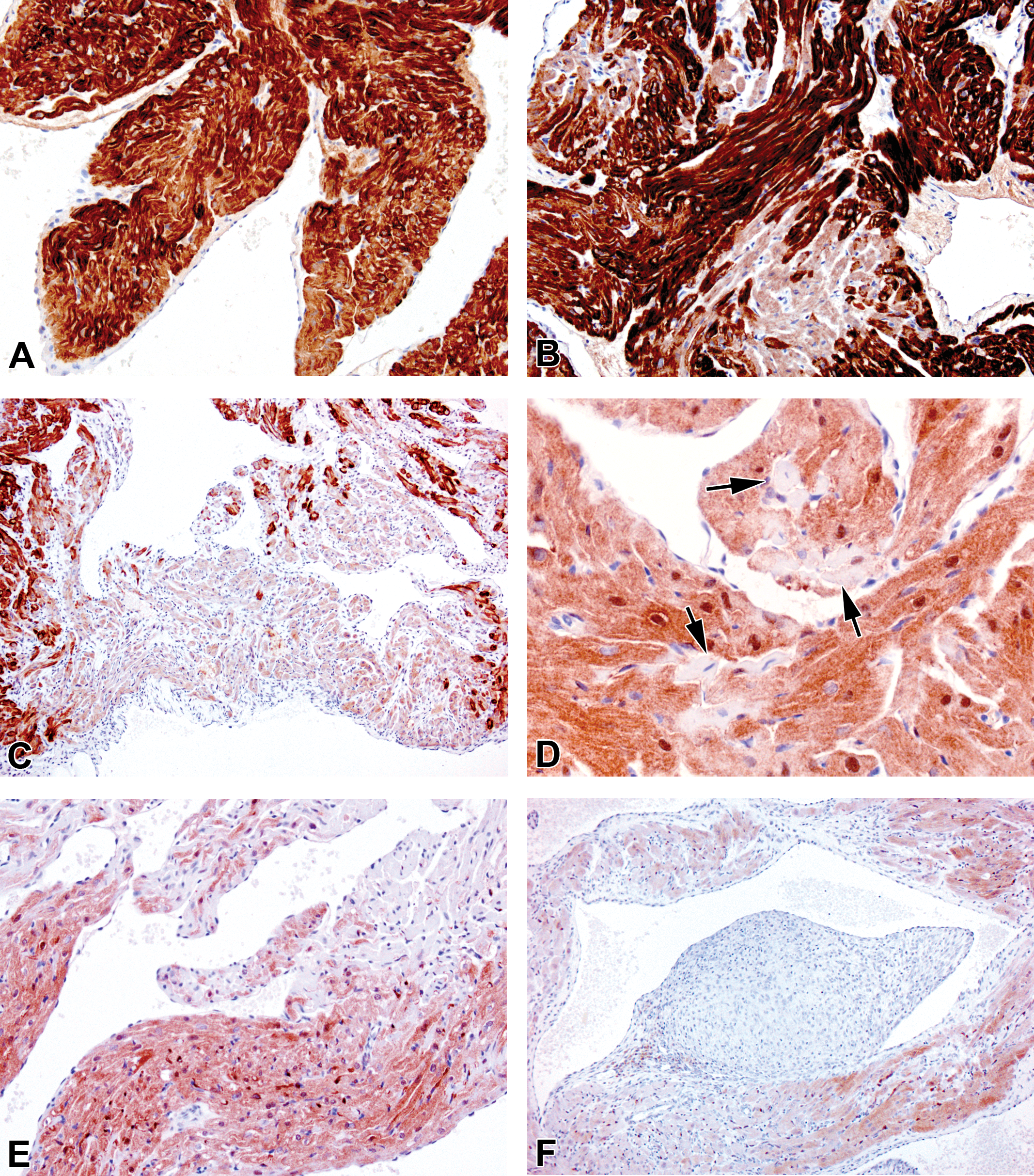
(A)–(C) Troponin I-immunostained, hematoxylin-counterstained left atrial sections from 7-day study with AZ10219759. (A) Vehicle-dosed, control animal showing diffuse troponin staining in all atrial myofibers. (B) Animal given 450-mg AZ10219759/kg/day for 7 days, showing multifocal, marked reduction of tissue staining and no single cell involvement. (C) Animal given 150-mg AZ10219759/kg/day for 7 days, showing multifocal, merging to diffuse, marked reduction of tissue staining. (D)–(F), FABP immunostained/hematoxylin-counterstained, left (D and F) and right (E) atrial sections from animals given 450-mg AZ10219759/kg/day for 7 days showing similar changes in atrial myofiber staining as illustrated with troponin. (D) Single myofibers show loss of staining (arrows). In (F), extensive loss of FABP staining is shown adjacent to the resolving thrombus shown in Figure 3C, in which FABP is absent. (Original objective lens magnifications—A, B, E, 20×; C, F, 10×; D, 40×).
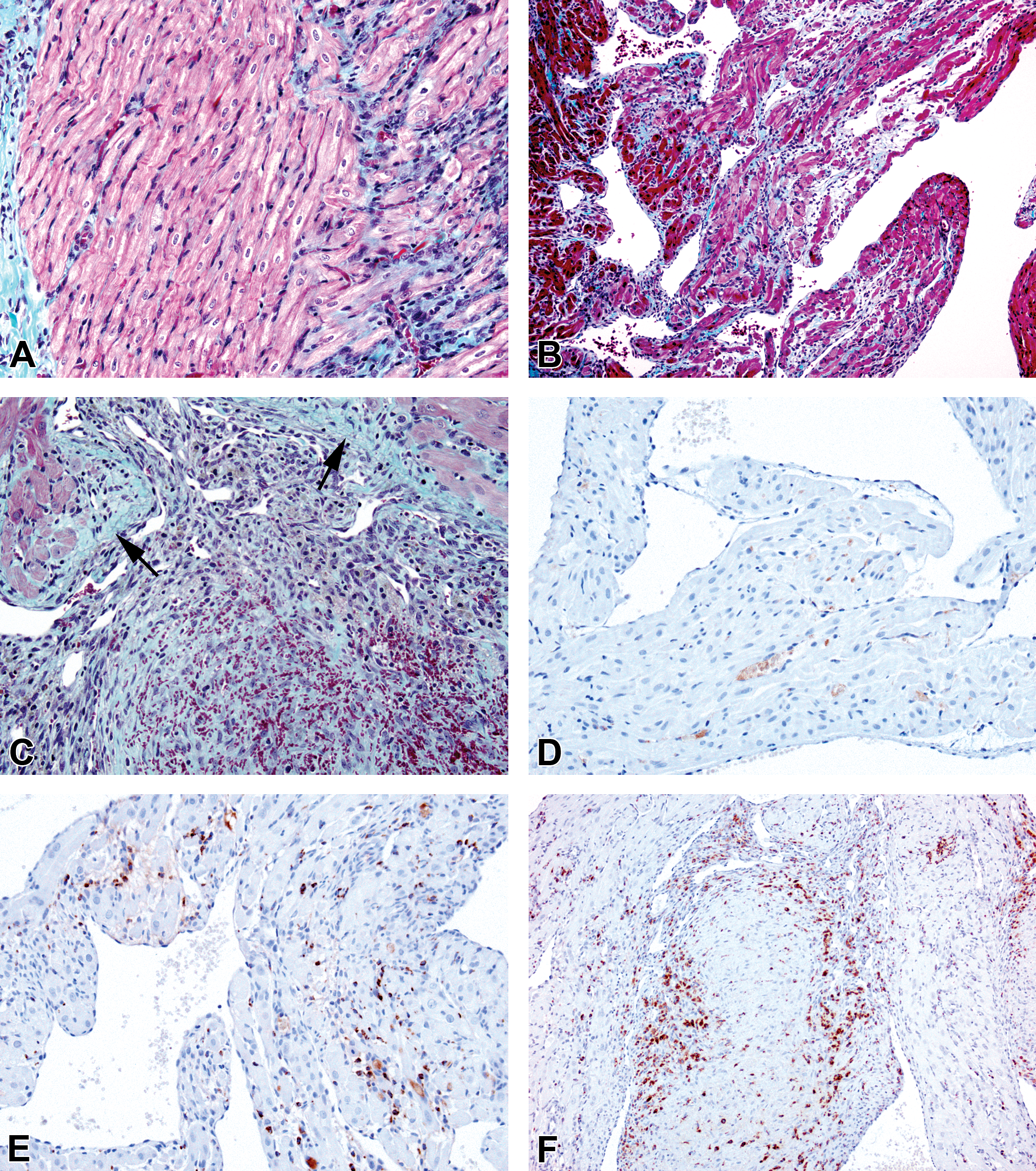
Left atrial sections from animals given 150-mg AZ10219759/kg/day for 7 days. (A)–(C) Masson Trichrome staining showing increased turquoise-stained collagen. (A) Abundant collagen, locally associated with an area of myofiber loss and inflammatory cell infiltration, with an adjacent area (shown in Figure 2B) being spared. (B) An extensive area of degenerating myofibers associated with inflammatory cell infiltration, edema, and increased collagen deposition enveloped by endocardium with activated endothelial cells showing rounding up. (C) Extensive collagen deposition in the resolving thrombus shown in Figure 3C and in adjacent subendocardial tissue (arrows). Micrographs (D)–(E), monocyte/macrophages visualized with ED1 immunostain in atrial sections from animals given either vehicle or 150-mg AZ10219759/kg/day for 7 days. (D) Right atrial section from vehicle-dosed animal showing absence of macrophage infiltration. (E) Left atrial tissue showing increased incidence and multifocal distribution of macrophages throughout atrial tissue. (F) Left atrial tissue showing resolving thrombus in Figures 3C and 5C, exhibiting substantial thrombus-associated macrophage infiltration and multifocal distribution of these elsewhere in atrial tissue. (Original objective lens magnifications—A, C, D, E, 20×; B, F, 10×).
Ultrastructural Pathology
Immersion-fixed, left ventricular myocardium from vehicle-dosed controls showed few, small sarcoplasmic lipid droplets located adjacent to cardiomyocyte mitochondria and clear separation in mitochondrial appearances between individual cells (Figure 6A). In drug-dosed animals, significant increases in numbers and size of cardiomyocyte lipid droplets were evident in some individual animals only, with other sarcoplasmic features being unaltered: substantial variation in the incidence of lipid droplets in individual cardiomyocytes was also apparent (Figure 6). Figure 6B illustrates the unremarkable sarcoplasmic lipid droplet incidence and normal left ventricular cardiomyocyte ultrastructural appearances in an animal (designated 4 in Table 2) given 150-mg AZ10219759/kg/day for 7 days with histological evidence of substantial right atrial myocardial degeneration and inflammation. By contrast, the decedent animal (animal 2 in Table 2) with histological evidence of moderately severe, bilateral atrial myocardial degeneration, inflammatory cell infiltration, and severe myocardial lipid (Oil Red O staining) exhibited substantial, large lipid droplet accumulation in both viable and necrotic cardiomyocytes (Figure 6C and D). As with the Oil Red O histochemical data, the ultrastructural appearances of ventricular cardiomyocytes showing absence of correlation between abundance and size of sarcoplasmic lipid inclusions with atrial pathological changes clearly illustrates the lack of concordance of steatosis and cardiac pathology (compare animals 2, 3, and 4, Table 2).
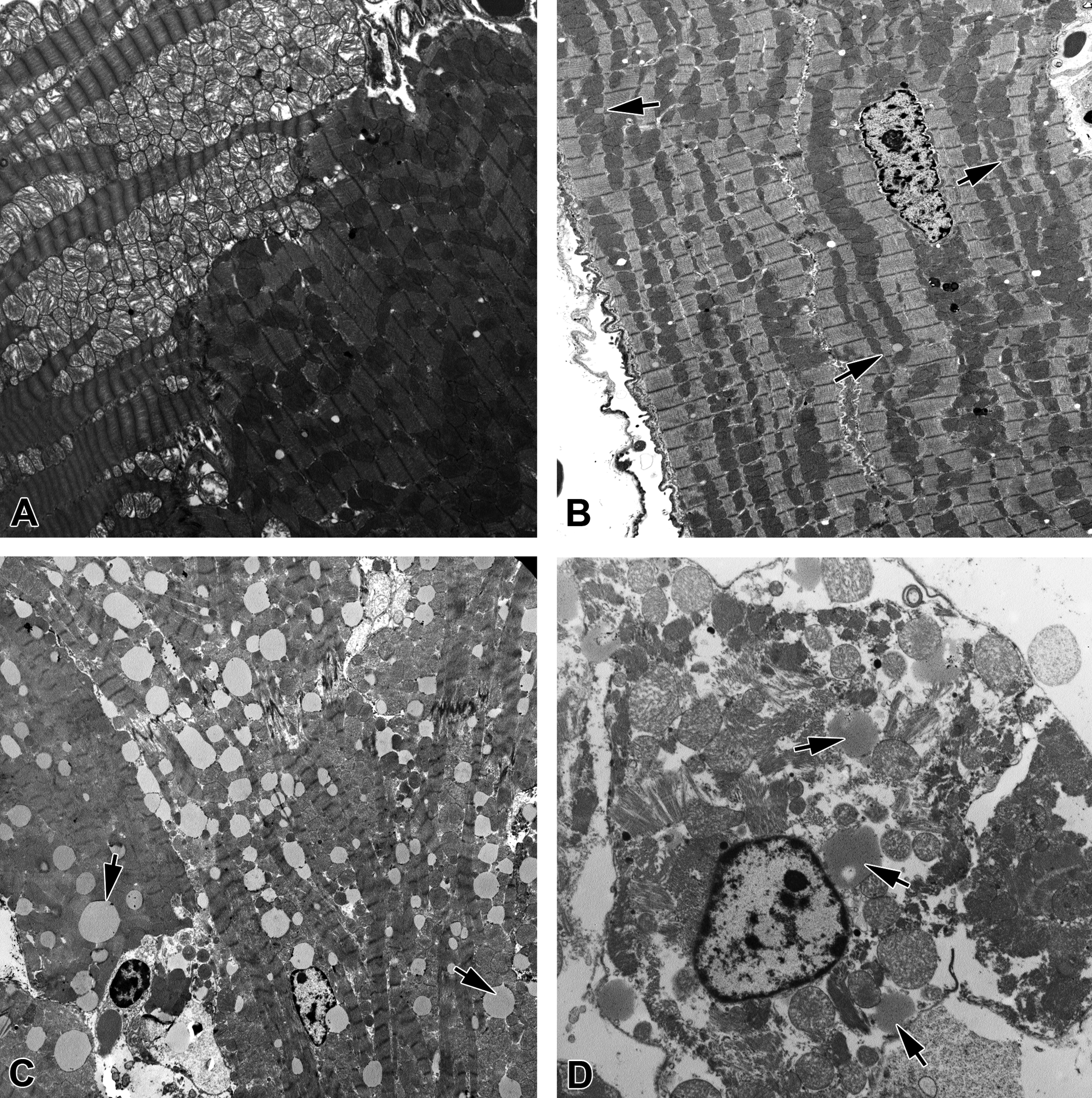
Transmission electron micrographs of left ventricular myocardium from (A) vehicle-dosed control animal showing adjacent myocytes displaying markedly different normal myocardial appearances due to lucent versus condensed mitochondrial structures and with small, cytoplasmic lipid droplets. (Note. This tissue was unstained for myocardial lipid on Oil Red O staining of formalin-fixed cryosections.) (B) Heart from an animal (designated 4 in Table 2) given 150-mg AZ10219759/kg/day for 7 days, which showed no Oil Red O histochemical positivity but substantial right atrial myocardial degeneration with inflammation of moderate severity. Note the few, small lipid droplets present (arrows). (C) Heart from an animal (designated 2 in Table 2) given 450-mg AZ10219759/kg/day and deceased on day 3, showing marked myocardial lipid deposition (arrows) in linear arrays within intact cardiomyocytes. Histological evidence of significant bilateral atrial myocardial degeneration and inflammation with reduced staining of muscle immunostains (troponin I and FABP) was present. (D) Micrograph from same animal as (C) showing a necrotic cell with sarcoplasmic disruption of contractile apparatus, abundant large lipid droplets (arrows) and rounded, effete mitochondria. (Original micrograph magnifications A, 5,000×; B, 4,000×; C, 3,000×; D, 8,000×).
Given the results from the studies conducted, no further development of this drug candidate was undertaken. No clinical biochemistry parameters were altered due to dosing with AZ10219759.
AZD7545: 28-Day Studies
Study 3: 28 days, drug development study
Three animals were deceased or terminated due to clinical condition on days 16, 24, and 25 (indicated in Table 3 as animals 1, 4, and 5, respectively). Variable, but sometimes substantial, cardiac pathology was noted in animals from all dosed groups. Myocardial steatosis was increased in both incidence and severity with its tissue distribution at lower severity grades (minimal and mild; see Figure 2C–F from animals given AZ10219759 for illustration of tissue appearance) being either multifocal or diffuse and always the latter at the highest grade observed (moderate) in a few animals (Figure 3B and E [representative illustrations from animals given AZ10219759]). A large majority of animals showed this change exclusively, with no other co-pathologies observed (nature of changes illustrated in Figure 2D and E). However, 4 animals given 600-/300-mg AZD7545/kg/day displayed a number of related co-pathologies that can be grouped as either “acute” or “chronic.” Two animals showing acute cardiac pathology (designated in Table 3 as animals 1 and 4) were terminated on days 16 and 24, respectively, and had moderately severe, diffuse (atrial, ventricular, and septal) myocardial vacuolation that was visible in H&E-stained sections (and equated to moderate diffuse myocardial steatosis on Oil Red O staining of cryosections of formalin-fixed myocardium), atrial myocardial necrosis, acute inflammatory cell infiltration with edema, endocarditis, and in 1 animal, left atrial thrombosis. Plasma samples at necropsy showed either no biochemical changes (animal 1) or 4.5× increased creatine kinase (animal 4: 964 IU/L vs. control group mean of 216 IU/L). In drug-dosed animals, significant increases in numbers and size of cardiomyocyte lipid droplets were evident in some individual animals only, with other sarcoplasmic features being unaltered: substantial variation in the incidence of lipid droplets in individual cardiomyocytes was also apparent (Figure 6). Two animals that showed chronic pathology (designated in Table 3 as animals 2 and 3) were terminated on day 29 following 28 days daily dosing and showed no adverse clinical signs, no clinical biochemical changes, and displayed cardiomyocyte loss with replacement fibrosis: mixed inflammatory cell infiltrates were absent but some mononuclear cells/macrophages were present and importantly, associated with minimal, diffuse myocardial steatosis. A fifth animal dosed with 600-/300-mg AZD7545/kg/day for 25 days and found deceased (indicated as animal 5 in Table 3) displayed moderate, diffuse myocardial steatosis with mild right atrial dilatation only. While mild, diffuse myocardial steatosis was common in animals dosed with AZD7545, the majority showed no other co-pathologies. However, 3 males given 30-mg AZD7545/kg/day for 28 days that displayed mild diffuse myocardial steatosis, and with no remarkable clinical biochemistry changes, on histopathological examination, only moderate right atrial dilatation (animal 6) and minimal right atrial mononuclear cell infiltration (animals 7 and 8) were noted. There were no salient biochemical changes in any other animals given either 30- or 100-mg AZD7545/kg/day for 28 days.
Study 3: AZD7545: 28-day, drug development study.
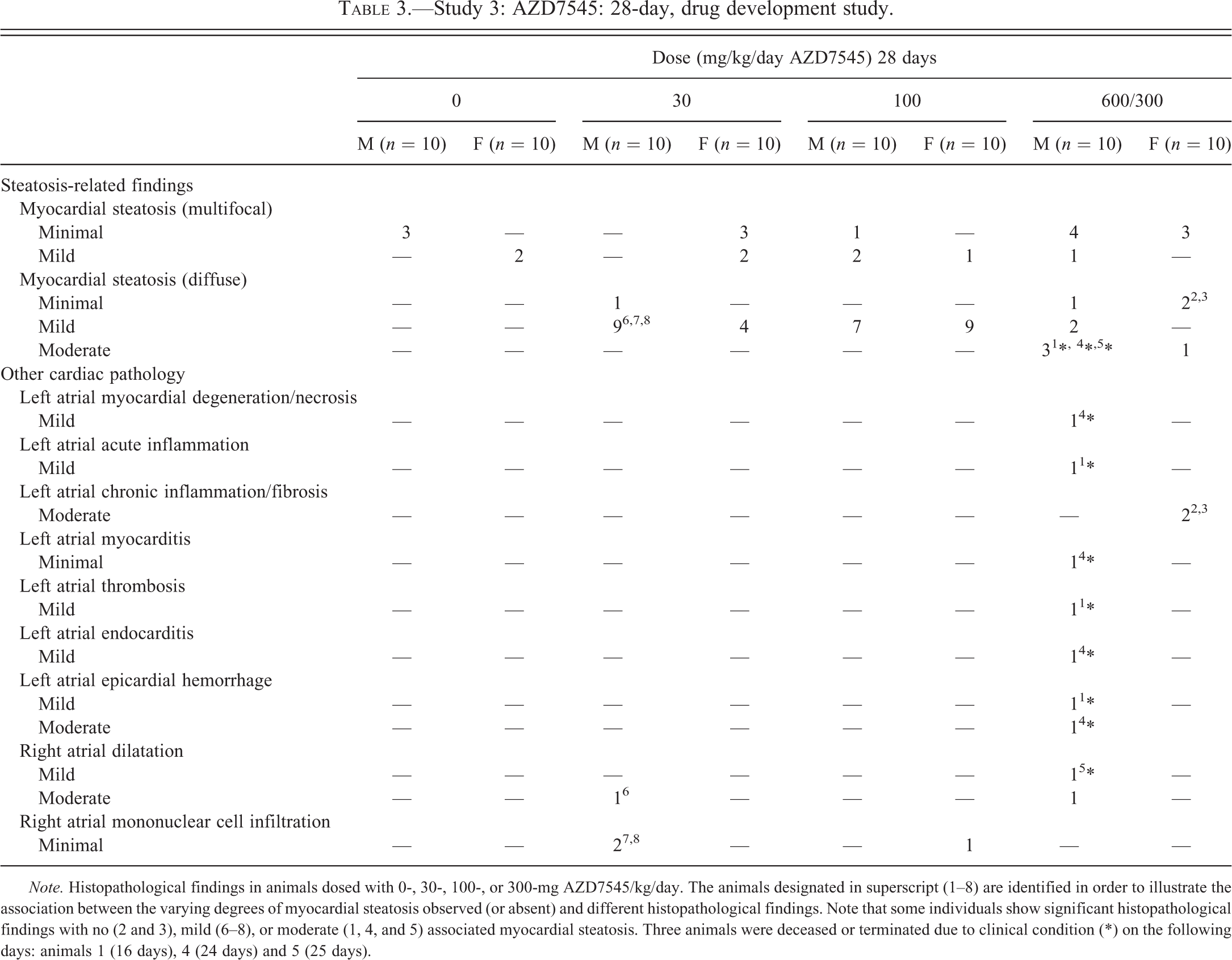
Note. Histopathological findings in animals dosed with 0-, 30-, 100-, or 300-mg AZD7545/kg/day. The animals designated in superscript (1–8) are identified in order to illustrate the association between the varying degrees of myocardial steatosis observed (or absent) and different histopathological findings. Note that some individuals show significant histopathological findings with no (2 and 3), mild (6–8), or moderate (1, 4, and 5) associated myocardial steatosis. Three animals were deceased or terminated due to clinical condition (*) on the following days: animals 1 (16 days), 4 (24 days) and 5 (25 days).
Study 4: 28 days, dose–response study
There were neither adverse clinical signs nor premature decedents noted in this study. However, myocardial steatosis was present at slightly greater incidence (Table 4), but not generally at greater severity in animals dosed with either 20- or 300-mg AZD7545/kg/day, by comparison with controls (Figure 2F; from an animal given AZ10219759, shown as illustration) although a minority of animals showed a diffuse rather than a multifocal distribution of myocardial steatosis. No other cardiac pathology was detected in any animal. No biochemical parameters were altered due to exposure to AZD7545 in this study.
Study 4: AZD7545: 28-day (dose–response) study.
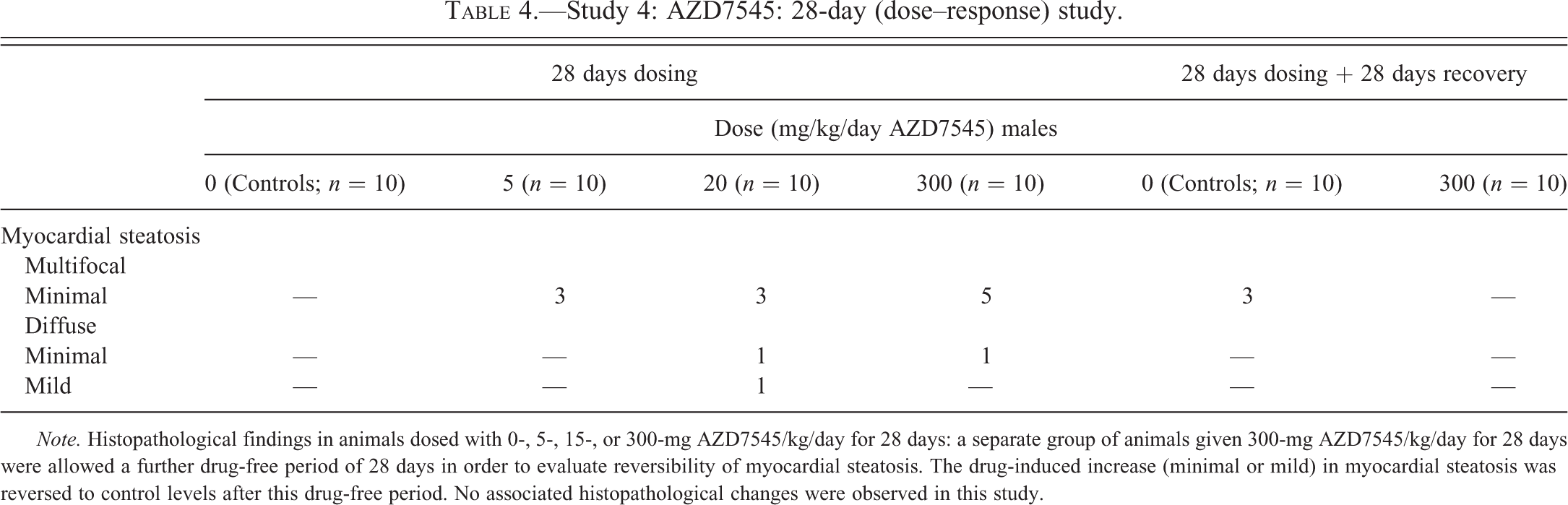
Note. Histopathological findings in animals dosed with 0-, 5-, 15-, or 300-mg AZD7545/kg/day for 28 days: a separate group of animals given 300-mg AZD7545/kg/day for 28 days were allowed a further drug-free period of 28 days in order to evaluate reversibility of myocardial steatosis. The drug-induced increase (minimal or mild) in myocardial steatosis was reversed to control levels after this drug-free period. No associated histopathological changes were observed in this study.
Discussion
The studies described were striking in the degree of variation in drug-induced myocardial steatosis and the generally, infrequent association of this feature with other cardiac pathologies. It is notable that in the 7-day investigative study with AZ10219759, no chronic histopathological findings were observed, all changes being characterized as displaying acute histopathology, that is, myocardial degeneration/necrosis, acute myocarditis, edema, endothelial activation (rounding up)/endocarditis or, of intermediate stages. Clearly, this absence of a shift from an acute to a chronic condition relates to the short duration of exposure, but its appearance provides persuasive evidence of early onset of significant morphological changes following several daily doses only, albeit in a minority of animals. Germane to this discussion of myocardial steatosis and its induction by PDHKIs are other, potential etiological factors. The absence of any adverse pharmacological influences of the drugs assessed in extensive in vitro screens of cardiac function strengthens our evidence-based conclusion that the myocardial steatosis and associated histopathological changes seen were due to drug-induced effects, primarily on cellular metabolism. Administration of PDHKIs to rats induced myocardial steatosis and in some instances, primarily unilateral or bilateral atrial and not ventricular pathology. Our studies show that the consequences of administration of PDHKIs are unpredictable with regard to the degree of dose-related induction of myocardial steatosis and are indicative of substantial and idiosyncratic individual animal susceptibility within a population of highly inbred rodents in which genetic diversity is expected to be lower than extensively outbred human populations. Data obtained from several studies with two molecules, AZD7545 and AZ10219759, illustrate the absence of first, an unequivocal dose–response relationship for myocardial steatosis and second, of clear evidence that marked, diffuse steatosis is associated consistently with cardiac (atrial) pathological alterations: indeed, there is no indication that a marked degree of steatosis is a prerequisite for such changes. The association between significant pathological changes in atrial morphology and Oil Red O myocardial staining (indicative of the vacuolation seen in conventional H&E-stained sections) was capricious, as those animals given AZD7545 in a 28 days duration study, which showed significant atrial pathology exhibited either minimal or marked Oil Red O staining, the former being associated with myocardial loss and replacement fibrosis and the latter, the acute changes characterized primarily by myocardial degeneration/necrosis, acute or mixed inflammatory cell infiltration with associated edema, endothelial cell activation “rounding up,” endocarditis, and atrial thrombi. It is evident that myocardial steatosis was induced by treatment with PDHKIs, but the absence of an association between degrees of myocardial steatosis and atrial pathology renders difficult interpretation of the significance of these changes in relation to their etiology. However, given the occurrence of the acute pathological changes in atria of some animals with moderate diffuse myocardial steatosis, it is likely that this initial damage is repaired, becoming manifest as the chronic pathological appearance. If generation of the acute atrial pathology is dependent on substantial myocardial lipid accumulation, the fact that, when observed, the chronic atrial pathology was associated with minimal steatosis only requires an explanation that is difficult to provide without consideration of temporal and transient myocardial lipid accumulation of varying severities within the same heart, for which we have no data. Importantly, loss of myocardial mass with associated monocyte/macrophage infiltration and fibrosis was notable at 7 days following treatment with either 150- or 450-mg AZ10219759/kg/day but was not observed with daily AZD7545 administration for similar duration (data not shown as no cardiac pathology noted in 7-day study with AZD7545). Plasma biochemical indicators of cardiomyocyte damage were not present in most animals, which suggest that timing of sampling likely prevented detection of alterations in biomarkers given that such changes might be expected in those animals in which atrial pathology was significant. The discussion regarding extrapolation of findings of drug-induced cardiac steatosis observed in animals to humans is based firmly in understanding the functional consequences, if any, of additional and excessive lipid deposition within cardiac myocytes (McGavock et al. 2006; Szczepaniak et al. 2003). The features of the PDHKI-induced cardiotoxicity resulted in significant difficulties in prediction of potential risk to humans and were important factors in decisions made to discontinue development of this class of drugs.
The relationship between cardiomyocyte metabolic perturbation and functional capability is pivotal in understanding cardiac functional integrity during and after lipid accumulation. In our studies, accumulation of myocardial lipid due to PDHKI administration varied in severity between minimal and moderate and in multifocal or diffuse distribution and potentially is indicative of some degree of variation in myofiber-specific, metabolically related functional differences. The notion that different cohorts of the cardiomyocyte population have distinct contractile capabilities related to specific metabolic and structural composition and may adapt in response to multiple influences is attractive and may go some way to account for our observations of greater or lesser lipid accumulation in individual fibers. In immunohistochemical studies of bovine and rabbit atrial and ventricular myocardium using anti-bovine atrial myosin antibodies, Sartore et al. (1981) reported significant regional differences between atria and ventricles in myofiber staining intensity and distribution. They also demonstrated that the weak ventricular myofiber staining in hypothyroid rabbits was reversed following thyroxine treatment and that propylthiouracil reversed this adaptive response. Further support for this notion has been reported in experiments in the cardiomyopathic hamster heart, in which decreased activities of mitochondrial creatine kinase (Awaji et al. 1990), myosin ATPase (Malhotra, Karell, and Scheuer 1985) and Na+-, K+-, sarcolemmal-, and SR-ATPase (Khuchua et al. 1989; Kjeldsen et al. 1988; Norgaard et al. 1987) have been seen. In focusing on oxidative and glycolytic metabolism using cytophotometric assessments of succinic dehydrogenase and glycerol-3-phosphatase dehydrogenase activities, respectively, in this same model, and myofibrillar ATPase as a marker of contractile activity, Punkt and Erzen (2000) demonstrated age-related differences between left and right ventricles and showed a more than 2-fold increase in succinic dehydrogenase activity at 120 to 190 days by comparison with earlier times in myopathic hamsters and also in normal myocardium. Left and right ventricular myofibrillar ATPase activity in the myopathic myocardium was reduced by up to 3-fold by comparison with that in normal myocardium at all times evaluated. It is noteworthy that the adverse functional consequences of myocardial lipid accumulations in animal hearts due to various causes (Sharma et al. 2004) are reversed following treatment with insulin-sensitizing drug treatment, providing further strong evidence of their deleterious association. Taken together, these data provide strong evidence that cardiomyocyte metabolic perturbation is associated with cardiomyocyte/cardiac contractile dysfunction and that different components of the heart may be more or less affected than others. It is a moot point, given our studies with PDHKIs were of limited duration, that extension of dosing may have led to significant cardiomyopathy.
Experimental evidence for PDHKI-induced cardiac contractile functional compromise is absent from our studies, but some elements of atrial histopathology in a few animals, namely atrial thrombi and endocardial, endothelial cell activation, or contraction (indicated by cellular rounding-up), strongly suggest such an effect and are indicative of turbulent flow conditions. In an extensive review of effects of disturbed flow on vascular endothelium, Chiu and Chien (2011) communicate that in vitro, endothelial cells exposed to disturbed flow of 0.5 dynes/cm2 mean shear stress were rounded in shape while those exposed to laminar flow (with cyclical bidirectional movement) of 20 dynes/cm2 were elongated and aligned parallel to the direction of flow. Numerous investigations provide substantial support for characteristic endothelial cell morphological appearances in regions of altered flow dynamics. Scanning electron microscopic studies of silver-stained rabbit aortic preparations demonstrated predictable appearances of elongated endothelial cell appearances in the straight regions of the vessel and rounded or polygonal cells at renal ostia and iliac bifurcations (Reidy and Langille 1980). Furthermore, Nerem, Levesque, and Cornhill (1981) demonstrated that vascular endothelial cell morphology was a sensitive indicator of the often complex pattern of blood flow, with long axes of these cells indicating primary flow direction.
The issue of the consequences of induction of myocardial steatosis on cardiac function is germane to considerations of possible administration of PDHKIs to humans and the association between them has been addressed in several ways, including high-fat diet feeding of rodents (Ouwens et al. 2005; Thakker et al. 2006), specific lipid metabolic gene transgenic or specific gene-deleted mice (Chiu et al. 2001; Chiu et al. 2005; Yagyu et al. 2003; Haemmerle et al. 2006; Glenn et al. 2011), and in genetic models of insulin resistance and obesity (Sharma et al. 2004; Listenberger and Schaffer 2002; Christoffersen et al. 2003; Buchanan et al. 2005; Zhou et al. 2000; McGavock et al. 2006). Several genetically modified mice have been created in order to evaluate the influence of key metabolic pathways on cardiac function such as diacyl glycerol acyl transferase ([DGAT], transgenic mouse overexpressing cardiomyocyte-selective DGAT1; Glenn et al. 2011), long chain acyl CoA synthase (transgenic mouse overexpressing cardiac-restricted long chain acyl CoA synthase [MHC-ACS]; Chiu et al. 2001; Lee et al. 2004), FABP (cardiac-specific, overexpression of fatty acid transport protein 1 transgenic mouse [FATP1]; Chiu et al. 2005), and lipoprotein lipase (cardiomyocyte-selective, GPI-anchored human lipoprotein lipase transgenic mice (LpL [hLpL GPI]; Yagyu et al. 2003); cardiac dysfunction measured in various ways, in association with myocardial steatosis was seen in all models. MHC-ACS transgenic mice show severe lipotoxic cardiomyopathy characterized by elevated cardiac triglyceride content, cardiomyocyte hypertrophy, and histological evidence of interstitial fibrosis associated with abnormal echocardiographic patterns due to consequences associated with cardiomyocyte-specific overexpression of the acyl CoA synthase (ACS) gene (Lee et al. 2004). They demonstrated severe diffuse myocardial steatosis by Oil Red O staining, in association with significant perturbation of left ventricular contractility assessed by echocardiography. Those animals made hyperleptinemic by treatment with recombinant adenovirus containing leptin cDNA were protected against myocardial triglyceride accumulation and the associated deterioration of left ventricular contractility, with function, morphology, and lipid biochemistry in these treated animals being normal. The transgenic mouse overexpressing cardiomyocyte-selective DGAT1 developed by Glenn et al. (2011) showed increased lipid accumulation without plasma dyslipidemia, hyperglycemia, or changes in body weight by comparison with controls. In these transgenics, the earliest adverse effect demonstrated by echocardiography was diastolic dysfunction with associated increased early mitral inflow velocity to late mitral inflow velocity ratio and decreased deceleration time, indicative of restrictive flow. At 52 weeks, significant systolic dysfunction was also present with histological evidence of elevated cardiac fibrosis with increased procollagen type 1A expression. Their studies demonstrated that cardiac lipid accumulation per se, in the absence of systemic metabolic perturbation resulted consequently in cardiac dysfunction. Further illustration of the multifocal susceptibility of myocardial lipid substrate metabolism in induction of myocardial steatosis and cardiac dysfunction is provided in FATP1 transgenic mice, which show 4-fold increased cardiomyocyte FFA uptake, 2-fold cardiomyocyte FFA accumulation, and at 3 months, echocardiographic indications of impaired left ventricular filling (with preserved systolic function) and bilateral atrial enlargement (Chiu et al. 2005). Multiple hemodynamic and Doppler imaging studies demonstrated that these mice displayed predominantly diastolic (atrial) dysfunction.
These examples illustrate the very significant adverse consequences of perturbation of cardiac metabolism on heart function induced through different routes. Crucially, our studies of PDHKIs show that myocardial steatosis and in a minority of animals, atrial pathology may be indicative of, but not evidence for some degree of cardiac functional compromise that was induced by relatively short duration, daily exposure to these molecules.
Clear similarities between myocardial steatosis with associated cardiac dysfunction are evident both in rodents and in the failing human heart (Sharma et al. 2004; McGavock et al. 2006; Szczepaniak et al. 2003; Sai et al. 2013). In human, elevated myocardial triglyceride content is associated with metabolic disorders such as insulin resistance and diabetes mellitus (McGavock et al. 2007; Utz et al. 2011) and increased myocardial lipids, and serum FFAs associated with compromised left ventricular (systolic) function in obese humans has also been demonstrated (Szczepaniak et al. 2003; Kankaanpaa et al. 2006). While elevated intramyocardial lipid content occurs in cardiac biopsies from obese or NIDDM heart failure patients, an unequivocal association between this and left ventricular functional efficiency remains to be demonstrated (McGavock et al. 2006). The question of whether or not intramyocardial lipid accumulation is an important determinant in dysfunction and is therefore a pathognomonic and diagnostic feature or merely an associated event of a different cellular metabolic perturbation is unclear with regard to the intergroup, variable incidence and severity of this feature as observed in our studies of the two PDHKIs investigated. However, given the evidence from different models, most notably those of cardiac-specific, lipid metabolic gene transgenic mice, that these PDHKIs alter cardiac metabolic substrate, and generate identical pathological outcomes, it is probable that they share significant single or multiple etiological factors. Certainly, observations of myocardial steatosis in assessment of safety of potential new drugs in animal studies should be regarded as significant cautionary information in relation to potential cardiac contractile dysfunction in both laboratory animals and human.
Footnotes
Acknowledgments
We are obliged for the expert contributions of staff from the Clinical Pathology, Pathology and Toxicology groups of AstraZeneca Pharmaceuticals, Alderley Park.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) received no financial support for the research, authorship, and/or publication of this article.