
Abstract
Establishing early diagnostic markers of harm is critical for effective prevention programs and regulation of tobacco products. This study examined effects of cigarette smoke condensate (CSC) on expression and promoter methylation profile of critical genes (DAPK, ECAD, MGMT, and RASSF1A) involved in lung cancer development in different human lung cell lines. NL-20 cells were treated with 0.1-100 μg/ml of CSC for 24 to 72 hrs for short-term exposures. DAPK expression or methylation status was not significantly affected. However, CSC treatment resulted in changes in expression and promoter methylation profile of ECAD, MGMT, and RASSF1A. For chronic studies, cells were exposed to 1 or 10 μg/ml CSC up to 28 days. Cells showed morphological changes associated with transformation and changes in invasion capacities and global methylation status. This study provides critical data suggesting that epigenetic changes could serve as an early biomarker of harm due to exposure to cigarette smoke.
Introduction
Lung cancer is one of the most frequent malignancies worldwide and the leading cause of cancer-related deaths in the Western world.1,2 Etiologically, tobacco smoking continues to represent the single most important risk factor for lung cancer development. 2 The exact underlying mechanisms of this malignancy require further delineation.3,4 Elucidation of the underlying mechanism of smoke-induced lung carcinogenesis can help define future strategies for early diagnosis, prognosis, treatment, and prevention of lung cancer 4 and inform the regulation of these products.
Epigenetic mechanisms of carcinogenesis manifest as heritable changes in the gene
expression without involving alterations in the underlying DNA sequence.5–7 Aberrant DNA methylation is the
best-studied epigenetic mechanism and causally implicated in human cancer.5,6 A global loss of DNA methylation
(hypomethylation) and a locus-specific gain in DNA methylation (hypermethylation)
are 2 distinct hallmarks of carcinogenesis.7,8 Aberrant DNA methylation occurs
predominantly in the context of 5’-CpG-3’ dinucleotides (CpGs).5–8 In mammalian genomes, the vast
majority of CpGs are normally methylated, for example 80% to 90% of CpGs in the
human genome are methylated.5–7 The remaining
methylation-free CpGs are found in stretches of >500 base pairs (bps) with a
guanine–cytosine (GC) content of >55% and an observed/expected CpG ratio of
A positive correlation between tobacco smoking and promoter hypermethylation in human lung cancer tissue has been demonstrated for many genes.16–20 However, the underlying mechanisms in tobacco-induced cancer and by which tobacco smoke disrupts a cell’s capacity to maintain the normal epigenetic code during the malignant transformation are yet to be fully determined. In this study, the effect of tobacco smoke in several lung cell systems was examined, including the expression levels and methylation status of a select group of genes. Cells were exposed to cigarette smoke condensate for various durations and doses. Genes (E-cadherin [ECAD], death-associated protein kinase [DAPK], O 6 -methyl-guanine-DNA methyltransferase [MGMT], and Ras-associated domain family 1, isoform A [RASSF1A]) were chosen based on their likely involvement in the genesis of lung cancer.20–24
Materials and Methods
Cell Lines and Treatment Conditions
The human lung cell lines, immortalized bronchial epithelial (NL-20), fibroblast
(LL29), and fibroblast (Hs.888), were obtained from the American Type Culture
Collection (Manassa, Virginia). The NL-20 cells were grown in Ham F12 medium
supplemented with 4% fetal bovine serum (FBS), 15 000 U penicillin, 15 000 U
streptomycin, 2 mmol/L
Total RNA Isolation and Quantitative Real-Time Reverse Transcriptase–Polymerase Chain Reaction
Total cellular RNA was isolated from the cells using the Qiagen RNeasy isolation kit (Qiagen, Valencia, California), according to the manufacturer's instructions. The concentration of RNA was determined by NanoDrop Spectrophotometer (ThermoScientific, Wilmington, Delaware). The OD260/OD280 nm ratios of all RNA samples were determined to be between 1.7 and 2.0 to ensure that all RNA samples are highly pure. The RNA integrity was verified by Experion (Bio-Rad, Hercules, California). The single-stranded complementary DNA (cDNA) used for analysis of DAPK, ECAD, MGMT, and RASSF1A was synthesized from 0.3 µg of purified total RNA using an Advantage RT-for-PCR reverse transcription kit (Clontech; Mountain View, California). Real-time polymerase chain reactions (PCR) were then performed using a SyberGreen PCR master mix on iQ5 Multicolor Real-time PCR Detection System (Bio-Rad). The primers for DAPK, ECAD, MGMT, RASSF1A, and GADPH were synthesized by Sigma-Aldrich Oligo Division (Woodlands, Texas). Primers were as follows: DAPK: forward 5’-CGAGGTGATGGTGTATGGTG-3’ and reverse 5’-CTGTGCTTTGCTGGTGGA-3’; ECAD: forward 5’-CGGGAATGCAGTTGAG GATC-3’ and reverse 5’-AGGATGGTGTAAGCGATGGC-3’; MGMT: forward 5’-TGCACAGCCTGGCTGAATG-3’ and reverse 5’-GGTGAACGACTCTTGCTGG AAA-3’; RASSF1A: forward 5’-GTCGTG CGCAAAGGCCT-3’ and reverse 5’-GCGCGGCAGCGGTAGT-3’. The reactions were carried out in triplicates in a 96-well plate in a total volume of 15 µL. Each reaction mixture contained 7.5 µL of SyberGreen PCR master mix, 3.8 µL of sterile nuclease-free water, 0.6 µL of forward primer (20 µmol/L), 0.6 µL of reverse primer (20 µmol/L). The PCR conditions were as follows: 50°C for 2 minutes, 95°C for 10 minutes, 95°C for 15 seconds, and 60°C for 1 minute (40 cycles). Quantification of the relative messenger RNA (mRNA) levels was carried out by determining the threshold cycle (CT), which is defined as the cycle at which the reporter fluorescence exceeds by 10 times the standard deviation (SD) of the mean baseline emission for cycles 3 to 10. Glyceraldehyde 3-phosphate dehydrogenase (GADPH) was used as an internal control. The mRNA levels of DAPK, ECAD, MGMT, and RASSF1A were normalized to those of GADPH, according to the following formula: CT (target) − CT (GADPH) = ΔCT. Thereafter, the relative mRNA levels of these genes after treatment were calculated using the ΔΔCT method: ΔCT (treatment) − ΔCT (vehicle) = ΔΔCT (treatment). The fold changes of mRNA levels were expressed as 2−ΔΔCT.
Bisulfite Modification and Methylation-Specific Polymerase Chain Reaction
The genomic DNA was extracted from the cells using the Qiagen DNA Isolation kit (Qiagen Inc) according to the manufacturer’s instructions. The DNA was modified with sodium bisulfite and analyzed according to the method described in the CpGenome DNA Modification kit (Intergene, Purchase, New York). The modified DNA was amplified with the primers for the methylated and unmethylated sequence of DAPK, ECAD, MGMT, and RASSF1A.24,25 The PCR was carried out at amplification conditions that consisted of 2.5 μL 10× PCR buffer, 0.25 mmol/L dNTPs, 0.25 pmol primers, and 2.5 U of Taq polymerase (Applied Biosystems, Foster City, California). After activation of the Taq polymerase at 95°C for 15 minutes, the PCR was performed in a GeneAmp 9700 thermal cycle (Applied Biosystems, Carlsbad, California) for 40 cycles consisting of denaturation at 95°C for 30 seconds, annealing at 62°C (DAPK, ECAD, and RASSF1A primer sets), or 66°C (MGMT primer set) for 1 minute, followed by a final 5-minute extension at 72°C. The PCR products were then loaded onto 3.5% low-range agarose gels, stained with ethidium bromide, and visualized with the ChemiDoc XRS system molecular imager (Bio-Rad). The CpGenome Universal ummethylated and methylated DNAs (Chemicon International, Inc, Temecula, California) were used as positive control for unmethylation and methylation, respectively. Template-free water was used as negative control.
Global DNA Methylation Quantification
Global methylation was conducted using a MethylFlash Methylated DNA Quantification kit (Epigentek Inc, Farmingdale, New York), an enzyme-linked immunosorbent assay (ELISA)-based colorimetric assay. The instructions were followed as outlined in the kit, using 100 ng of genomic DNA. The methylated fraction of DNA is detected using the capture and detection antibodies and then quantified colorimetrically by reading the absorbance in a microplate spectrophotometer. Relative quantification was determined by normalizing the readings to the positive control provided with the kit. Results of the CSC exposures are presented as fold changes relative to the untreated cells.
Invasion Assay
The BD BioCoat Matrigel Invasion Chambers (BD Biosciences; Bedford, Massachusetts) was used to measure acquisition of an invasive phenotype in vitro. The chambers consisted of a BD Falcon TC Companion Plate with Falcon cell culture inserts that contains an 8-μm-pore-size membrane with a thin layer of Matrigel basement membrane matrix. Briefly, the chambers were removed from −20°C storage and allowed to come to room temperature. Medium (0.5 mL insert and 0.5mL well) was added to rehydrate the matrigel for 2 hours at 37°C in the presence of 5% CO2. The media was removed from the wells and replaced by the media with serum (chemoattractant), followed by 0.5 mL of NL-20 untreated, 1 μg/mL of CSC and 10 μg/mL of CSC cell suspension (2.5 × 104 cells) to the matrigel inserts. The chambers were incubated in a tissue culture incubator, at 37°C, 5% CO2 atmosphere for 22 hours. The noninvading cells were removed and the filter stained according to instructions provided in the assay. Cells were counted using a Nikon microscope.
Statistical Analysis
The Prism IV software (GraphPAD Software, San Diego, California) was used for graphical analyses. Data were analyzed for statistical significance using 1-way analysis of variance (ANOVA) and Student t test. Differences in P values of <.05 were considered statistically significant.
Results
The effect of CSC on cell growth was assessed in cultured lung and fibroblast cell lines. In these experiments, the dose range of CSC was 1 μg/mL to 100 μg/mL. Duration of the exposure was up to 72 hours. Under these conditions in each of the 3 cell lines examined, inhibition of cell growth increased with the dose of CSC and duration of exposure (Figure 1). Overall, the extent of inhibition did not exceed 20%.
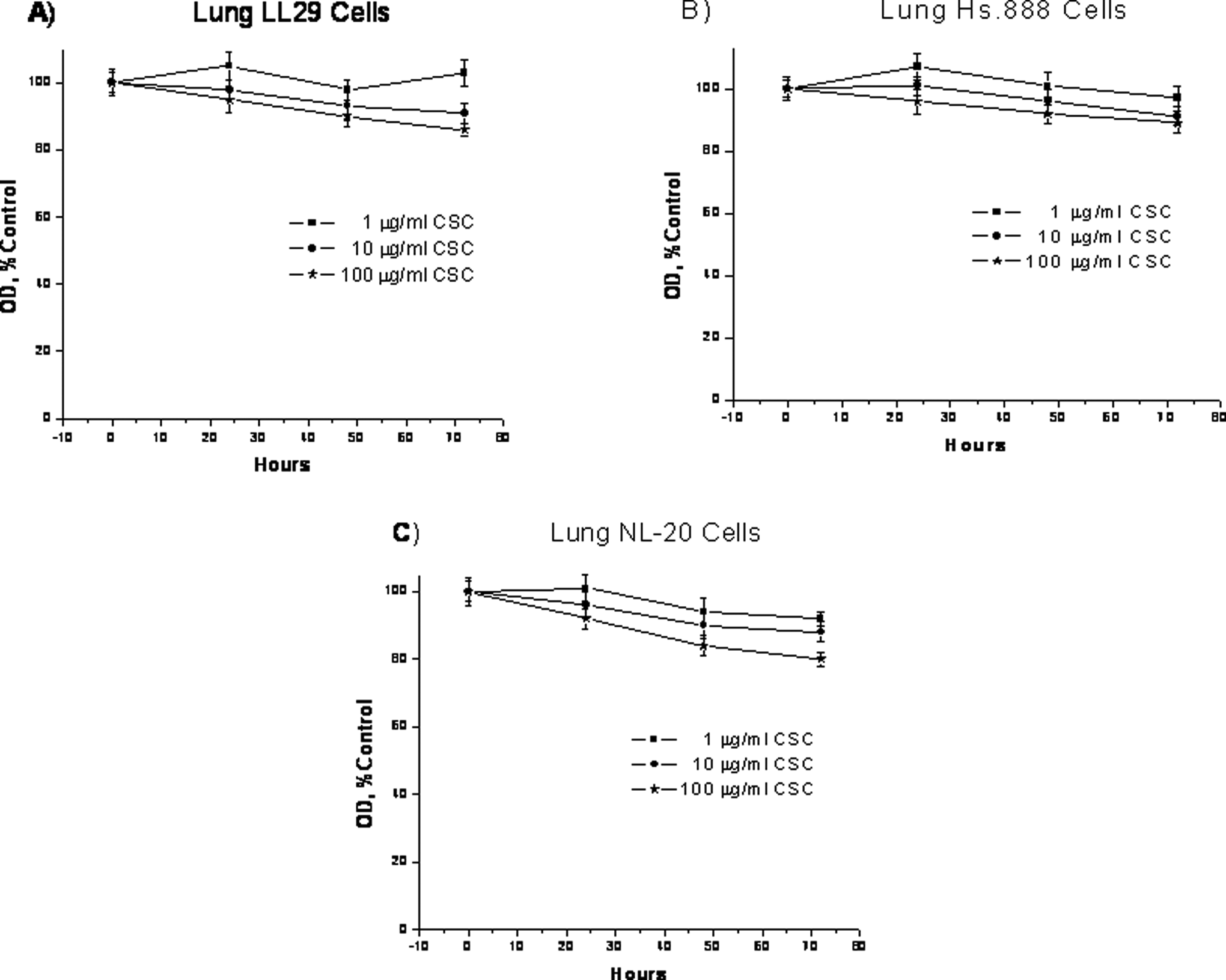
Effect of cigarette smoke condensate on lung cells proliferation. The LL29, Hs.888, and NL-20 cells were treated with 1, 10, or 100 μg/mL cigarette smoke condensate for 24, 48, or 72 hours and assayed by MTT techniques. Data are presented as mean ± standard deviation of at least 3 determinations. NL-20 indicates immortalized bronchial epithelial lung cells; LL29, fibroblast lung cells; and Hs.888, fibroblast lung cells.
Gene expression levels and methylation status were differentially affected by short-term exposure to CSC. In LL29 and Hs.888 fibroblast cell lines, expression levels and methylation status were not significantly affected by CSC exposure for any of the genes examined under the conditions employed (Figure 2 and Table 1). Cells were exposed to CSC, 10 μg/mL or 100 μg/mL, for 24 to 72 hours. The DAPK expression in NL-20 lung cells, a bronchial epithelial cell line, was also not significantly affected (Figure 3), nor was its methylation status (Table 1). However, the ECAD in this cell line showed a significant decrease in the expression after 72 hours with both dosages of 10 µg/mL and 100 µg/mL CSC (Figure 3). This decrease in expression correlated with the hypermethylation of the ECAD promoter at both doses (Table 1). The MGMT expression increased only at a CSC dose of 100 µg/mL, which was found for all exposure times (Figure 3). This correlated to an unmethylated profile of the promoter at this concentration (Table 1). The RASSF1A promoter profile was methylated in the control samples (Table 1). However, when treated with 10 µg/mL or 100 µg/mL CSC, an unmethylated promoter profile was noted. This correlated with increased expression of RASSF1A at both the 10 µg/mL and 100 µg/mL doses (Figure 3). No changes were noted at 0.1 µg/mL or 1 µg/mL of CSC in the NL-20 lung cells for these exposure times (data not shown).
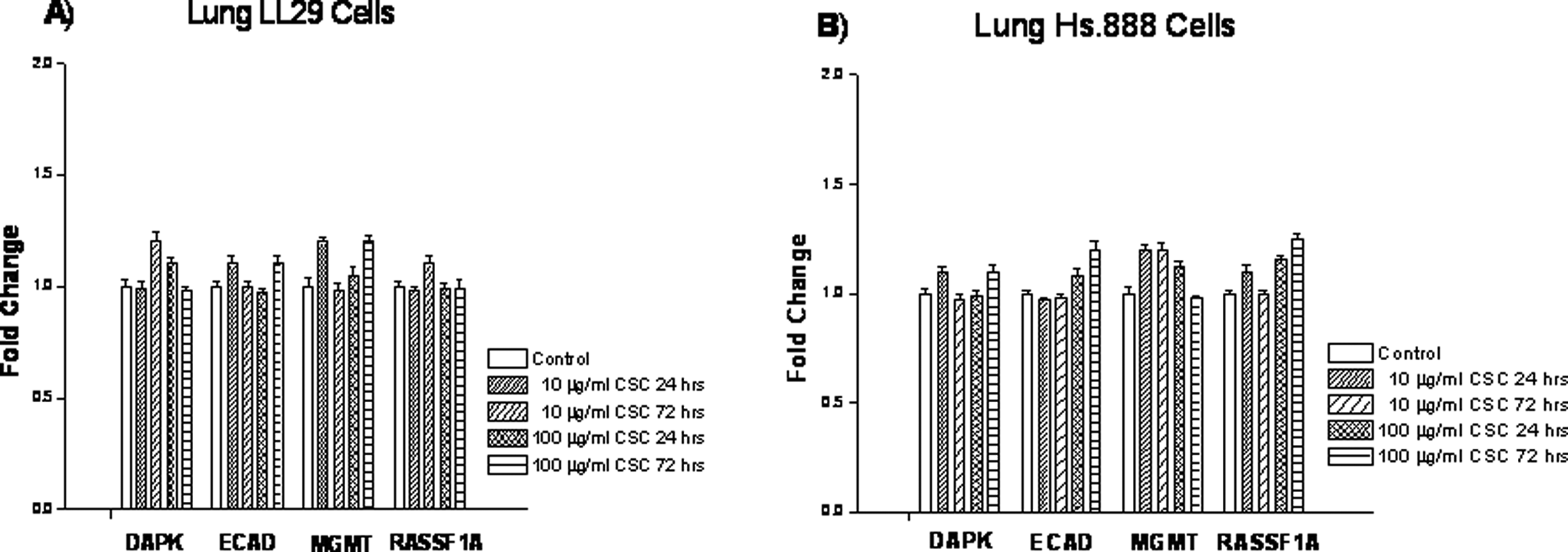
Effect of cigarette smoke condensate on expression levels of death-associated protein kinase, E-cadherin, O 6 -methyl-guanine-DNA methyltransferase, and Ras-associated domain family 1, isoform A in LL29 and Hs.888 lung cells. Cells were treated with 10 or 100 μg/mL cigarette smoke condensate for 24 or 72 hours. Expression levels were determined by quantitative real-time reverse transcriptase–polymerase chain reaction. Data are presented as mean ± standard deviation of at least 3 determinations. LL29 and Hs. 888 indicate fibroblast lung cells.
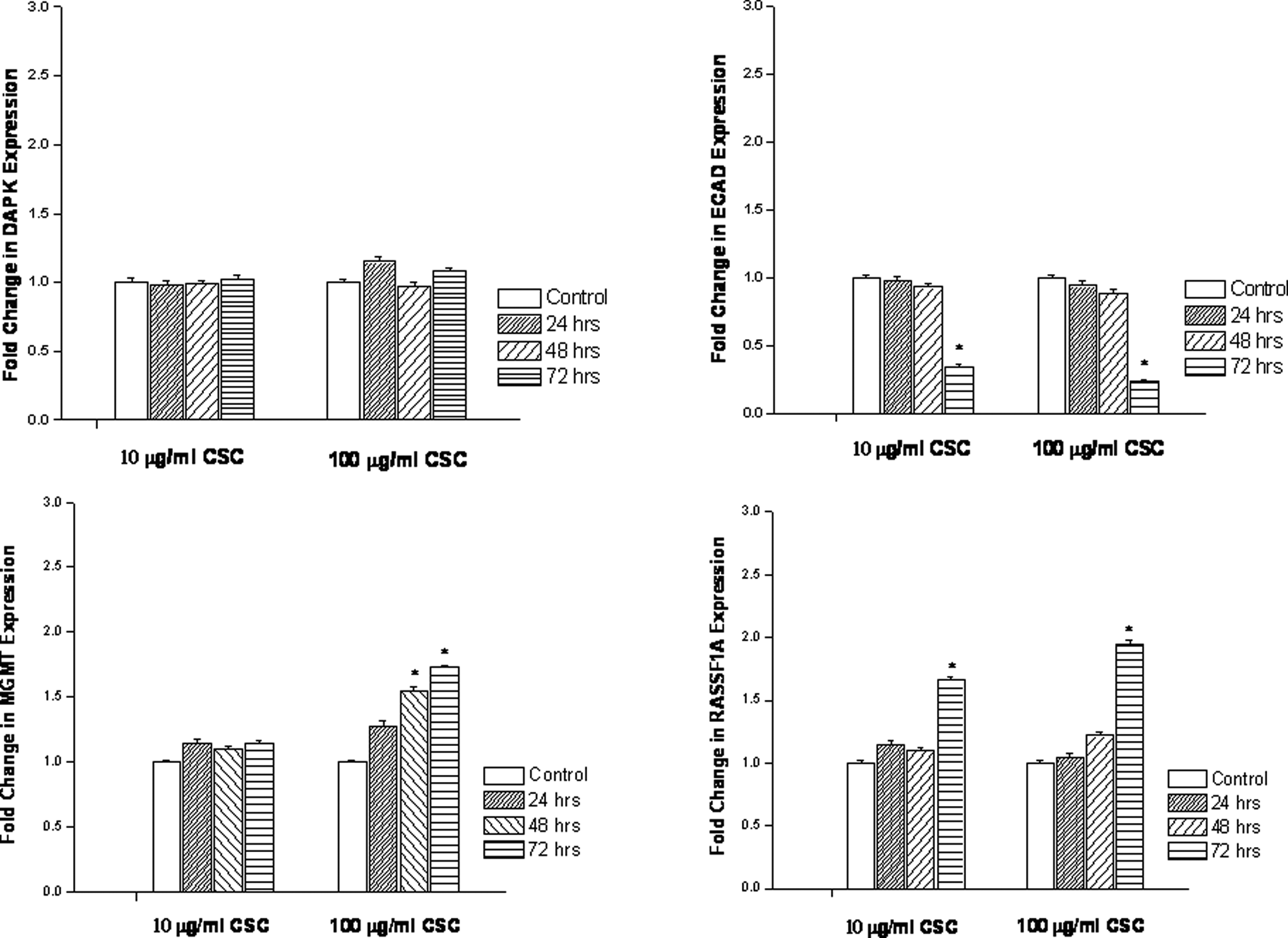
Effect of cigarette smoke condensate on expression levels of death-associated protein kinase, E-cadherin, O 6 -methyl-guanine-DNA methyltransferase, and Ras-associated domain family 1, isoform A in immortalized bronchial epithelial lung cells. Cells were treated with 10 or 100 μg/mL cigarette smoke condensate for 24, 48 or 72 hours. Expression levels were determined by quantitative real-time reverse transcriptase–polymerase chain reaction. Data are presented as mean ± standard deviation of at least 3 determinations. *Denotes a significant difference compared to the controls.
Effect of Cigarette Smoke Condensate on Methylation Status in Lung Cells.
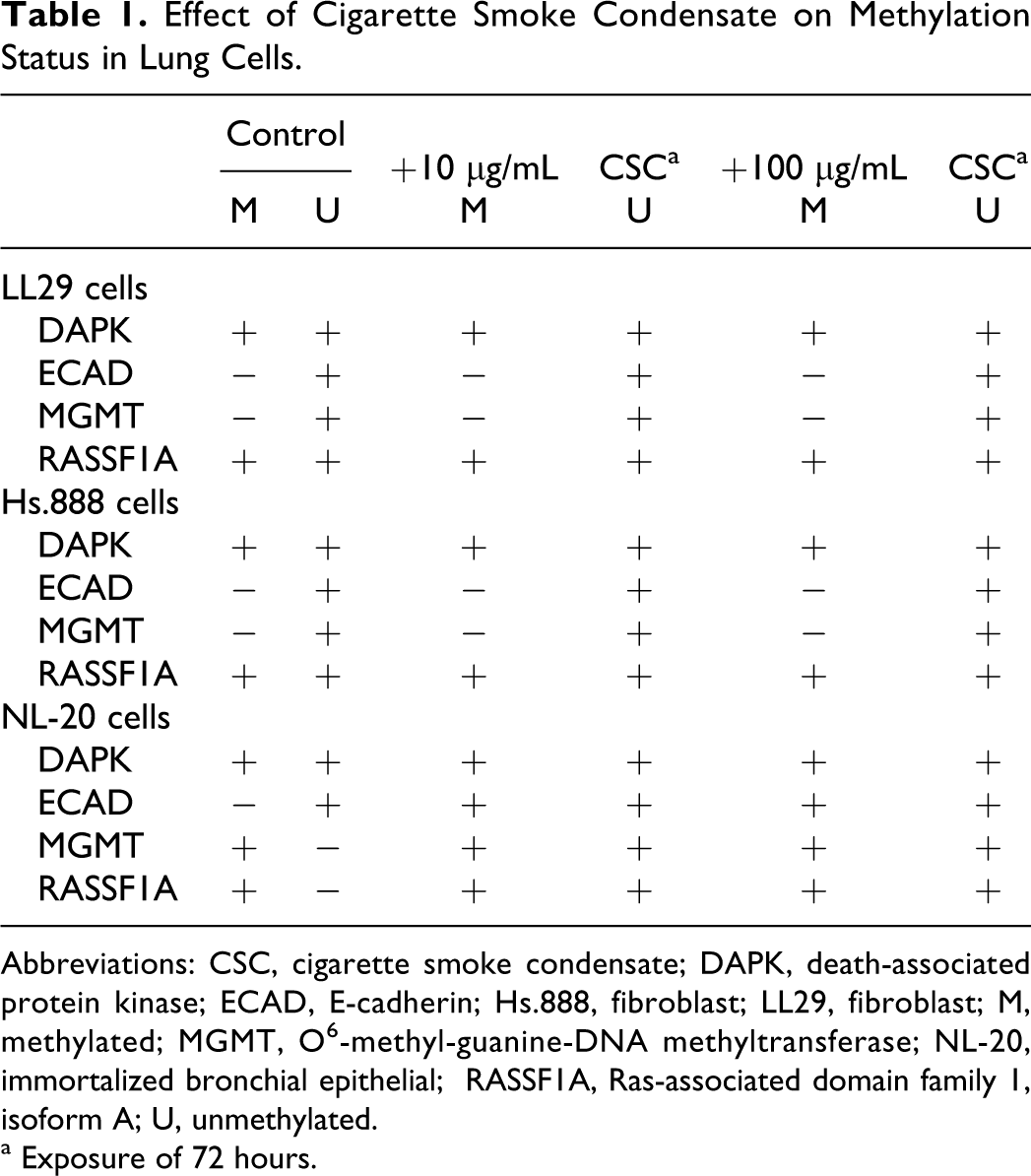
Abbreviations: CSC, cigarette smoke condensate; DAPK, death-associated protein kinase; ECAD, E-cadherin; Hs.888, fibroblast; LL29, fibroblast; M, methylated; MGMT, O 6 -methyl-guanine-DNA methyltransferase; NL-20, immortalized bronchial epithelial; RASSF1A, Ras-associated domain family 1, isoform A; U, unmethylated.
a Exposure of 72 hours.
The NL-20 lung cells were also chronically exposed to CSC (1 μg/mL or 10 μg/mL) for 7, 14, and 28 days. Promoter methylation status for the 4 genes was similar after 28-day exposures to methylation status shown at 72 days, except the RASSF1A promoter profile was methylated after exposure for 28 days (Table 1 and Figure 4). Morphological changes associated with the cell transformation, such as focal areas and loss of contact inhibition, were observed after CSC treatment for 14 days (Figure 5). To determine the effect of CSC on cell invasion, the invasive capacities of NL-20 lung cells without and with CSC exposures of 7, 14, and 28 days at a dose of 1 μg/mL or 10 μg/mL were examined. The CSC was found to stimulate the NL-20 lung cell invasion (Table 2). The expression level of ECAD in these cells was significantly decreased after chronic exposure to CSC for 28 days (Figure 6). The effect of chronic exposure of CSC on global methylation was also assessed. Methylation was measured in an ELISA-based assay after exposures of 7, 14, and 28 days at a dose of 1 μg/mL or 10 μg/mL. The CSC decreased global DNA methylation in NL-20 lung cells (Figure 7). This decrease was dose and duration dependent.

Effect of cigarette smoke condensate on methylation status of death-associated protein kinase, E-cadherin, O 6 -methyl-guanine-DNA methyltransferase, and Ras-associated domain family 1, isoform A in immortalized bronchial epithelial lung cells. A representative sample of immortalized bronchial epithelial lung cells is shown. Cells were treated with 1 or 10 μg/mL cigarette smoke condensate for 28 days. Methylation was determined by bisulfite modification of the genomic DNA and MSP using primers for the unmethylated or methylated promoter sequence. US indicates unmethylated positive control; MS, methylated positive control; UC, unmethylated control; UC, methylated control; U, unmethylated; M, methylated.
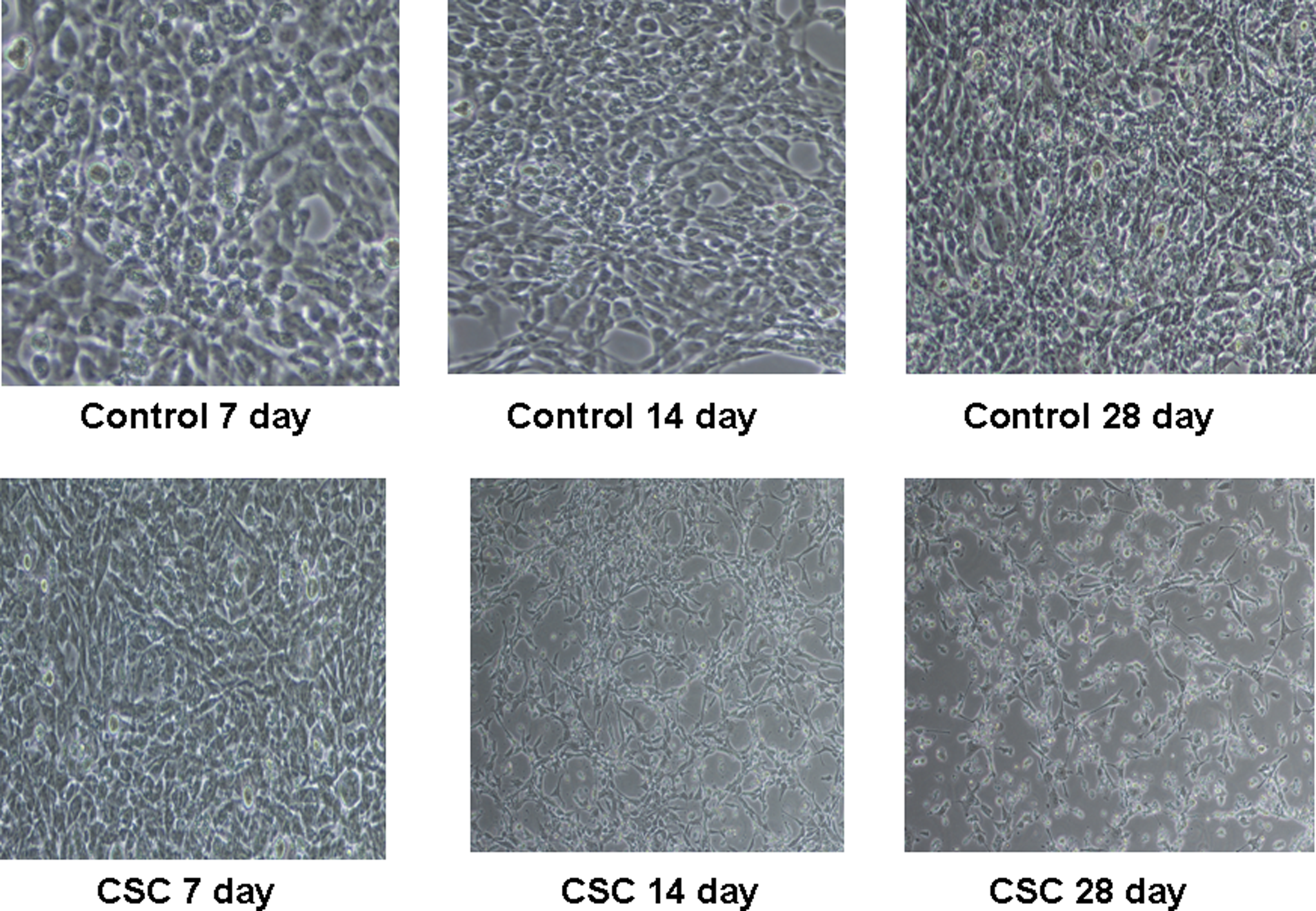
Morphology of immortalized bronchial epithelial lung cells exposed to cigarette smoke condensate. Photograph of controls for days 7, 14, and 28. Also shown are photographs of morphological changes noted with 10 μg/mL of cigarette smoke condensate for 7, 14, and 28 days of chronic treatment.
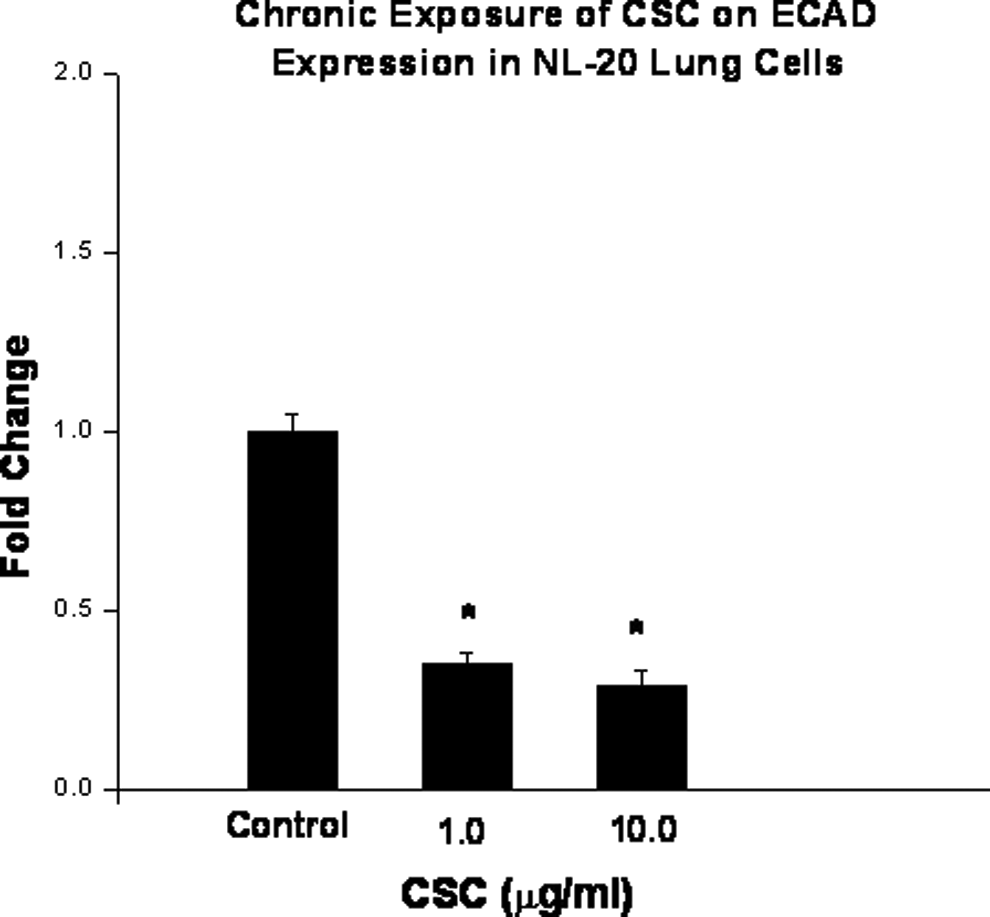
Effect of chronic exposure of cigarette smoke condensate on E-cadherin expression in immortalized bronchial epithelial cells. Cells were treated with 1 or 10 μg/mL of cigarette smoke condensate for 28 days. Expression levels were determined by quantitative real-time polymerase chain reaction. Data are presented as mean ± standard deviation of at least 3 determinations. *Denotes a significant difference compared to the controls.
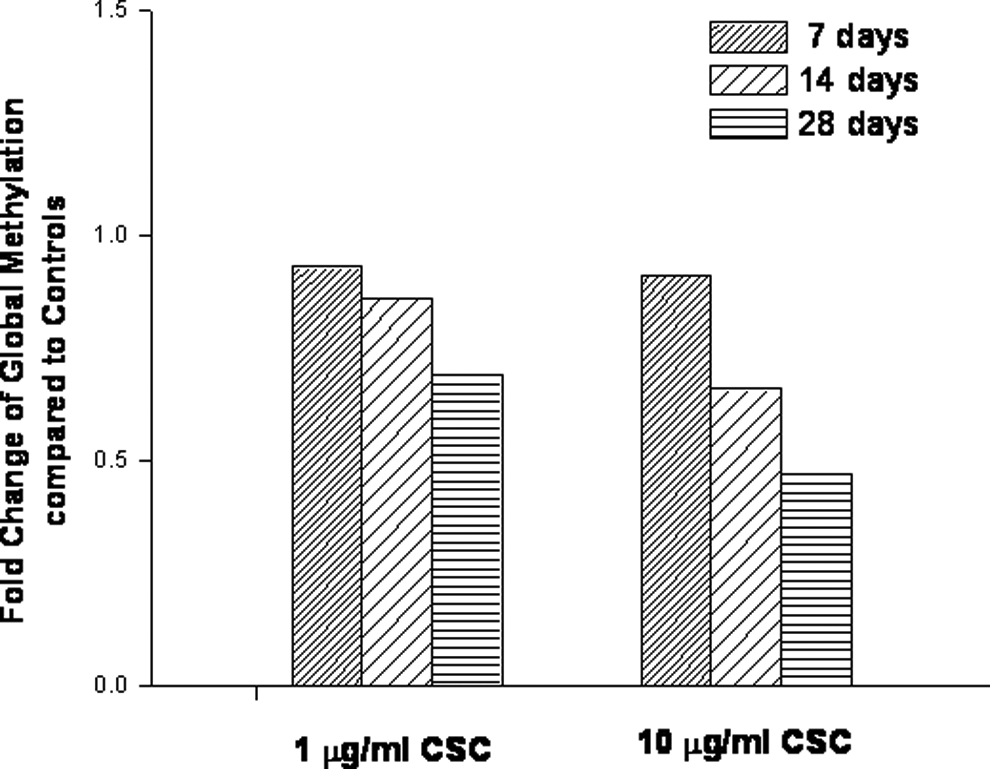
Effect of chronic exposure of cigarette smoke condensate on global DNA methylation. The immortalized bronchial epithelial cells were treated with 1 or 10 μg/mL cigarette smoke condensate for 7, 14, or 28 days. Genomic DNA was extracted. Global methylation status was quantified in a microplate-based enzyme-linked immunosorbent assay. Values were derived as fold change in treated cells compared to the untreated cells (controls) in duplicate determinations.
Effect of Cigarette Smoke Condensate Chronic Exposure on Immortalized Bronchial Epithelial Lung Cells Invasion.

Abbreviation: CSC, cigarette smoke condensate.
Discussion
The vast majority of lung cancers are directly attributable to cigarette abuse1,2; furthermore, a substantial number of lung cancers in never-smokers are due to environmental tobacco exposure (passive smoking). 4 Continued efforts are necessary to better define the mechanisms contributing to tobacco-induced pulmonary carcinogenesis and to identify novel molecular targets for the treatment and prevention of this tobacco-related disease as well as molecular markers for the assessment of risk. Studies have implicated a relationship between aberrant DNA methylation and tobacco-related lung cancer.12–15 However, mechanistic studies are yet to establish the exact nature of this relationship by finding the sequence of events that lead to global DNA demethylation and locus-/gene-specific gain of DNA methylation, which may, in turn, contribute to lung cancer development.
In the present study, an in vitro model system was employed to gain insight regarding alterations potentially contributing to the promotion and progression of tobacco-lung cancers. Due to the difficulties in conducting such studies in vivo, investigations of the effects of in vitro exposure of human lung cell lines to cigarette smoke offer a rational approach. This study provides critical data showing epigenetic regulation of critical genes involved in DNA repair, invasion, and tumor suppression of lung cells treated with CSC. Specifically, CSC treatment increased expression of RASSF1A and MGMT in NL-20 lung cells after short-term exposure. For each gene, hypomethylation increased. On the other hand, the ECAD expression decreased with the CSC treatment, which correlated with the hypermethylation of the promoter. These results are in contrast to those in a recently reported study that investigated tobacco-induced epigenetic events in vitro. 26 Expression and methylation of MGMT and ECAD were unchanged after CSC treatment, and RASSF1A expression was found to be reduced, which coincided with promoter hypermethylation. It should be noted that this study was conducted in a different lung cell line, HBEC, and the exposure duration was 5 months at doses of 50 µg/mL and 100 µg/mL. However, in another study of in vitro CSC exposure in BEAS-2B lung cells, CSC was found to reduce the ECAD expression, 27 as demonstrated in the current study. Cells were chronically exposed for 1 month at a dose of 20 µg/mL. The DAPK is involved in DNA damage-induced apoptosis and has been shown to be inactivated via hypermethylation in a number of studies involving non-small cell lung cancer.22,23 The MGMT is involved in DNA repair, 20 and RASSF1A encodes an RAS effector that has been identified as a tumor suppressor of many different cancer types. 24 The loss of expression of ECAD, a Ca2+-dependent adhesion molecule responsible for mediating intercellular contacts in morphogenesis and tissue structure maintenance, has been implicated in a number of cancers. 21 Increased ECAD promoter methylation has been shown in smokers using bronchial brushings. 25 Furthermore, silencing of the E-CAD by promoter hypermethylation has been reported in several cancer types including lung cancer. 15
The effects of longer term exposures of CSC in NL-20 lung cells were also examined in the current study. The observed morphological changes suggest that CSC has a transforming effect, as noted in chemical-induced cellular transformation properties. 28 These cells showed focal areas and loss of contact inhibition, which also indicated that they had begun to develop invasive properties. Phenotypic change has been observed in BEAS-2B cells chronically exposed with CSC. 27 Because invasion contributes prominently to the tobacco-induced pulmonary carcinogenesis, the effect of cigarette smoke on the invasive behavior of NL-20 lung cells was tested after exposures of 7 days, 14 days, and 28 days. The CSC stimulated cell invasion in this system. The ECAD expression in these cells remained significantly decreased after the 28-day exposure. The ECAD has responsibility for mediating intercellular contacts in morphogenesis and tissue structure maintenance, 21 and loss of ECAD expression is a hallmark of epithelial-to-mesenchymal transition (EMT).29,30 Chronic exposure of these cells to CSC decreased global DNA methylation. Global loss of DNA methylation is also associated with carcinogenesis, which is thought to contribute to oncogenesis by reactivation of the latent retrotranspoons, inductions of genomic instability, and activation of proto-oncogenes.31,32 These results are in agreement with a previous in vitro study in which CSC treatment was shown to produce hypomethylation of D4Z4, NBL2, and LINE-1 repetitive DNA sequences, surrogate markers of global DNA methylation. 26
In summary, this study demonstrated that short-term or long-term exposure of CSC exerts changes in the lung cells that have been shown in the lung cancer in vivo. The increase in MGMT and RASSF1A could be in response to induced damage to the cells. The MGMT expression may represent the cells’ attempt to repair damage; however, if the cells escape repair, these cells can begin to transform to cancer cells. The RASSF1A, the tumor suppressor, also increased to suppress transformation or altered cells becoming cancer cells. This study also showed that CSC can exert its effects through epigenetic mechanisms. The in vitro approach provides a useful model in establishing causal linkages between tobacco smoke exposure and events leading to potential harm in humans.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.