
Abstract
Toxicologic pathologists contribute significantly to the development of new biopharmaceuticals, yet there is often a lack of awareness of this specialized role. As the members of multidisciplinary teams, toxicologic pathologists participate in all aspects of the drug development process. This review is part of an initiative by the Society of Toxicologic Pathology to educate scientists about toxicologic pathology and to attract junior scientists, veterinary students, and veterinarians into the field. We describe the role of toxicologic pathologists in identifying candidate agents, elucidating bioactive pathways, and evaluating efficacy and toxicity in preclinical animal models. Educational and specialized training requirements and the challenges of working in a global environment are discussed. The biopharmaceutical industry provides diverse, challenging, and rewarding career opportunities in toxicologic pathology. We hope that this review promotes understanding of the important role the toxicologic pathologist plays in drug development and encourages exploration of an important career option.
Introduction
Toxicologic pathology is a rewarding and challenging discipline that touches the daily lives of nearly everyone. From testing new drugs before they reach human patients to determining toxic effects of oil spills on wildlife, toxicologic pathologists make major contributions in a variety of work settings. Yet, despite their wide-ranging influence, toxicologic pathologists work primarily behind the scenes, ensuring the safety of food, chemicals, biopharmaceuticals, and consumer products without attracting great notice. The Society of Toxicologic Pathology (STP) recently has initiated an outreach effort to raise awareness among colleagues and to attract new scientists into a field with fascinating career opportunities. This review is one component of that outreach effort and describes the work of industrial clinical and anatomic toxicologic pathologists, as well as the training and background necessary to join the profession. The authors hope to convey their enthusiasm for toxicologic pathology so that others may better understand the contributions made to solving global human, animal, and environmental health problems in satisfying careers that challenge both ingenuity and intellect.
In industry, toxicologic pathologists contribute unique skills in all phases of the development and evaluation of new chemicals, food components, agrochemicals, biopharmaceuticals, and medical devices.1,2 They perform hands-on macroscopic and microscopic evaluation of animal organs and tissues (“bench pathology”), interpret changes in composition of body fluids such as blood and urine, and integrate findings from animal studies into conclusions about the safety of new products.1,2 Career development opportunities to fit most temperaments and ambitions exist, ranging from basic research positions to business management of pathology and toxicology departments.
Industrial toxicologic pathologists comprise a substantial portion of professional groups such as the American College of Veterinary Pathologists (ACVP). A recent ACVP survey reported that large numbers of veterinary pathologists are employed in industrial settings as toxicologic pathologists, with 44% (283) of respondents listing “industry” as their primary work sector. 3 Within this industrial group, 59% (177) worked for biopharmaceutical companies, 25% (74) worked in contract research organizations (CROs, laboratories that provide fee-based services to other companies), and the remaining 16% (32) were employed in chemical, agricultural, consulting, consumer products, or other industries. 3
The perspective of this review emphasizes the biopharmaceutical industry, reflecting the primary employment sector of the contributing authors. The subsequent sections describe the specialized education, training, and experience that uniquely qualify toxicologic pathologists to identify and interpret toxicologically significant effects on animal studies in the context of drug development.
Training and Certification in Toxicologic Pathology
The skills necessary to identify toxic effects and to determine their relevance to human risk are gained from education, specialized training, and on-the-job experience in toxicologic pathology, as well as extensive, ongoing continuing education. There are many pathways to the broad training and experience necessary to practice toxicologic pathology, and most include formal programs. 4 - 6 In the United States and Canada, toxicologic pathologists typically have degrees in medicine or veterinary medicine, followed by residency or graduate training in anatomic or clinical pathology, and usually board certification (ACVP or American College of Pathologists). Similarly, in Europe, toxicologic pathologists may be veterinarians with board certification in anatomic (European College of Veterinary Pathology) or clinical pathology (European College of Veterinary Clinical Pathology). Additional instruction is often provided by other toxicologic pathology professional groups, such as continuing education courses provided by STP and training modules presented by the British Society of Toxicologic Pathologists (BSTP).
Formal certification processes are not harmonized globally. A global recognition system for proficiency of toxicologic pathologists involved in regulated nonclinical toxicity studies is in current discussions by the International Federation of Societies of Toxicologic Pathologists (IFSTP) and its member organizations across North America, Europe, and Asia.1,2 Recently, international recommendations for training toxicologic pathologists for work in regulatory-type, nonclinical studies were put forth by IFSTP. 7
In the absence of formal university degree programs in toxicologic pathology per se, comprehensive training in this field is gained by active involvement in the early development of new therapeutic agents. At least 1 year of on-the-job training that includes mentoring and peer review by more experienced pathologists is generally considered necessary to acquire the fundamental skills of toxicologic pathology. Experience obtained by evaluating different classes of compounds, animal species, and study designs allows the nascent toxicologic pathologist to further sharpen these skills. In addition, self-study and attendance of continuing education courses offered at regional, national, and international conferences are needed to develop and maintain proficiency.
Knowledge of Comparative Anatomy/Physiology and Human Risk Assessment
Risk assessment is the analysis of toxicity issues that arise during the course of animal studies and determining whether these issues are relevant to humans. It is one of the most important contributions a toxicologic pathologist makes in the biopharmaceutical and chemical industries. To accurately assess human risk based on the results from animal studies, the toxicologic pathologist must have extensive knowledge of comparative anatomy and physiology of several species including rodents (typically the rat and mouse), nonhuman primates, and dogs, and even that of humans. The contribution of the toxicologic pathologist is critical in mitigating both financial and medical risks during the progression of a biopharmaceutical candidate. Lack of knowledge of comparative anatomy and physiology might allow progression of compounds that could be harmful to humans or, alternatively, could halt progression of potentially promising and life-saving compounds.
Training in comparative anatomy and physiology can begin with undergraduate human physiology courses and anatomy laboratories that provide basic knowledge about processes conserved across species, such as cell physiology, cell-to-cell signaling, and multisystem homeostatic pathways (eg, those regulating blood pressure). In veterinary school, more intensive comparative anatomy training takes place, typically with detailed dissections of all organ systems in multiple species, ranging from cat to cow. In-depth physiology courses covering multiple species provide extensive knowledge of respiratory, reproductive, endocrine, musculoskeletal, gastrointestinal, and neurologic physiology.
Postveterinary training in pathology provides detailed study of normal and abnormal pathology of the various systems and tissues in animal species ranging from fish to farm animals to research rodents. This postgraduate period also often provides the trainee an opportunity to interact with medical colleagues in roles such as the pathology advisor on research projects involving nonclinical species or as an invited instructor in medical school or graduate school courses.
Throughout this training, it becomes apparent that while many of the physiologic processes that occur at the subcellular, cellular, or systems level are similar between species, some are very different. Induction of urinary bladder tumors in male Harlan Sprague-Dawley rats after administration of muraglitazar, an antidiabetic agent, illustrates the importance of this training. 8 Investigative work supported a nongenotoxic mechanism of tumor formation involving chronic mucosal injury and proliferation secondary to treatment-related increases in urinary solids, including calcium phosphate precipitate, which has been shown to be directly cytotoxic to rat urothelial cells. 9 - 11 The finding of tumors specifically in male rats was likely due to several factors including their quadriped stance, which allows for pooling of sediment-rich urine in the ventral bladder and subsequent physical damage with each bladder contraction during urination, the extremely high osmolality of rodent urine, and the high levels of protein present in the urine of male rats (Figure 1).12,13 The bipedal stance in humans and major differences in basic urine composition between rats and humans, along with extensive investigative work that supported the hypothesis, allowed this finding to be presented to regulatory authorities as a sex- and species-specific phenomenon that was determined to be of negligible risk to humans. 14 To obtain this favorable clinical risk assessment, detailed knowledge of species- and strain-specific differences in urine composition and micturition was critical.
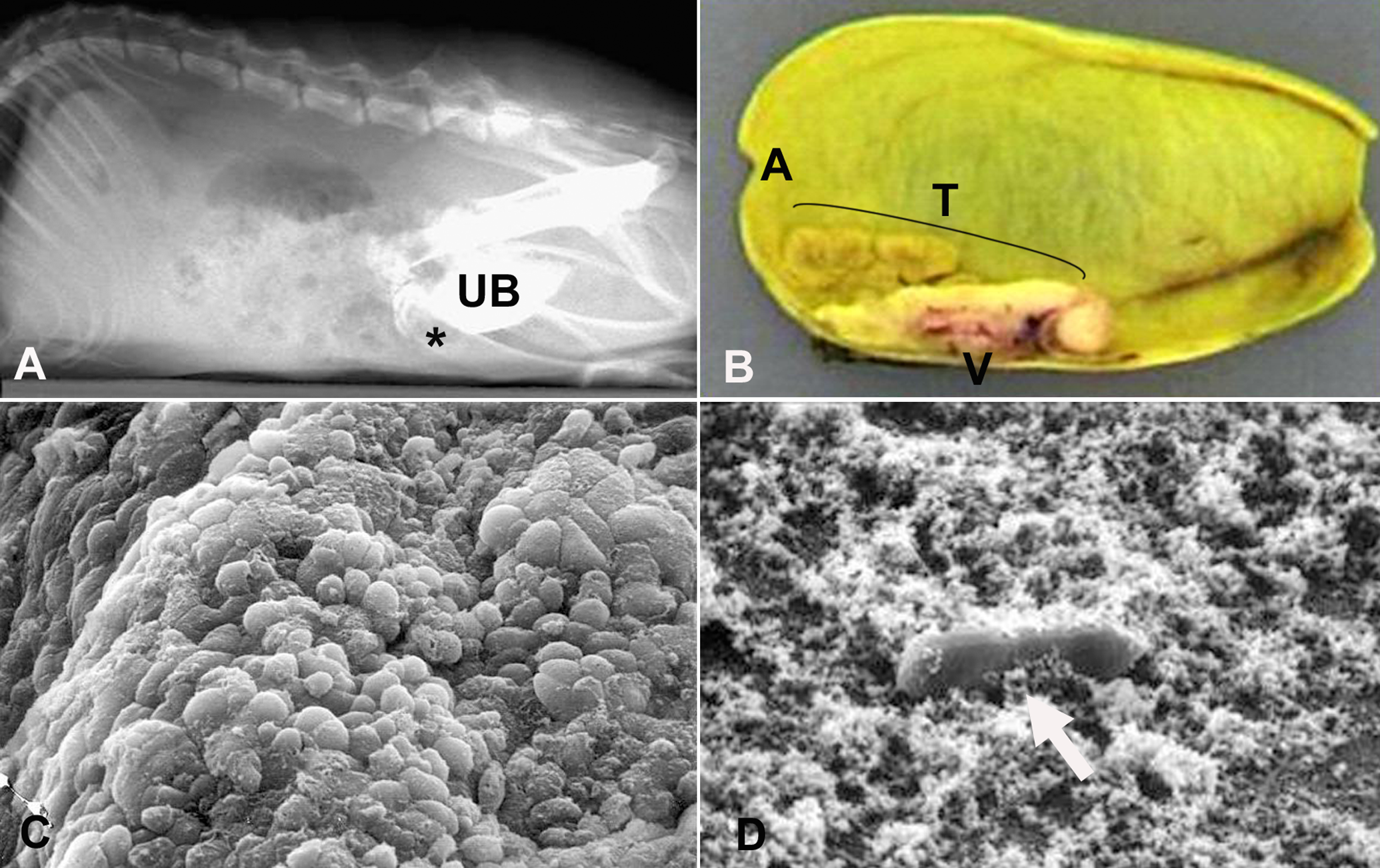
Effects of muraglitazar administration in the urinary bladder of male Sprague-Dawley rats. A, Anterioventral (*) orientation of urinary bladder (rat in ventral recumbent position). B, Urinary bladder tumor, also in anterioventral location. C, Diffuse hyperplasia of urinary bladder mucosa, scanning electron microscopy. D, Urine sediment containing abundant flocculent calcium phosphate precipitate and a normal MgNH4PO4 (struvite) crystal (white arrow), scanning electron microscopy. UB indicates urinary bladder; A, anterior; V, ventral; T, tumor.
Another well-known example of an important species difference involves liver enzyme induction and its role in thyroid hyperplasia in rats. Uridine diphosphate-glucuronosyltransferase (UDP-GT) enzymes can be induced by some xenobiotics leading to increased thyroid hormone clearance and subsequent upregulation of thyroid-stimulating hormone (TSH). These increases in TSH can lead to proliferative lesions in the thyroid gland, ranging from hyperplasia to carcinoma. Uridine diphosphate-glucuronosyltransferase is more readily induced in rodents, making this phenomenon more common in rats than other nonclinical species.15,16 However, there is no evidence that humans administered UDP-GT-inducing compounds are at an increased risk of proliferative changes in the thyroid gland. 16 Knowledge of this mechanism is critical in assessing the clinical relevance of such findings in nonclinical studies in rats.
The specialized training that toxicologic pathologists receive in comparative anatomy and physiology, and their extensive knowledge of processes that may vary between different species and strains, as well as between animals and humans, allow them to provide invaluable input into the significance of pathologic findings noted in nonclinical studies and to contribute to the progression of a biopharmaceutical candidate or chemical.
The Drug Development Process and its Language
The biopharmaceutical industry uses its own language filled with complicated jargon that can obscure its simple purpose of producing therapeutic agents with 3 attributes—quality, safety, and efficacy. Quality is a characteristic of the chemical substance of a potential new drug and is primarily the responsibility of chemists who use analytical methods to ensure purity, stability, and other physical properties. Safety and efficacy pertain to the in vivo effects of a potential drug and are assessed in the laboratory, in animal studies, and ultimately in clinical trials in humans. Toxicologic pathologists test safety and efficacy by examining the effects of new agents in experimental animals. As a basis for understanding the following discussion of how toxicologic pathologists contribute to drug development, this section provides a high level overview of the process and defines key words and phrases used in this industry.
The drug development process occurs in 2 major phases: drug discovery and drug development. In the discovery phase, therapeutic targets are identified and early drug candidates are screened for both safety and efficacy. The development phase, in contrast, focuses primarily on safety. Lead candidates (ie, those compounds selected for further development) progress through rigorous government-regulated nonclinical safety studies in animals (often also referred to as preclinical studies) to assess human risk and to estimate safe human starting doses for clinical trials, that is, studies to assess safety and efficacy in humans (Figure 2).
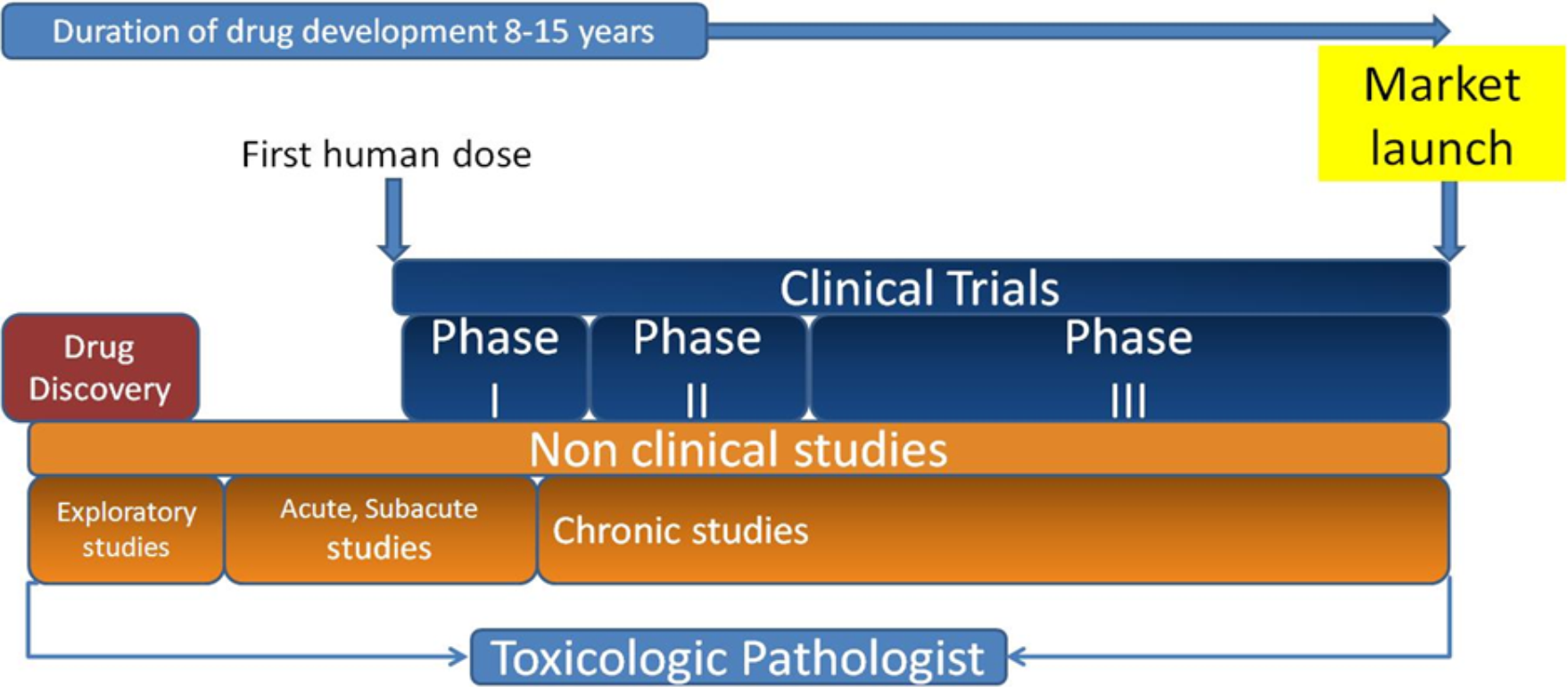
Overview of the drug development process indicating the different phases of nonclinical and clinical development and the involvement of toxicologic pathologists during this process. Figure modified from Schultze et al. 4
During both phases of drug development, a potential new drug may be referred to in various ways: test compound or molecule, test article or item, new chemical entity, potential new drug, or drug candidate. A compound is referred to as a drug only after it has been demonstrated as safe and efficacious to the satisfaction of regulatory agencies such as the US Food and Drug Administration (FDA), European Medicines Agency (EMA), or Japanese Ministry of Health, Labor, and Welfare (MHLW), and is approved for marketing.
The goal of nonclinical studies is to establish a toxicity profile for a drug candidate by identifying the organ systems that it affects (ie, target organs) and by establishing a safety margin, which in very general terms is the ratio of a level of a compound found to be safe in animal studies compared to its predicted efficacious level in humans. These concepts are more fully discussed in subsequent sections of this review.
It is important to distinguish between dose (the amount of compound administered) and plasma exposure (the amount of compound that is absorbed and retained), as there is not always a linear relationship between the two. Safety and efficacy are more accurately linked to exposure. The relationship between dose and exposure is elucidated by toxicokinetic evaluation, a key component of nonclinical studies that provides analytical data describing the time-dependent behavior of a compound for each administered dose, and defines parameters such as highest and lowest circulating levels or measures of how quickly a compound is eliminated or accumulates. This information often explains adverse effects observed by toxicologic pathologists and may be used to alter dosing regimens to minimize toxicity or to improve efficacy.
Toxicologic Pathology—Essence of the Job
Toxicologic pathologists are uniquely trained and qualified to assess the toxicologic significance of the effects of test compounds in experimental animals. There are 2 recognized subspecialties of toxicologic pathology characterized by the types of specimens evaluated. In anatomic pathology, tissues and organs primarily are assessed using microscopic evaluation (often referred to as histopathologic or morphologic evaluation). In clinical pathology, the cellular and chemical composition of blood, serum, urine, and other bodily fluids are studied using microscopy and analytical chemistry tools. 17
In nonclinical studies, both anatomic and clinical pathology data are used to fully characterize potential toxicities associated with a test compound. Test compound-related histopathologic findings are described in detail, using standardized terminology and a semi-quantitative scale for grading the severity of findings. Often additional methods are applied to tissue sections that allow further understanding of the biochemical nature of any changes that are seen. A few examples include special histochemical stains such as trichrome to identify collagen, immunohistochemical methods with antibodies to label antigens of interest, or in vivo expression markers such as in situ hybridization to localize messenger RNA (mRNA) for cellular proteins.
Additionally, test compound effects are evaluated by looking at clinical pathology parameters such as alterations in numbers and types of bone marrow or circulating blood cells; levels of proteins, electrolytes, and tissue-specific enzymes in blood and urine that reflect specific organ status; and functional parameters such as blood clotting times. Clinical pathology data are usually available during, or soon after, the end of the in-life portion of a nonclinical study and can provide the first clues about the identity of target organs. For example, significant serum elevations of the liver enzyme alanine aminotransferase would signal the need to look for corroborating evidence of liver cell damage during histopathologic evaluation. The clinical pathology data set is common to both nonclinical and clinical studies and thus can bridge these study types to provide significant information about safety and efficacy.
Toxicologic pathologists develop critical interpretative expertise for differentiating test compound effects from background findings known to occur in a particular species. For example, toxic effects seen in the eye during a nonclinical study in monkeys would likely stop the development of an otherwise promising test compound. However, optic atrophy, a striking histologic change shown in Figure 3 is known to occur in normal monkeys.18,19 An experienced toxicologic pathologist will recognize that this finding may be a background change, unrelated to test compound administration, and will follow-up with a review of the incidence and severity, looking for a relationship to dose and plasma exposures. Observing this change in a few monkeys, evenly distributed throughout low-, mid-, and high-dosage groups, supports the idea that it is a background finding. If, however, nerve atrophy occurs in a higher percentage of monkeys in higher dosage groups or is significantly more severe in these groups, a connection to test compound cannot be ruled out.
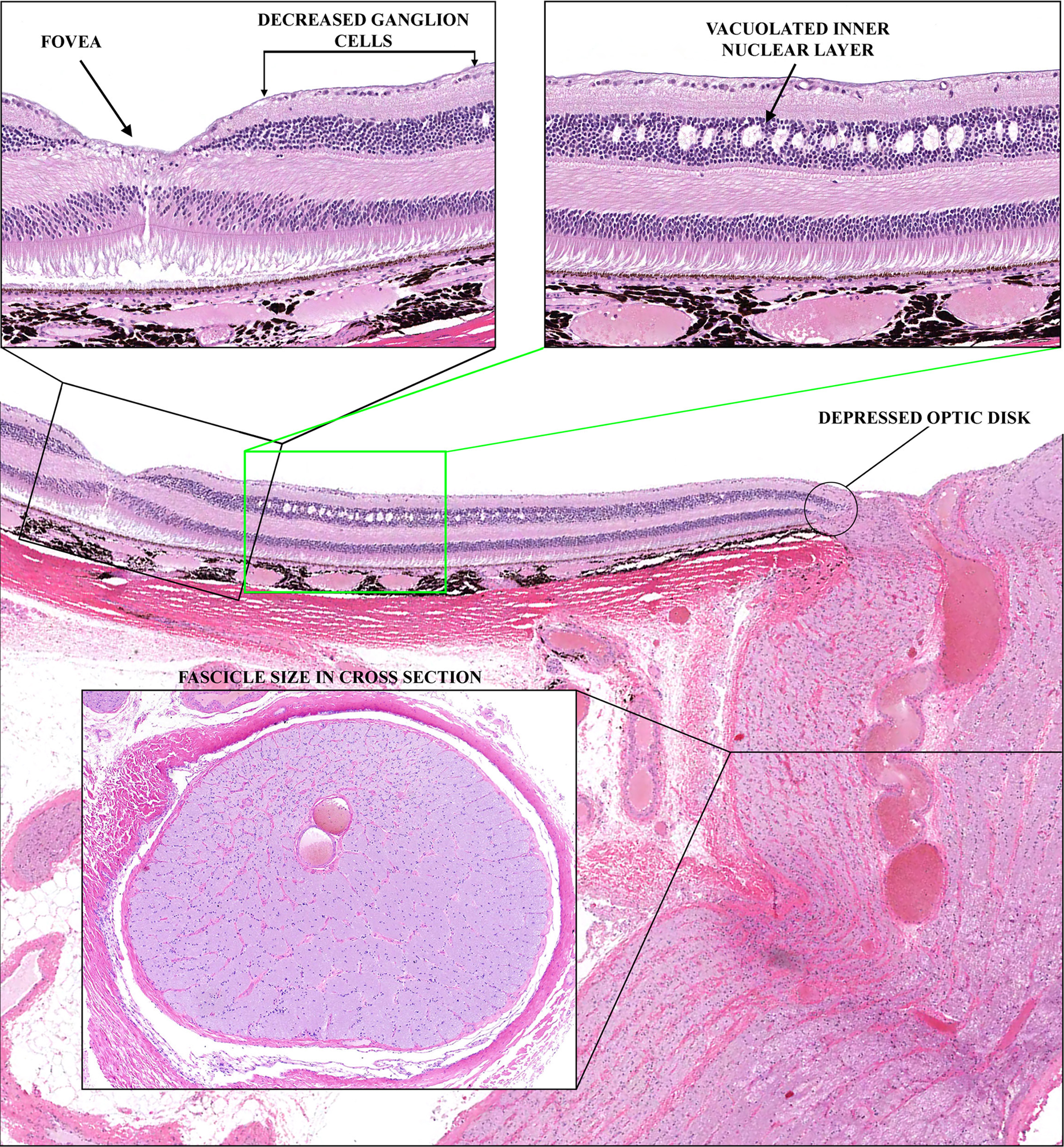
Microscopic characteristics of bilateral optic atrophy, a finding in normal cynomolgus monkeys. Features include variable fewer ganglion cells in the macular region of the retina, vacuolization of the inner nuclear layer, depressed optic disk, and smaller nerve fascicles. 6
Historical data may be used to place pathologic findings into context, 20 helping toxicologic pathologists to assess the relevance of findings in nonclinical studies and to interpret their potential significance to human safety. Demonstrating that even the highest incidence of a study finding falls within the historical rates may minimize concern.
Histopathology peer review is a typical practice in toxicologic pathology to ensure quality and scientific integrity of study results and conclusions. Final microscopic findings in a pathology report represent the consensus of the study pathologist and peer reviewer. 21 Serious concerns based on the findings of the toxicologic pathologist will be communicated to toxicology team representatives and senior management to decide whether potential risk to humans is sufficient to justify the termination of a compound. Toxicologic pathologists may serve on project teams that make these decisions, and as team members may participate in generating documents for regulatory filings and may attend teleconferences or face-to-face meetings with the FDA or other health authorities.
Thus, anatomic and clinical toxicologic pathologists use their expertise not only to identify and describe findings in nonclinical studies, but, more importantly, to determine their significance, taking into account species, age, and known background findings. This assessment is key to determining whether a test compound is sufficiently safe to justify spending time and monetary resources toward its continued development, or whether it may be more prudent to allocate those resources instead to a compound with a better chance of success.
Toxicologic Pathologists in Drug Development
As previously mentioned, there are 2 major phases in developing new therapeutic agents—drug development and drug discovery. The aim of discovery studies is to rapidly assess compound toxicities. Since these studies are not intended to be submitted for government review, they are often referred to as “unregulated” and do not adhere strictly to regulatory requirements that can add substantially to study costs and constrain the flexibility needed to fully explore the mechanisms of toxicity. The requirements for development (“regulated”) studies are established by governmental regulations enacted to ensure that the study data are of dependable quality, and that all aspects of study conduct are reliably documented. The following 2 sections describe in further detail the role of the toxicologic pathologist in discovery and development studies and highlight some differences between the two.
Drug Discovery
Launching a new drug is a costly process, with a very low probability of success. Approximately 10 to 15 years are required to develop a new drug, at a cost of $1.2 to $1.3 billion. Only 1 in 5 drug candidates that reach clinical studies is approved for marketing, and only 1 in 5 of these will return the research and development (R&D) costs that were expended to develop it.
22
Thus, establishing efficacy and identifying safety liabilities as early as possible in the drug development cycle (ie, in the discovery phase) are linked to the profitability and continued existence of a biopharmaceutical company.23,24 To support early discovery goals, toxicologic pathologists participate in a wide range of investigative study types designed to
25
validate and evaluate therapeutic targets, differentiate on-target from off-target effects, select relevant species for nonclinical studies, and identify and verify biomarkers of safety and efficacy.
Each of these activities is discussed further below
Target validation demonstrates that drug candidate interaction with cellular molecules is linked to desired biological effects in efficacy models, such as obese mice for the study of diabetes. In early in vivo efficacy studies, the traditional role of the toxicologic pathologist has centered on providing and interpreting morphologic data in conjunction with the results from cell-based assays. Linking these 2 types of data sets is key to understanding target modulation. For example, pathologists may correlate in vitro angiogenesis assay findings to histomorphologic assessment of tumor appearance and vascular density in animal cancer models to assess the efficacy of antiangiogenic oncology therapeutic agents.
Differentiating on-target from off-target effects
On-target effects are a consequence of the desired activity of a molecule, while off-target effects are related to the chemical nature rather than the pharmacologic activity of a compound. Distinguishing these 2 types of effects is important in accurately predicting human toxicity. The risk of an on-target effect is gauged by whether it occurs at far higher exposures than those expected at the anticipated efficacious dose in humans. A compound with serious off-target effects (eg, cardiotoxicity) may be rescued if chemists can alter its molecular structure to remove toxic side effects without eliminating efficacy. Often, however, it is determined that a core structure is inherently toxic, and a new chemical series must be pursued. The toxicologic pathologist is a key participant in these assessments by placing any adverse study findings in the context of the normal activity of the therapeutic target.
Selection of relevant species for establishment of toxicity profiles
Counterintuitive as it may seem, toxicity profiles to establish safety can only be created when there is demonstrable toxicity in animal species. In the absence of toxicity, it is not possible to calculate safety margins or to identify target organs. For this reason, regulatory agencies require testing in rodent and nonrodent species that are sensitive to the compound effects. A toxicologic pathologist collaborates with toxicologists to ensure that the correct species is chosen in early studies by correlating anatomic and clinical pathology evidence of toxicity with dose and plasma levels of the test compound.
Genetically modified animal models such as transgenic or knockout mice are often chosen for discovery, nonclinical toxicity, and pharmacokinetic studies. A key contribution of toxicologic pathologists is to ensure that these models accurately reflect human disease states and expected pharmacology.26,27 This assessment is essential in distinguishing on-target and off-target toxicities and allows the prediction of target-based responses, providing valuable direction to discovery chemists. The toxicologic pathologist working in discovery is uniquely equipped to provide a “whole animal” perspective within a team of chemists, biologists, and pharmacologists that otherwise might become molecule-, target-, or single organ-focused to the detriment of advancing efficacious new compounds. Familiarity with a broad range of animal models makes the discovery pathologist an integral player in the development of hypothesis-driven experimental protocols to investigate the mechanisms of toxicity, providing the necessary link between understanding species-specific target distribution and physiologic or pathologic responses to target modulation.
Identification and verification of biomarkers
Biomarkers are sensitive tools to detect early signals of both toxicity and efficacy. Toxic side effects in animals are less likely to halt a drug development program if there is a means to monitor similar effects in humans. The toxicologic pathologist assists in identifying suitable biomarkers, which may be as straightforward as liver enzyme activity measured in a standard clinical chemistry panel in serum, or as complex as an electrochemiluminescence assay to assess endogenous biochemicals in urine as indicators of kidney function. 28 Early identification of pharmacodynamic biomarkers to be applied in clinical trials can accelerate delivery of new drugs to patients, and toxicologic pathologists working in these areas play a critical role in ensuring the development of such assays.
To carry out these activities, discovery toxicologic pathologists correlate the results from specialized techniques such as immunohistochemistry (Figure 4), immunofluorescence, in situ hybridization, and reverse transcription-polymerase chain reaction (RT-PCR), with observations made from tissue sections or cell preparations. 29 These correlations not only provide valuable information about the mechanisms of action for potential new drugs but also may help to determine the mechanisms of toxicity. Similarly, the discovery pathologist provides insight into relationships between tissue and gene expression changes. Laser capture microdissection can be used to compare gene expression changes, 30 for example, in renal glomeruli versus tubules, or to characterize temporal aspects of tissue changes (eg, acute necrosis vs fibrosis). Thus, the discovery pathologist is a critical member of teams, helping to link molecular data on potential drug targets to physiologic outcomes.
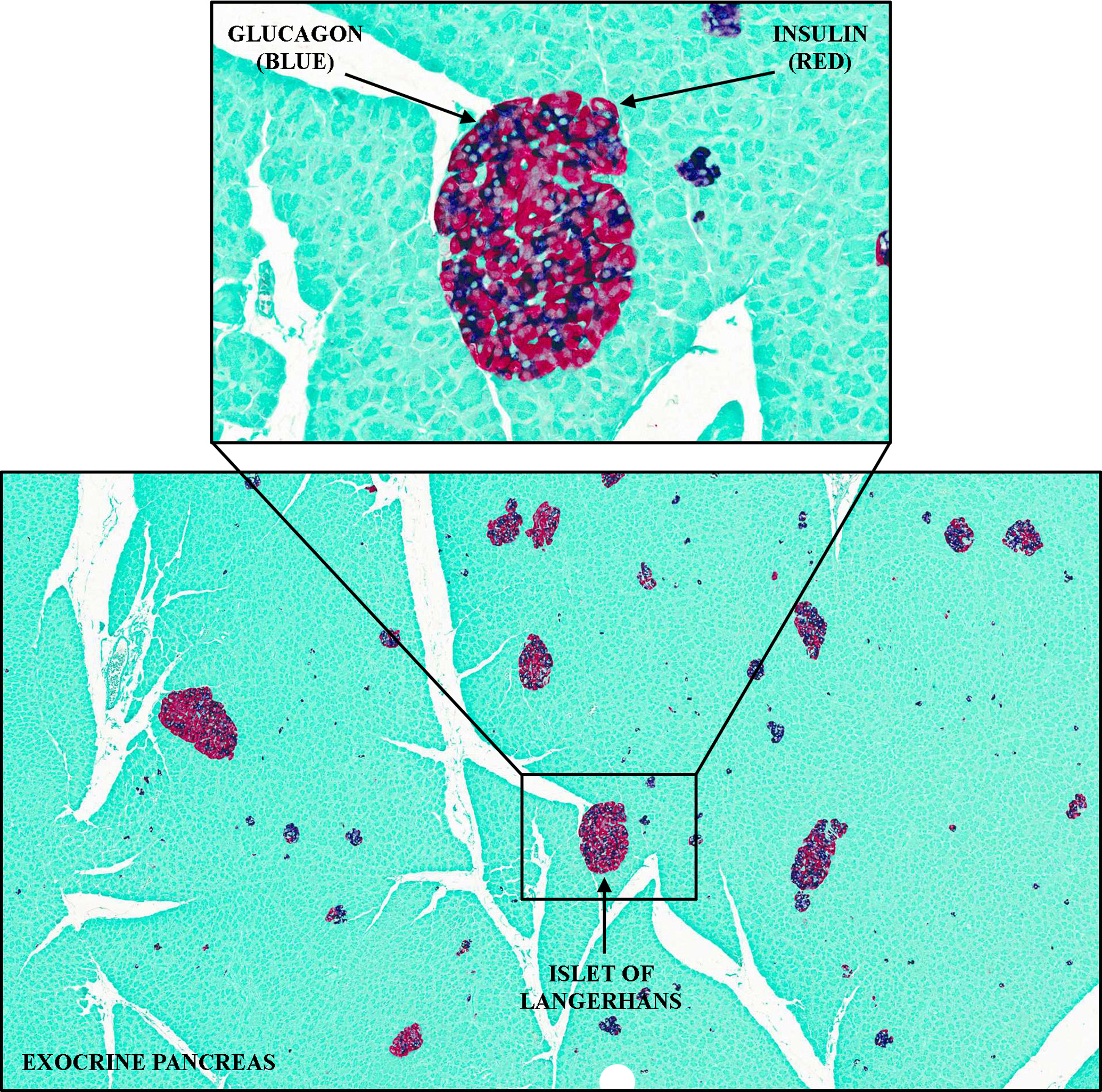
Dual immunohistochemical labeling for insulin and glucagon in the pancreas of cynomolgus monkey, identifying 2 hormone-producing populations of endocrine cells in the islets of Langerhans; glucagon-producing cells (blue) and insulin-producing cells (red).
Decisions that can be made in the discovery phase expedite drug candidate development at reduced cost, allowing available resources to be focused on molecules with greater probability of success. Chemical scaffolds or formulations associated with unacceptable toxicity early in drug development can be identified and eliminated quickly. Early understanding of adverse effects can lead to understanding of ways to mitigate (perhaps by dosing less frequently or at lower doses more often) and to monitor toxicity. The discovery pathologist can then transfer this knowledge from early exploratory studies to development phase animal studies, ultimately contributing to the success of molecules as they travel through the drug development pipeline.
Drug Development
The primary purpose of the development phase is to prepare for the first human testing of a potential new drug. Building on discovery studies, drug development studies establish safe dose levels, identify target organs of toxicity, and determine compound levels at which toxicity occurs. A safety margin (the ratio of the plasma exposure of a compound that does not cause adverse effects in animals to the predicted effective exposure in humans) must be established. Acceptable safety margins vary depending on the intended patient population and the target disease. For the patients with cancer, relatively severe toxic effects may be tolerated if the result is improved survival. For children with attention-deficit disorder, however, very high safety margins must be established.
Toxicologic pathologists work with other scientists in the biopharmaceutical sector to ensure the safety of humans by carrying out a series of studies that meet criteria established by the governments of many countries. The goal of development studies is to support regulatory filings that enable the initiation of clinical dosing in humans to establish adequate plasma exposures and safety of new therapeutic compounds. In the United States, this request for permission to begin clinical studies is an Investigational New Drug (IND) Application; in Europe and other major regions, it is a Clinical Trial Application (CTA).
Toxicologic pathologists evaluate anatomic and clinical pathology data from a series of nonclinical studies of increasing duration, beginning with acute, single-dose studies, then repeated dose range-finding studies (often 2 weeks in duration), followed by subacute (4 and 13 weeks) and chronic (26 and 52 weeks) repeated dose studies. The duration and dosing regimen for all of these studies are based upon intended clinical-dosing schedules and patient populations. For example, 2- to 4-week nonclinical studies usually support single-dose human phase I trials. In later clinical phases, the duration of nonclinical studies must be equal to that of the human clinical trials.31,32 These studies are usually performed in 1 rodent (mouse or rat) and 1 nonrodent (dog, nonhuman primate, or minipig) species. For most drug candidates, these are followed by carcinogenicity studies (2-year bioassays in rat and mouse) preceded by their dose range-finding subacute studies (4-week and 13-week duration). Recently, regulatory approval to carry out carcinogenicity studies in genetically modified mice has resulted in the possibility of decreasing the in-life duration of a study from 2 years to 6 months, which in turn reduces resource needs substantially. While there is great enthusiasm for use of these models to replace the standard 2-year mouse carcinogenicity bioassay, new historical databases and reference points will need to be established to evaluate the results.33–38
The role of the toxicologic pathologist during drug development is to identify target organ toxicity by evaluating clinical pathology, macroscopic, and microscopic changes, and to interpret these findings within the context of overall study results, including toxicokinetics and in-life observations.39,40 This assessment results in the establishment of a no-observed-adverse-effect-level (NOAEL), which is used to calculate safe starting doses of the compound in clinical trials.
As in the discovery phase of drug development, further investigations into the mechanisms of target organ toxicity may occur, especially if observed toxicity is suspected to be species-specific and therefore of questionable relevance to human safety. Detailed descriptions of microscopic changes may also help to identify biomarkers that could be used to monitor toxicity.
Toxicologic pathologists employed in the food and chemical industry assess human risks using studies similar to those used in the biopharmaceutical industry, and the requirements for toxicity data and type of studies are defined by regulatory agencies. In the United States, the Environmental Protection Agency (EPA) defines testing requirements in the Toxic Substances Control Act, and the Office of Chemical Safety and Pollution Prevention has developed a series of harmonized test guidelines for use in the testing of pesticides and toxic substances. In Europe, the Registration, Evaluation, and Control of Chemicals (REACh) provides requirements for toxicity information that must be obtained prior to registration. Study types typically include acute studies (defining an acutely toxic dose by ingestion, absorption, and inhalation), although these studies typically have only limited pathology end points. In addition, carcinogenicity studies of similar design to those used for biopharmaceuticals are key to evaluating human carcinogenic risk following exposure to a given chemical. Chronic repeated dose studies, (eg, a 1-year dog study) may be required to determine the long-term toxic effects. The resulting chemical safety assessment establishes maximum acceptable daily intakes for pesticide residues and food additives and threshold limits for occupational exposure. Occupational exposure limits for chemicals, which must also be established for new drugs during drug development, are regulated by various government agencies including Occupational Safety and Health Administration (OSHA), National Institute of Occupational Safety and Health, the American Conference of Governmental Industrial Hygienists, the American Industrial Hygiene Association, state-specific regulations, EPA in the United States, and REACh in Europe.41–44
A recent example of the critical role of toxicologic pathologists in human safety is illustrated by their involvement in investigating contamination of human and pet food with melamine. This nitrogen-rich chemical is used in the production of a variety of inedible products such as plastics, dyes, fertilizers, and fabrics but also was used illegally to increase the nitrogen level of food substances, making them appear to be protein rich. Melamine was tested in rat toxicity studies and deemed to be of low human risk. In rats, the main finding was urolithiasis (the presence of kidney stones), but there was no evidence of microscopic damage to renal tubules. 45 Urinary bladder proliferative changes were seen in chronic rat studies but were considered to be due to irritation by stones rather than a direct toxic effect of melamine on bladder epithelium. 46 Despite this evidence of low toxicity and the expectation that human exposure levels would be very low, adulteration of infant milk formula and pet food resulted in kidney failure and death in cats and dogs in Europe and the United States47,48 in 2004 and 2007 and to lethal melamine-associated nephrotoxicity in children in 2008. This led to the collaboration of toxicologic and diagnostic pathologists to elucidate the mechanism of this toxicity. While the exact mechanism remains unclear, it is likely that multiple factors (cyanuric acid exposure, volume depletion, low urine pH) in combination with melamine intoxication contributed to the toxicity, which may also explain why the toxicity was not predicted by studies in rats.45,49,50
Ultimately, the toxicologic pathologist is a key member of a multidisciplinary team that performs regulatory agency-mandated safety testing of drugs and chemicals. The ability to interact effectively with study directors, toxicologists, clinicians, technical staff, clinical veterinarians, Institutional Animal Care and Use Committee representatives, other pathologists, contract laboratory personnel, project managers, regulatory agency staff, management, chemists, and pharmacokineticists is critical to successfully carrying out this important role (Figure 5).
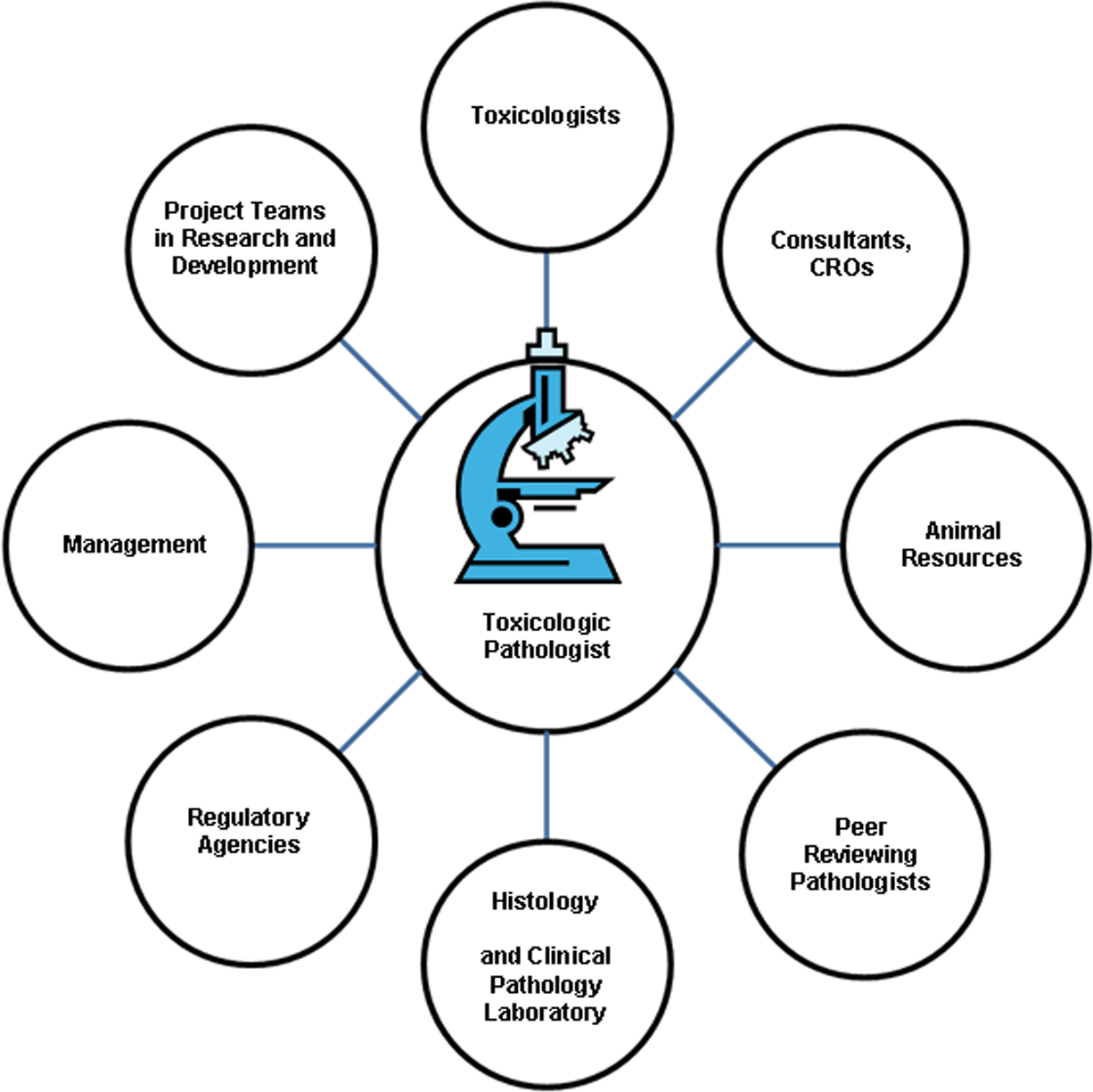
Interactions a toxicologic pathologist may have as a member of a multidisciplinary team for safety testing of drugs and chemicals.
Development of Veterinary Drugs
Development of products for veterinary use must satisfy similar criteria and go through a process like that required for drugs developed for human use. Specifically, veterinary products must be of high quality, efficacy, and safety. 51 Therefore, the role of the toxicologic pathologist in the development of veterinary medicinal products does not significantly differ from that for the development of human medicines. The expertise of the toxicologic pathologist is critical during the entire process, from drug discovery into clinical stages of development, and must include extensive knowledge of comparative anatomy and physiology for both laboratory animal species as well as for the target species, that is, the species in which the product is intended to be used.
In general, drug development studies, whether for human or veterinary medicines, are conducted to enable predictions about health risks of human patients, exposed workers, or consumers, and also to exclude the effects of no relevance to human health. 51 The development of veterinary drugs is subjected to rigorous legislative requirements, which may differ slightly between countries. In some areas of the world, veterinary products may be partially regulated by agricultural, environmental, and/or health agencies.52–59
In addition, for veterinary products, there are regulatory requirements to ensure that any residues persisting in tissues after slaughter of food animals, secreted into milk, or that may find their way into foods like honey, are not toxic to consumers and do not pose unacceptable risks to users. 56
Studies to predict safety and efficacy of veterinary products are similar to those for human products and include evaluation of toxicity, pharmacology, microbiology, consumer safety, user safety, and target animal safety (TAS). Many of these assessments overlap. For example, the results of toxicity tests may have relevance for TAS, while adverse effects in target animal studies may impact the overall safety profile and hence both consumer and user safety assessments.53,56
Because some adverse effects become evident only when products are tested in the target species, veterinary products must be tested in the species in which they are intended to be used. Several guidance documents exist for TAS studies.52,56 Studies are conducted using the proposed commercial formulation and dosing regimen at commercial doses and multiples of that dose to identify a safety margin. Carcinogenicity studies are not required for veterinary drugs; however, sponsors of veterinary drugs intended for use in food animals must demonstrate a lack of genotoxic and carcinogenic potential prior to product approval. 56
Thus, as for human drug development, the contributions of toxicologic pathologists are pivotal to ensuring the safety of animals and humans.
Regulatory Interaction
Studies evaluating the safety profile of new drug candidates and chemicals, as well as food, medical devices, and cosmetics, are highly regulated by government agencies that oversee drug development, benefit-risk balance, marketing authorization, and postauthorization steps.17,60 Agencies responsible for regulating human exposure to marketed products include the FDA, EMA, EPA, OSHA, and the Consumer Product Safety Commission.61,62 These agencies (and others) perform scientific review of nonclinical animal studies and supporting data, including pathology assessments.
In addition, biopharmaceutical industries must also follow government regulations/guidelines for the proper care and use of research animals. These are administered in the United States under the authority of the Animal Welfare Act.63,64 Many industry facilities are accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, International (AAALAC), an internationally recognized organization that provides comprehensive peer review of animal care and use programs. The AAALAC evaluates implementation of the Guide for the Care and Use of Laboratory Animals. 65
Conduct and reporting of nonclinical safety studies, including pathology data, is governed by international Good Laboratory Practices (GLP) Regulations.66,67 The GLPs provide legally mandated requirements for nonclinical study conduct, such as adequately trained personnel, adequate laboratory, and animal-holding facilities, demonstrated animal quality, regular equipment maintenance, and formal procedures to ensure data integrity.
An important role in a GLP-regulated nonclinical study is that of the study director who represents the single point of control and is legally responsible for all aspects of the study, from technical conduct to data interpretation and analysis. A key study director responsibility is making certain that every activity in the study is accurately and completely recorded, including unforeseen events or deviations from a written protocol that must be in place prior to the study start. An independent quality assurance auditor (and regulatory authorities) must be able to reconstruct an entire study and its conclusions from archived study records. While toxicologic pathologists may serve as study directors, they are more likely to serve as study pathologists, where they are responsible for the interpretation of clinical and anatomic pathology study data and for placing these data into context so that, in collaboration with the study director, conclusions about the safety of the test compound may be drawn.
Pathology data represent a critical component of the nonclinical safety package submitted to regulatory agencies.
68
Anatomic and clinical pathology data are typically reviewed for: diagnostic terminology; incidence, magnitude, and descriptive characteristics of findings; alignment of summarized and individual animal data; interpretation of the pathology findings relative to study design and their biological significance (eg, test compound-related, adverse, pharmacological); and validity of control data on the basis of concurrent controls and/or historical controls.
Regulatory review may even include the examination of specific microscopic slides or clinical pathology reports.68,69
If a follow-up review is requested by a regulatory authority, study pathologists may be asked to clarify the findings or to substantiate the interpretation of their significance. Reviewers may request additional experiments to better characterize the mechanisms of toxicity. Importantly, for marketed compounds or those being tested in clinical trials, any findings suggesting the potential for significant human risk must be communicated to regulatory agencies within 15 days. 70 This includes both new findings and new information about the previously known findings that indicate there may be additional risk to humans.
In addition to following regulatory requirements, toxicologic pathologists often shape these requirements through collaborations that advance both the science and the practice of the field. 69 Position papers are written to establish consistent practices, such as guidelines for routine pathology evaluation of the immune system, 71 the use of genetically modified mice for carcinogenicity studies, 33 generation of regulatory guidelines for establishing starting doses and dose escalations from nonclinical studies for clinical trials, 72 and the selection and interpretation of clinical pathology indicators of hepatic injury in preclinical studies. 73 Position papers and regulatory guidelines are important in establishing expectations for nonclinical drug development, as well as for generating, updating, and improving current procedures in industry.
Professional toxicologic pathology societies, as well as toxicologic pathology specialty groups within other pathology societies, play a key role in establishing and interpreting pathology-related regulatory guidelines for successful design and conduct of GLP studies. This work is often carried out in collaboration with other professional societies, such as the Society of Toxicology, the American College of Toxicology, and the Society of Quality Assurance.
Reduce, Refine, and Replace
Toxicologic pathologists are involved in additional aspects of drug development, which may appear to be peripheral to the process but which contribute importantly to all animal research. One of these aspects is the current global initiative to improve the care and use of experimental research animals. Not only is this initiative important for humane reasons, but it is also important from a financial standpoint, because the high cost of research animals contributes to the high cost of new drugs. Thus, the toxicologic pathologist plays a critical role in minimizing the use of animals, which not only enhances development efficiency while reducing development costs but also supports the philosophy of the “Three Rs” of animal use currently being promoted throughout the biopharmaceutical industry. The Three Rs include
Replacement of animals in scientific studies, for example, by in silico modeling and in vitro assays
Refinement of animal use by minimizing the potential for suffering and distress, and
Reduction of animal use, through improved study design, use of more specific animal models, and by adding investigative components to standard toxicity studies.
The toxicologic pathologist can contribute to the Three Rs in several phases of discovery. One significant area is target profiling, that is, defining the distribution of a receptor or ligand target, first through literature and database searches and then by evaluating target tissue distribution in normal or genetically modified animals by immunohistochemistry or mRNA characterization. 24 Target profiling also identifies potential targets of toxicity for which assays can be developed to screen compounds for toxicity before nonclinical safety assessment studies begin. This information can be used to predict potential consequences of altering pharmacologic activity of a target and to efficiently select lead compounds with minimized toxicities, refining animal use in nonclinical safety studies.
Another strategic area in discovery that has led to replacement and reduction of animal use is the application of systems pathology in which targets are validated and biomarkers identified based on differential gene and protein expression profiles in disease states. 74 A comparative approach can be used to determine which animal model best emulates human disease conditions. Firsthand knowledge of basic disease mechanisms makes the toxicologic pathologist an invaluable team member in the use of systems pathology, and increased use of gene profiling has led to more predictive animal modeling, reducing overall animal use. 30
After targets have been identified and expected gene profiles are characterized, the pathologist can play a further important role in refining animal use in the conduct and interpretation of small-scale investigative studies that are used to evaluate potential efficacy in disease models, providing early proof of principle. Early insights into potential safety issues identified in investigative studies can then be used to develop end points in definitive nonclinical studies; provide cross-species data to further refine animal model selection; and better correlate pharmacology and toxicology data, resulting in better lead compound selection decisions.25,75 As a consequence of these pilot studies, more informed decisions can be made regarding lead selection with a better awareness of dose selection and potential safety issues for definitive development studies, further refining animal use.
During the development phase, toxicologic pathologists directly contribute to refinement and reduction of animal use by reviewing study designs and protocols to ensure that high-quality and appropriate animal models are used for all studies, that proposed study protocol and procedures reflect current standards of pathology practice, that data collection is maximized from each animal, and that study objectives are achievable while using fewer animals. Further, because data from animals found moribund or dead can be difficult to interpret, pathologists contribute to refining end point criteria for studies, ensuring data integrity and minimizing animal distress.
Finally, the ability of the highly skilled toxicologic pathologist to correlate, integrate, and interpret study findings, often requiring discrimination between normal variation or spontaneous changes and compound-related changes, ultimately results in the reduction of animal use. Decisions can be made early in the development process to continue or terminate further studies with a lead compound based on the identified safety profile. Earlier decisions improve drug development efficiency, reduce development costs, and ultimately lead to more considered and selective use of animals in toxicologic studies.
Globalization and Transition to the Future
For most of the last century, Europe and North America have been the primary source of biopharmaceutical R&D and have also represented the major markets for these products. The role of toxicologic pathology in drug development and the qualification expectations for toxicologic pathologists are well established on these continents.
In the last decade, the shift of biopharmaceutical R&D to Asia is expected to have a profound effect on drug development and toxicologic pathology. Numerous CROs have already been established in China and India to meet R&D needs, and for conduct of studies to be submitted to North American and European markets, as well as in emerging markets. Regardless of the destination of the regulatory submissions, the need remains for qualified pathologists to evaluate tissues and clinical pathology data to test compound-related effects and to create study reports acceptable to diverse regulatory agencies. One of the biggest challenges of the current trend toward globalization of drug development will be harmonizing the practice of toxicologic pathology regulatory guidelines across the globe to incorporate different education programs and cultures.1,2,4,5,31,34–38 Initial steps have been taken toward harmonization of veterinary specialties in Asia, including a planning meeting held in December 2010 for an Asian Board of Veterinary Specialties that could ultimately include an Asian College for Pathology with residency programs and proficiency examinations. 76
In India, both veterinary and toxicologic pathology have been established as disciplines, but the number of certified pathologists is small. In China, neither veterinary nor toxicologic pathology has been well developed as a specialty in veterinary medicine, nor are there societies dedicated to the advancement of these disciplines. Thus, there is an unmet need for toxicologic pathologists in these countries. Some of this need is being met by a limited pool of returning expatriates trained in Europe or North America; but in the long term, the success of Asian pharmaceutical or contract research enterprises will be linked to the development of local expertise. This presents a great opportunity for toxicologic pathologists from North America and Europe to participate in training pathologists from these countries and to assist in developing professional societies to support the development of toxicologic pathology.
Globalization of biopharmaceutical companies has brought about a need for collaboration across geographically distant sites. Portions of a study may be performed by scientists at multiple facilities, a process that historically has entailed either shipping of irreplaceable study materials (ie, tissue sections on glass microscope slides) or travel by pathologists. Digital imaging systems allow ready access to high-resolution, whole-slide images that have been scanned remotely, even at facilities on different continents. Thus, the real-time review of tissue specimens and related data by pathologists in multiple locations is possible, facilitating timely assignment of diagnoses, expert opinion solicitation, and information-sharing opportunities, as well as the opportunity to work from home. The ability to evaluate clinical pathology data electronically also facilitates global collaboration.
Summary
The biopharmaceutical industry is an ideal setting for anyone drawn to challenge. The environment is fast paced, with tight timelines and ever-changing priorities. However, the ability to succeed as a toxicologic pathologist in this field is generously rewarded. In the short term, compensations are monetary. In the longer term, knowledge that this work can improve the quality of life for a patient with cancer, a child with a debilitating infectious disease, or a loved one with mental illness, and the satisfaction of active involvement in improving health globally provide intangible rewards that make the challenging job worthwhile.
Footnotes
Acknowledgments
The authors thank Eric Schultze (Eli Lilly), Mike Tomlinson (Nova Pathology), and Aaron Sargeant (Charles River Laboratories) for manuscript review; Mark Dominick (Bristol-Myers Squibb Company) for Figure 1, Robert Leedle (Covance) for Figure 3, Thomas P. Pienkowski (Covance) for immunohistochemistry (Figure 4), Steven R. Van Adestine (Covance) for photomicrography (Figures 3 and ![]() ), and Gerard Gagne (Abbott Laboratories) for final figure preparation.
), and Gerard Gagne (Abbott Laboratories) for final figure preparation.
The authors declared no conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors received no financial support for the research, authorship, and/or publication of this article.