
Abstract
Liver diseases are often associated with hyperglycemia, inflammation, and oxidative stress. These conditions, commonly associated with diabetes mellitus and obesity, facilitate the formation of advanced glycation end products (AGEs). These products are known to impair protein function and promote inflammation. Accumulation of AGEs such as N ε-(carboxymethyl)lysine (CML) is related to chronic liver diseases and their severity. Although several reports suggest a crucial role of AGEs in liver failure, there is little investigation on the direct effects of reducing sugars, precursors of AGEs, and on the onset and progression of liver failure. In this work, we investigate the effects of intravenously administrated glycolaldehyde (GA), a short-chain aldehyde, on oxidative parameters in the liver of Wistar rats. Animals received a single injection of GA (10, 50, or 100 mg/kg) and were sacrificed after 6, 12, or 24 hours. Levels of protein carbonyl, lipid peroxidation, and reduced thiol were quantified. The activities of catalase, superoxide dismutase, and glyoxalase I were also assessed. The amount of CML was quantified with specific antibody. There was an increase in oxidative stress markers in the liver of GA-treated rats. Glycolaldehyde induced a decrease in the activities of all enzymes assayed. Also, all tested doses led to an increase in CML content. Our data suggest that GA might play an important role in liver diseases through the impairment of antioxidant defenses and generation of AGEs.
Introduction
The liver plays a main role on metabolism. Regulation of glucose levels and synthesis of lipoproteins and fatty acids are among its most important functions. Impairment of liver function is known to be crucial in diabetes, cirrhosis, and steatohepatitis. Several models of liver disease suggest a crucial participation of oxidative stress in liver failure. 1,2 This role is corroborated in many reports by the beneficial effects of antioxidant therapy. Treatment with N-acetylcysteine reduces fibronectin deposition and attenuates oxidative damage in rats with dimethylnitrosamine-induced liver fibrosis. 3 Moreover, some plant extracts act as hepatoprotectors in different animal models, mainly by reducing oxidative damage. 4,5
Another common feature of liver diseases is the increase in advanced glycation end products (AGEs). Advanced glycation end products are formed through the reaction of reducing sugars and amino groups, forming a Schiff base that rearranges to more stable Amadori products and later lead to AGEs. 6 Liver transplantation in patients with cirrhosis lowers the levels of plasma N ε-(carboxymethyl)lysine (CML). 7 Plasma CML levels are also correlated with the severity of cirrhosis. 8 Advanced glycation end products exert its actions through the activation of the receptor for AGEs (RAGE), which promotes the migration of activated hepatic stellate cells (HSCs), the main extracellular matrix-producing cells in the liver. Recent report demonstrates that AGEs upregulate RAGE in quiescent and activated HSCs, leading to reactive oxygen species (ROS) production via the activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. 9
Glycolaldehyde (GA) is a by-product of nonenzymatic glycosylation 10 and of the myeloperoxidase in neutrophils. 11 It rapidly reacts with protein amino groups leading to the formation of AGEs such as CML and GA-pyridine. 12 We previously demonstrated that GA promotes protein carbonylation and impairs fibrinogen coagulation. 13 Although there is evidence demonstrating a pathophysiological role for GA, its physiological concentrations have not been determined yet. It has been estimated that GA concentration ranges from 0.1 to 1 mmol/L. 14,15
Despite a huge interest in elucidating the mechanisms that directly link AGEs and many diseases, and several evidences of the involvement of oxidative stress in these pathologies, little is known about AGEs precursors, such as GA, and their effect on oxidative stress and biochemical parameters. Thus, in this study we aimed to evaluate the acute effects of a single intravenous injection of GA on oxidative parameters in the liver of Wistar rats.
Materials and Methods
Animals and Chemicals
Adult male Wistar rats (280-320 g) were obtained from our own breeding colony. They were caged in groups of 5 with free access to water and food and were maintained on a 12-hour light–dark cycle (lights on at 7
Treatments
Animals were anesthetized (ketamin 100 mg/kg and xylazin 10 mg/kg) and treated with a single injection of GA via the dorsal vein of the penis, in different doses (10, 50, and 100 mg/kg) in a volume range of 120 to 150 µL (n = 7 for each dose). Control group received 130 µL of NaCl 0.9% (n = 7).
Oxidative stress and antioxidant enzymes analysis
Animals were sacrificed at 6, 12, or 24 hours after injection. Blood was collected and the plasma separated. The liver was dissected out in ice and immediately stored at −80°C for posterior analysis. Immediately before the analysis, the organs were homogenized in PBS 10 mmol/L with Potter-Elvehjem apparatus and centrifuged (1000g, 10 minutes at 4°C) to remove cellular debris. Supernatants were used to all biochemical assays described herein. Protein content was quantified using Lowry protein assay. 16 Samples for quantification of CML content were diluted in phosphate saline buffer containing 0.05% sodium azide, 0.5% Triton X-100, and a protease inhibitor cocktail (pH 7.4).
Measurement of Protein Carbonyl
The oxidative protein damage was measured by the quantification of carbonyl groups based on the reaction with 2,4-dinitrophenylhydrazine (DNPH). Proteins were precipitated by the addition of 20% trichloroacetic acid (TCA) and resuspended in 10 mmol/L DNPH and the absorbance read at 370 nm. 17 Results were expressed as nmol carbonyl/mg protein.
Measurement of Thiobarbituric Acid Reactive Species
As an index of lipid peroxidation, we detected thiobarbituric acid reactive species (TBARS) formation through a hot and acidic reaction. This is widely adopted as a method for measurement of lipid redox state, as previously described. 18 Briefly, the samples were mixed with 0.6 mL of 10% TCA and 0.5 mL of 0.67% thiobarbituric acid and then heated in a boiling water bath for 25 minutes. Thiobarbituric acid reactive species were determined by absorbance in a spectrophotometer at 532 nm. We have obtained TBARS concentration in the samples from a calibration curve that was performed using 1,1,3,3-tetramethoxypropane as standard, which was subjected to the same treatment as that applied to the supernatants of the samples. Results are expressed as nmol TBARS/mg protein.
Measurement of Total Reduced Thiol Content
To quantify the content of reduced thiol, samples were diluted in 10 mmol/L phosphate buffer (pH 7.4), followed by the addition of 0.01 mol/L 5,5′-dithionitrobis 2-nitrobenzoic acid (DTNB) in ethanol. The intense yellow color was developed and read at 412 nm after 20 minutes. A blank sample was run simultaneously, except for the absence of DTNB. Protein thiol content was calculated after subtraction of the blank absorbance, utilizing the molar extinction coefficient of 13 600/mol/L per cm. 19
Assay of Enzymes Activities
Catalase (CAT)
20
activity was measured as previously described.
21
The rate of decrease in absorbance at 240 nm was used as an index of H2O2 degradation by catalase. One unit of CAT was considered the amount of enzyme needed to degrade 1 µmol/min H2O2 at 25°C. Superoxide dismutase (SOD) activity was assessed by quantifying the inhibition of superoxide-dependent epinephrine autooxidation in a spectrophotometer at 480 nm.
22
To determine glyoxalase I (GLO) activity, we quantified the rate of formation of S-
Enzyme-Linked Immuno Sorbent Assay for CML
The wells of a microtiter plate were coated overnight with 0.1 µg protein in 0.1 mL 50 mmol/L sodium carbonate buffer (pH 9.6). Wells were washed three times with washing buffer (PBS containing 0.5% Tween 20) and then incubated with 0.5% gelatin for 3 hours to block nonspecific binding. The wells were washed again with washing buffer and incubated with 100 µL anti-CML (2G11) for 1 hour. After being washed three times, the wells were incubated with 100 µL of peroxidase-conjugated secondary antibody for 60 minutes. The reactivity of peroxidase was determined by incubation with o-phenylenediamine dihydrochloride (OPD) for 30 minutes. The reaction was stopped with addition of 50 μL 3 mol/L sulphuric acid. Absorbance was read at 492 nm. 24
Statistical Analysis
Results were expressed as mean ± standard error of the mean (SEM). Data were analyzed by 1-way analysis of variance (ANOVA) followed by Newman-Keuls' multiple comparisons test using software Prism 2.01 (GraphPad, San Diego, California). A P value <.05 was considered statistically significant.
Results
Redox Status
All doses of GA induced protein carbonylation. These modifications were persistent up to 24 hours (Figure 1 ). Damage to lipids was also assessed and GA promoted an increase in the levels of TBARS. Figure 2 shows the levels of lipid peroxidation in the liver of treated rats. Along with protein damage, reduced thiol content was also quantified. As presented in Figure 3 , animals treated with GA showed lower levels of reduced thiols. This was observed only 12 and 24 hours after injection.
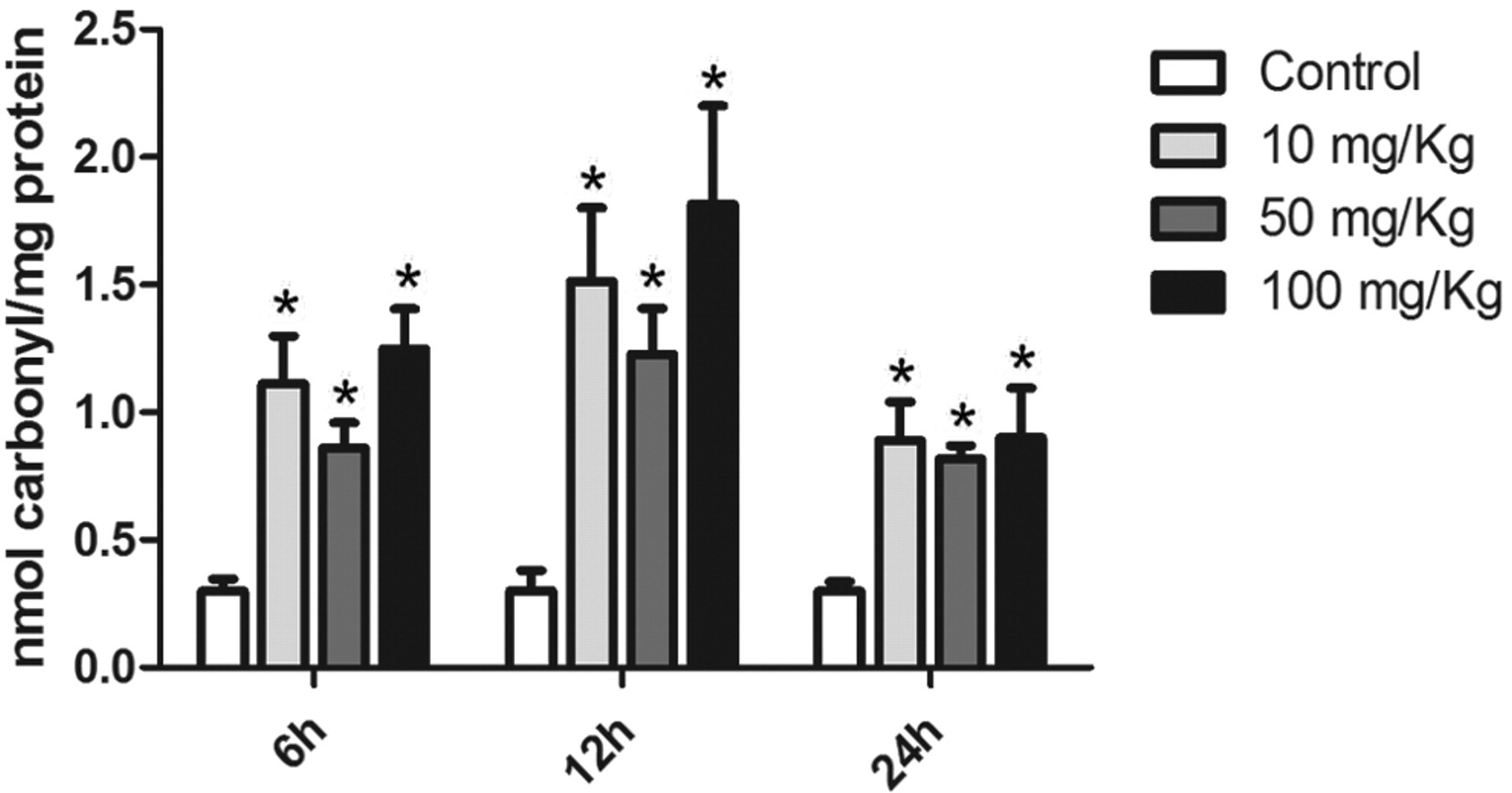
Circulating GA induces protein carbonylation. Glycolaldehyde was administered intravenously at the following concentrations: 0, 10, 50, or 100 mg/kg. Liver was surgically removed after 6, 12, or 24 hours. Data presented as mean ± SEM (n = 7). *Different from respective control, P < .05. GA indicates glycolaldehyde; SEM, standard error of the mean.
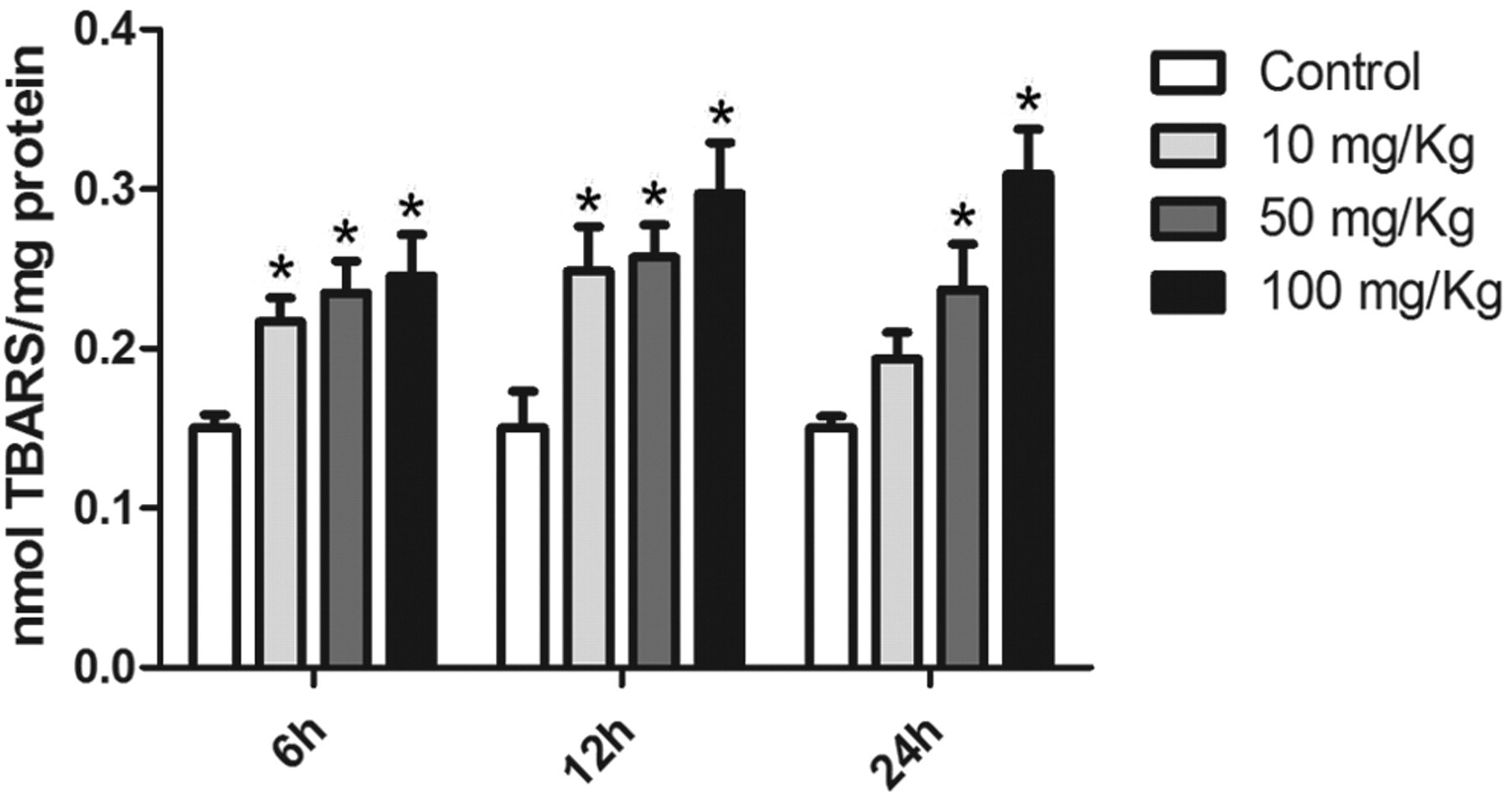
Lipid peroxidation increases after GA injection. The levels of thiobarbituric acid reactive species (TBARS) were assayed as an index of lipid peroxidation. At all doses, GA promoted lipid peroxidation, which was persistent up to 24 hours after injection. Data presented as mean ± SEM (n = 7). *Different from respective control, P < .05. GA indicates glycolaldehyde; SEM, standard error of mean.
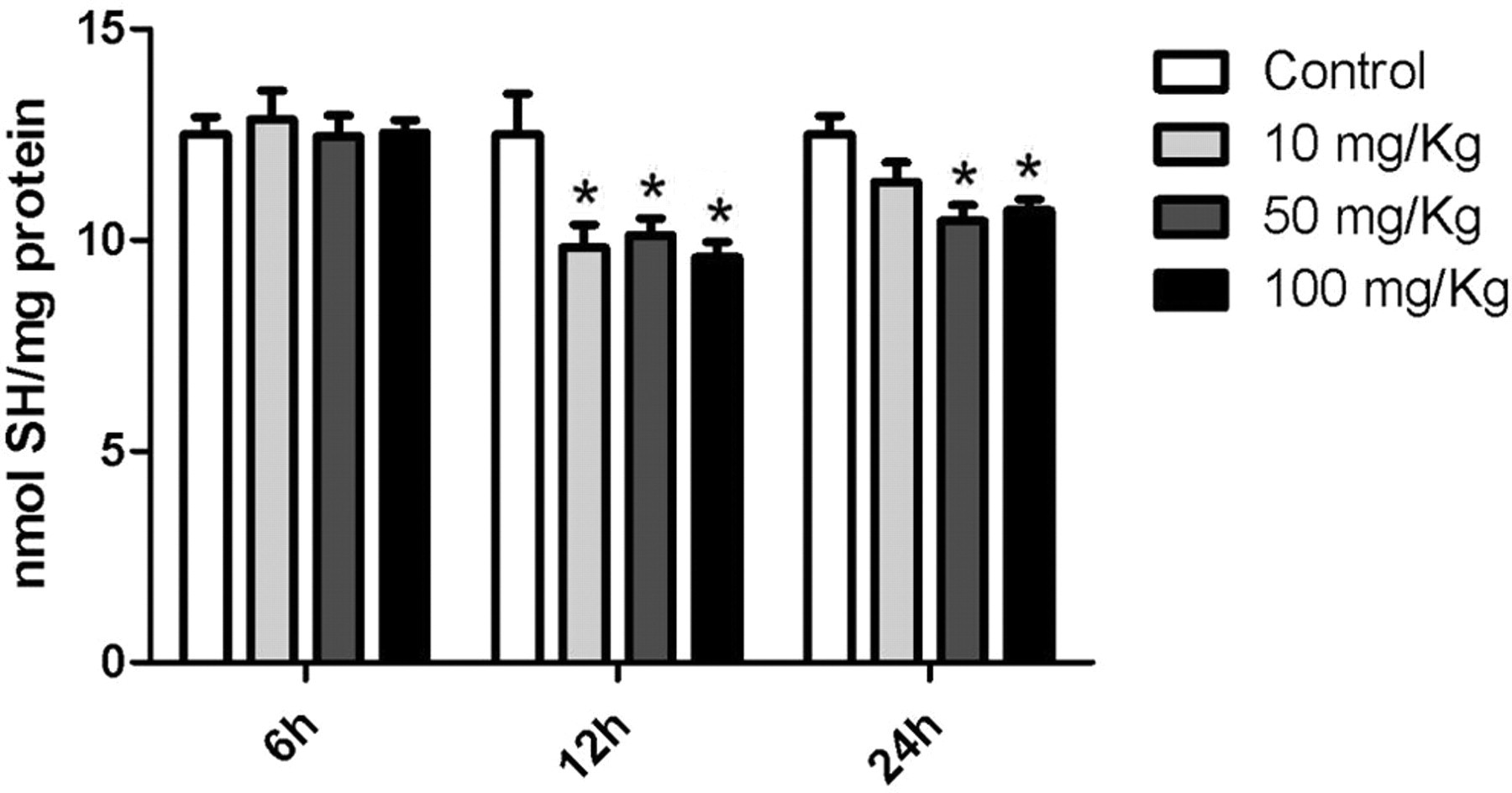
Glycolaldehyde promotes oxidation of thiols in the liver of Wistar rats. Animals received a single injection of GA. Samples were incubated with DTNB for 20 minutes. Absorbance was read at 412 nm. Data presented as mean ± SEM (n = 7). *Different from respective control, P < .05.GA indicates glycolaldehyde; DTNB, 5,5′-dithionitrobis 2-nitrobenzoic acid; SEM, standard error of the mean.
Assay of Enzymes Activities
All enzymes assayed were downregulated by the treatment with GA. Superoxide dismutase, which catalyzes the dismutation of radical superoxide anion radical into oxygen and hydrogen peroxide, had a decrease in its activity 6 and 12 hours after GA injection (Figure 4A ). Catalase, responsible for dealing with hydrogen peroxide, was also decreased during the same times (Figure 4B), although the lower dose had no effect at 6 hours. The activity of GLO was lower in comparison to the control group only 12 hours after GA injection (Figure 4C). Although all concentrations of GA could affect enzyme function, activities were restored 24 hours after injection.
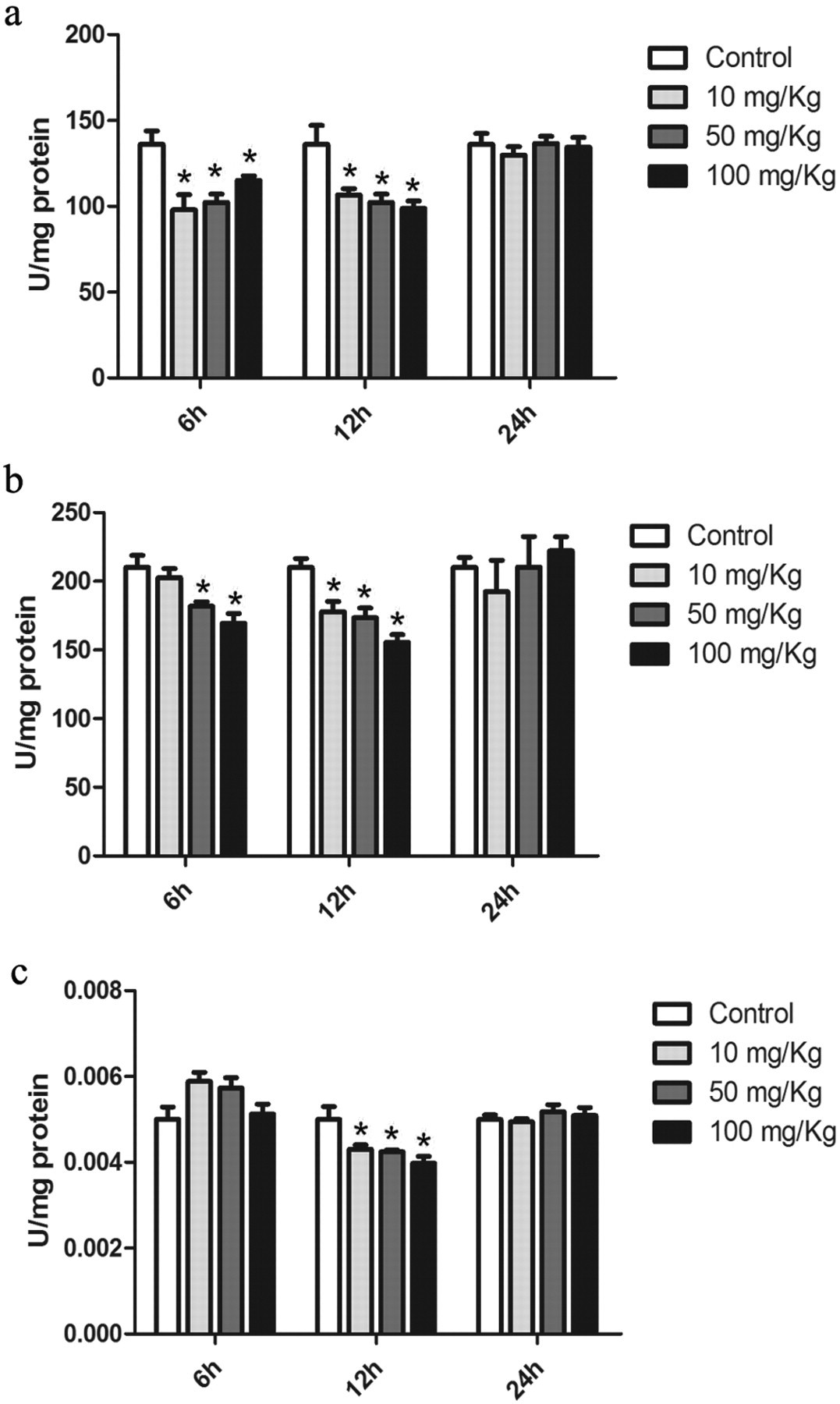
Glycolaldehyde decreases enzyme activities in the liver. Wistar rats were killed 6, 12, or 24 hours after GA injection. Superoxide dismutase (A) and catalase (B) had a decrease in activity 6 and 12 hours after injection. Glyoxalase I (C) was only altered 12 hours after GA injection. Data presented as mean ± SEM (n = 7). *Different from respective control, P < .05. GA indicates glycolaldehyde; SEM, standard error of the mean.
N ε-(carboxymethyl)lysine Content
The intravenous administration of GA promoted the formation of the ADE CML (Figure 5 ). All tested concentrations of GA induce the formation of CML. After 12 and 24 hours of injection, the amount of CML quantified in the liver was dose dependent.
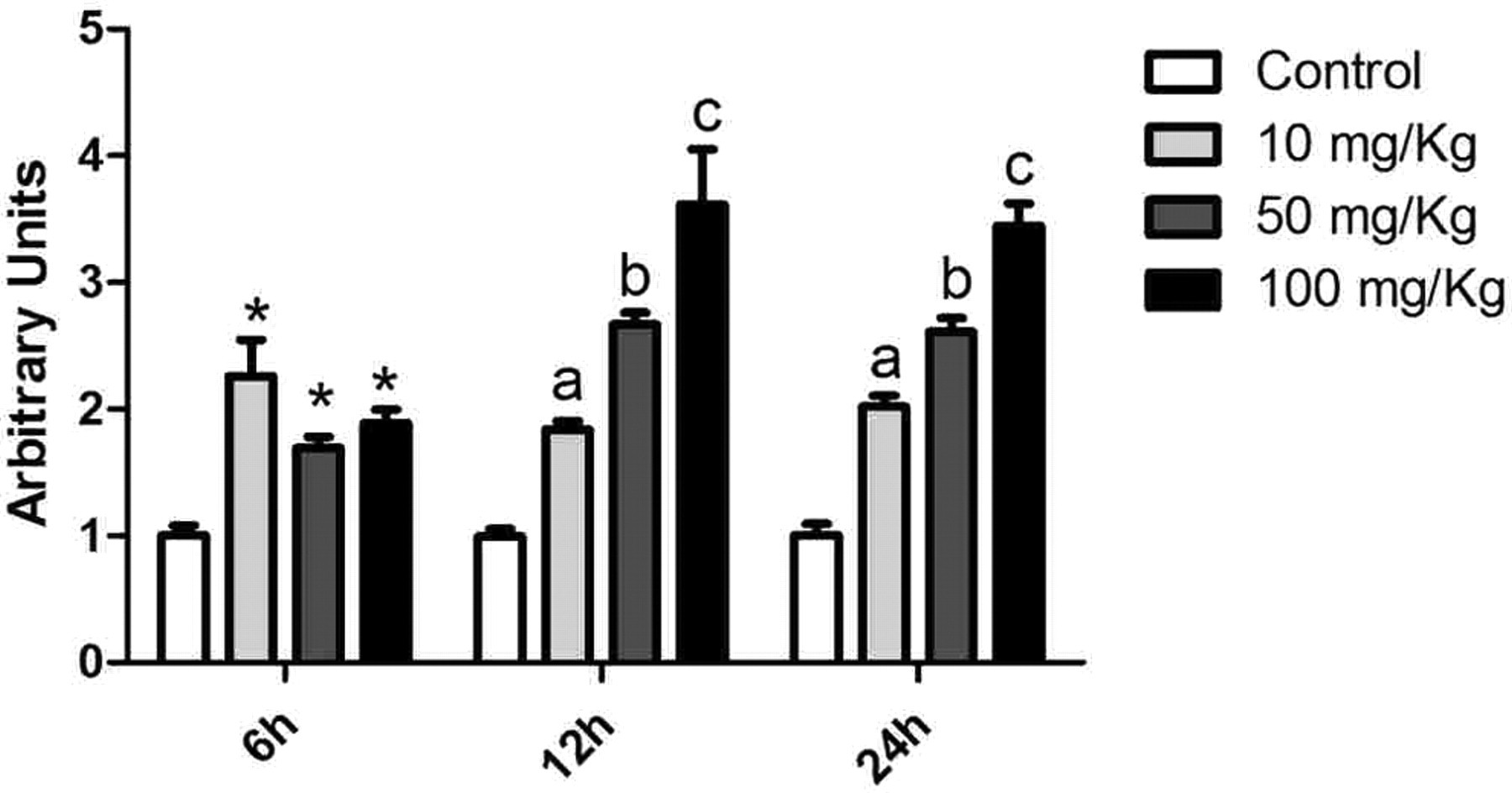
N ε-(carboxymethyl)lysine content in the liver. A specific antibody (2G11) against CML was incubated with 0.1 µg protein. Peroxidase-conjugated secondary antibody was added and the reactivity was determined by incubation with OPD. Data are presented as mean ± SEM (n = 7). At the 6-hour time point, each treatment resulted in significantly higher levels of CML than control (*P < .05). At the 12- and 24-hour time points, each treatment resulted in significantly higher levels of CML than control, and further, increasing doses resulted in significantly higher levels of CML (a = P < .05 compared to control; b = P < .05 compared to the 10 mg/kg treatment group, c = P < .05 compared to the 50 mg/kg treatment group). CML indicates N ε-(carboxymethyl)lysine; OPD, o-phenylenediamine dihydrochloride; SEM, standard error of the mean.
Discussion
Liver disease is often associated with fatty acid accumulation and cirrhosis. Such conditions are closely related to diabetes mellitus and metabolic syndrome. These conditions favor the formation of AGEs and the establishment of oxidative stress. In this work, we show that intravenously administrated GA induces oxidative damage to proteins and lipids and also decreases antioxidant enzymes activities. The doses of GA were chosen in order to obtain concentrations of circulating GA ranging from 1 to 10 mmol/L, according to the estimated blood volume of the animals. 25
Glycolaldehyde is a short-chain aldehyde that reacts mainly with lysine and arginine residues.
26
Glycolaldehyde also reacts with cysteine residues,
27
which might explain the lower levels of reduced thiols in the rats treated with GA (Figure 3). Moreover, GA modulated the activities of SOD, CAT, and GLO. The lower activity of SOD could raise the levels of anion superoxide and lead to direct inhibition of CAT.
28
Methylglyoxal, which reacts with arginine and lysine just as GA, inhibits liver SOD in vivo and in vitro.
29
This could explain the inhibition of SOD induced by GA administration. Furthermore, the observed inhibition of GLO might lead to increased levels of MG that can inhibit SOD. Glyoxalase I converts the hemithioacetal adduct between GSH and MG into S-
The observed raise in CML levels can partly explain the high levels of protein carbonylation, as CML has a carbonyl group in its structure. Nonetheless, it remains unclear the extent of protein carbonylation that is due to the formation of CML and to other oxidative processes. It seems, although, that the lower activities of antioxidant enzymes might have favored the observed redox imbalance. Previous reports from our group demonstrated that circulating GA induces oxidative damage in the kidney and heart but not CML accumulation. 35,36 This susceptibility of the liver might be due, partly, to higher iron (Fe2+) concentrations of the organ. Xiao and colleagues 37 reported that AGE formation is accelerated in the presence of Fe2+.
In summary, our results show that circulating GA induces an oxidative state in the liver, which might contribute to the development of chronic liver diseases such as steatosis and cirrhosis. Cumulative events of glycoxidation, which raise the levels of GA and other aldehydes, might also contribute to the onset of common liver dysfunction observed in these complications, as well as in diabetes. For the first time, it is shown that GA can promote accumulation of CML in the liver. With circulating levels ranging from 1 to 20 mmol/L, GA induced oxidative damage to proteins and lipids and modulated the activities of SOD, CAT, and GLO. The cumulative effects of such events, combined with genetic predisposition and other environmental conditions, might lead to the progression of liver dysfunction and culminate in liver disease. Further work is necessary to elucidate the molecular mechanisms of short-chain aldehydes in liver pathologies.
Footnotes
Acknowledgments
Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and Rede Instituto Brasileiro de Neurociência (IBN-Net) 01.06.0842-00.
The author(s) declared no conflicts of interest with respect to the authorship and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research and/or authorship of this article: This work was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and Rede Instituto Brasileiro de Neurociência (IBN-Net) # 01.06.0842-00.