
Abstract
The ubiquitin-proteasome system plays an essential role in a broad range of cellular signaling pathways. Ubiquitination is a posttranslational protein modification that involves the action of an enzymatic cascade (E1, E2, and E3 enzymes) for the covalent attachment of ubiquitin to target proteins. The emerging knowledge of the molecular mechanisms and correlation of deregulation of the ubiquitin system in human diseases is uncovering new opportunities for therapeutics development. The E3 ligase RNF8 acts in cooperation with the heterodimeric E2 enzyme Ubc13/Uev1a to generate ubiquitin conjugates at the sides of DNA double-strand breaks, and recent findings suggest RNF8 as a potential therapeutic target for the treatment of breast cancer. Here, we present a novel high-throughput screening (HTS)–compatible assay based on the AlphaScreen technology to identify inhibitors of the RNF8-Ubc13 protein–protein interaction, along with a follow-up strategy for subsequent validation. We have adapted the AlphaScreen assay to a 384-well format and demonstrate its reliability, reproducibility, and suitability for automated HTS campaigns. In addition, we have established a biochemical orthogonal homogeneous time-resolved fluorescence (HTRF) assay in HTS format and a cellular microscopy-based assay allowing verification of the primary hits. This strategy will be useful for drug screening programs aimed at RNF8-Ubc13 modulation.
Introduction
Ubiquitination is a highly hierarchical enzymatic cascade including the E1-activating enzyme, E2-conjugating enzymes, and E3 ligases, while removal of ubiquitination is done by deubiquitinating enzymes (DUBs). The proteasome is necessary for degradation of cellular proteins and to recycle the ubiquitin pool. The ubiquitin-proteasome system (UPS) is associated with many pathological disorders, including cancer and neurodegenerative disorders. 1 Therefore, developing assays to identify small molecules that target specific components of the UPS is of major clinical interest. The E3 ubiquitin ligase ring finger protein RNF8 is essential in the cellular response to DNA double-strand breaks (DSBs). This process is a highly regulated signaling cascade that is controlled via the recruitment of DSB repair factors to the break, alongside with different posttranslational modifications. In a first step, the ATM kinase phosphorylates components of the early DNA repair machinery. This phosphorylation cascade then leads to the recruitment and activation of RNF8.2–4 The RING domain of RNF8 interacts with the ubiquitin-conjugating enzyme heterodimer Ubc13/Uev1a, which in turn ubiquitinates H1-type linker histones and thereby recruits RNF168.5,6 RNF168 ubiquitinates H2A-type histones to permit the recruitment of both the mediator protein 53BP1 and the repair factor BRCA1.7–10 Importantly, the ubiquitin signal is attenuated by the Ubc13 inhibitory factor, ovarian tumor domain protease (OTU) ubiquitin binding 1 (OTUB1).11,12 Here, the crystal structure of the RNF8-Ubc13 interaction 13 and the molecular basis of the RNF8-Ubc13 protein–protein interaction (PPI) inhibition by OTUB1 give good evidence that it is indeed possible to target the RNF8-Ubc13 interaction.
Lately, increasing evidence appeared that RNF8 is involved in the resistance to PARP1 inhibitors, a therapeutic treatment for breast and ovarian cancer. 14 Moreover, it could be demonstrated that RNF8 is overexpressed in highly metastatic breast cancer cell lines and positively correlates with lymph node metastases and poor survival time.14,15 These findings support the hypothesis of RNF8 being a potential clinical target. For this purpose, we have established a high-throughput screening (HTS)–compatible assay in this work to identify small-molecule inhibitors for pharmacological inhibition of the RNF8-Ubc13 PPI. Furthermore, we describe a strategy for the validation and profiling of the primary hit compounds in biochemical and cell-based secondary and counterscreening assays.
Materials and Methods
Plasmids
DNA fragments encoding human OTUB1, OTUB1 (100-end), Ubc13, and RNF8 (RING) (1–402 bp) were amplified from cDNA by PCR with specific primer pairs (
Cell Culture
Adherent U2OS cells (ATCC) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) growth medium containing 4.5 g/L
Protein Purification
Bacterial expression of GST fusions of RNF8 (RING), RNF8 (RING) I439D, Ubc13-Flag-His, Ubc13 A98D-Flag-His, OTUB1-Myc, and OTUB1 T134R-Myc were purified by affinity chromatography.
For some proteins (OTUB1 100-end, Ubc13-Flag-His, and Ubc13 A98D-Flag-His), the GST tag was cleaved off by thrombin (Novagen) and further purified via gel filtration. Expression and purification of MBP-RNF8 (RING) was done from pETM-40. In detail, all protein constructs were transformed into BL21-CodonPlus-(DE)-RIPL (Agilent, Santa Clara, CA) cells, grown in Luria-Bertani (LB) medium with antibiotics to OD600 = 0.6, and expression was induced with 1 mM IPTG (Fisher Scientific, Waltham, MA) O/N at 21 °C. Cells were harvested and lysed by sonication. GST-RNF8 proteins were lysed in buffer containing 50 mM HEPES pH 8.0, 150 mM NaCl, 2 mM DTT, 10% glycerol, 1 µM ZnCl2, and a cOmplete EDTA-free protease inhibitor cocktail tablet (Roche Applied Science, Penzberg, Germany). GST-OTUB1 and GST-Ubc13 were resuspended and lysed in 1× phosphate-buffered saline (PBS), 400 mM NaCl, 5 mM DTT, and a cOmplete EDTA-free protease inhibitor cocktail tablet. All GST-protein lysates were incubated with equilibrated gluthathione sepharose B (GE Healthcare, Chalfont St. Giles, UK) for 2 h at 4 °C. Proteins were eluted with 50 mM glutathione and desalted on a 5 mL desalting column (GE Healthcare) in lysis buffer. Similar approaches were taken for the other proteins. His-OTUB1 was lysed in 50 mM TRIS pH 7.5, 300 mM NaCl, and 20 mM imidazole and the cleared lysate incubated on a Ni2+ sepharose column (GE Healthcare) and eluted with 250 mM imidazole. MBP-RNF8 (RING) was lysed in 50 mM HEPES pH 8.0, 150 mM NaCl, 2 mM DTT, 10% glycerol, and 1µM ZnCl2. The cleared lysate was incubated on an amylose resin (New England Biolabs) for 2 h at 4 °C and eluted with 10 mM Maltose (Sigma-Aldrich, St. Louis, MO). For some proteins, the GST tag was cleaved off on the resin with 50 units of thrombin in PBS O/N at 21 °C, eluted with PBS, and subjected to gel filtration (Superdex 75, GE Healthcare).
Screening Instruments
The HTS platform consists of integrated instrumentation for plate and liquid handling. Experiments were performed using a Sciclone G3 Liquid Handler from PerkinElmer (Waltham, MA) with a Mitsubishi robotic arm (Mitsubishi Electric, Tokyo, Japan, RV-3S11) and a Flexdrop dispenser (PerkinElmer). 16 The Amplified Luminescent Proximity Homogeneous Assay Screen (AlphaScreen) and time-resolved fluorescence resonance energy transfer (TR-FRET) assays were performed in white 384-well Optiplates (PerkinElmer, product no. 6007299), and the signals were detected on the EnVision Multilabel Reader (PerkinElmer).
AlphaScreen and HTRF reagents
The AlphaScreen detection systems (PerkinElmer) used in this study consist of glutathion donor beads and nickel chelate acceptor beads (AlphaScreen Histidine [Nickel Chelate] Detection Kit, PerkinElmer, product no. 6760619C). The homogeneous time-resolved fluorescence (HTRF) antitag reagents toolbox consisting of Eu3+ cryptate-conjugated mouse monoclonal antibody maltose binding protein (Cisbio, Bedford, MA, product no. 61MBPKAA) and XL665-conjugated mouse monoclonal antibody anti-Flag was used (Cisbio, product no. 61FG2XLA).
Assay Development and HT Adaptation of AlphaScreen PPI Assay
The assays for all PPI pairs were developed so that protein concentrations for GST-RNF8 (RING) and GST-OTUB1-Myc were identical (30 nM). Also, the concentrations of Ubc13-Flag-His and GST-Ubc13-Flag-His were identical in all AlphaScreen PPI assays (20 nM). Proteins and beads were diluted in assay buffer containing 1× PBS (pH 7.4), 0.5% bovine serum albumin (BSA), and 0.1% Tween-20. The RNF8-Ubc13 AlphaScreen experiments were performed in white 384-well Optiplates as follows: (1) dispensation of 40 µL of RNF8-Ubc13 protein mix into white 384-well plates using a robotic liquid handler; (2) transfer of 0.6 µL of DMSO into each well using a compound transfer station with a nanoliter head yielding a final assay concentration of 1% v/v DMSO; (3) addition of 10 µL each of GSH donor and nickel chelate acceptor beads (18 µg/mL, 3 µg/mL final), followed by incubation for 1 h at room temperature in the dark; and (4) reading of the assay plates using laser excitation at 680 nm, with emission detected at 520–620 nm in an EnVision 2102 Multilabel Reader (PerkinElmer). The quality and robustness of the assay was calculated using Z′ factor and signal window (SW).
TR-FRET Counterassay Development
All components in the assay (with final concentrations of 20 nM MBP-RNF8 [RING], 64 nM Ubc13-Flag-His, or 64 nM Ubc13 A98D-Flag-His, 1.3 nM α-MBPEu+, and 13 nM α-Flag-XL665) were diluted in assay buffer containing 1× PBS (pH 7.4), 0.5% BSA, and 0.3% Tween-20. A total of 40 µL/well of the final mixture was transferred into a 384-well Optiplate. The assay plates were incubated at room temperature for 1 h. Fluorescence at 615 and 665 nm was measured simultaneously using the EnVision 2102 Multilabel Reader.
siRNA Treatment, Cell Lysates, and Western Blot
A total of 1.5× 105 U2OS cells/well were seeded in six-well plates. The day after, siRNAs were transfected using Lipofectamine RNAiMAX (Life Technologies) according to the manufacturer’s protocol. Per well, 250 µL of OptiMEM (Life Technologies) + 7.5 µL of RNAiMAX and 250 µL of OptiMEM + 3 µL of siRNA (10 µM) were used. The following siRNA sequences were used (Eurogentec, Liège, Belgium): siOTUB1 CCGACUACCUUGUGGUCUA, siRNF8 GGA GAUAGCCCAAGGAGAA, and siUbc13 UUCUGGAA GGAA UAGUUCAAGUUUA.
Forty-eight hours later, medium was changed. Twenty-four hours later, adherent cells were washed once with cold 1× PBS and lysed using lysis buffer (20 mM HEPES, 350 mM NaCl, 1 mM MgCl2, 0.5 mM EDTA, 0.1 mM EGTA, 20% glycerol, and 1% NP-40) containing protease (Complete; Roche, Mannheim, Germany) and phosphatase inhibitor cocktails (PhosStop; Roche). The whole cell lysates were subjected to 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene fluoride (PVDF) membrane (Immobilon-P; Millipore, Billerica, MA). Three percent BSA in 0.05% PBS-T was used for blocking. The membranes were incubated with primary antibody (α-OTUB1 polyclonal rabbit [1:1000; A302-917A; Bethyl Laboratories, Montgomery, TX], α-RNF8 [B-2] monoclonal mouse [1:1000; sc-271462; Santa Cruz Bio-technology, Santa Cruz, CA], α-Ubc13 monoclonal mouse [1:1000; 37-1100; Invitrogen, Carlsbad, CA], or α-actin [I-19] goat [1:1000; sc-1616; Santa Cruz Biotechnology]). After incubation with the corresponding secondary antibody, signals were visualized with the enhanced chemiluminescence system (Amersham ECL Plus Western blotting detection system; GE Healthcare).
Foci Formation
A total of 10,000 U2OS cells/well were seeded in poly-
Results and Discussion
In order to develop a biochemical in vitro assay that could reflect the binding of RNF8 to Ubc13, we expressed and purified RNF8 and Ubc13 and several known nonbinding variants, that is, GST-RNF8 (RING), GST-RNF8 (RING) I439D, Ubc13-Flag-His, and Ubc13 A98D-Flag-His. Highly pure proteins were generated as assessed by SDS-PAGE, followed by Coomassie staining ( Fig. 1A ). It is proposed that only the C-terminal RING domain of RNF8 directly interacts with Ubc13, and that single point mutations in RNF8 (I439D) or Ubc13 (A98D) would disrupt this PPI. 13 To establish a suitable in vitro HTS system for primary screening, the AlphaScreen system was used. Therefore, glutathione (GST) donor beads binding GST-RNF8 (RING) and nickel chelate (Ni-NTA) acceptor beads targeting Ubc13-Flag-His were applied to the reaction mix. With this strategy, we were able to set up a highly robust and specific AlphaScreen assay for the RNF8-Ubc13 PPI ( Fig. 1B ). Mutations in the predicted binding site at either side of the PPI pair (RNF8 [RING] I439D or Ubc13 A98D) drastically impaired the AlphaScreen signal ( Fig. 1B ). As chemical small-molecule libraries are typically stored in DMSO, we tested the DMSO tolerance of this assay. The assay should allow a DMSO concentration up to 1%, as this is an essential criterion for future screening campaigns. We could demonstrate that DMSO concentrations up to 5% had hardly any impact on the AlphaScreen signal intensity of the RNF8-Ubc13 PPI pair ( Fig. 1C ). In the next step, we tested the adaptability and reproducibility of the assay for high throughput. To this end, all dispense steps and the DMSO transfer (1% final concentration) were performed by automation in a 384-well format (see Materials and Methods) to mimic screening conditions. By comparison of plate-to-plate (n = 3) and day-to-day (n = 2) experiments, we can reveal that our assay is reliable and reproducible across plates and screen days ( Fig. 1D ). The assay quality was determined by calculation of the Z′ factor and SW for all assay plates and always exceeded 0.9 and 20, respectively ( Fig. 1E ). Thus, the assay fulfills relevant screening criterions and is therefore suitable for small-molecule HTS campaigns. To finally confirm that a known Ubc13 binder (OTUB1) can compete for RNF8-Ubc13 interaction in the AlphaScreen system, we purified an untagged OTUB1 100-end variant. As expected, untagged OTUB1 100-end blocked the binding of GST-RNF8 (RING) to its Ubc13-Flag-His binding partner. Importantly, OTUB1 100-end was able to reduce the AlphaScreen signal in a dose-dependent manner ( Fig. 1F ). Together with the fact that a single point mutation is sufficient to abrogate the RNF8-Ubc13 interaction, these data support the notion that a small molecule could indeed block the RNF8-Ubc13 binding.
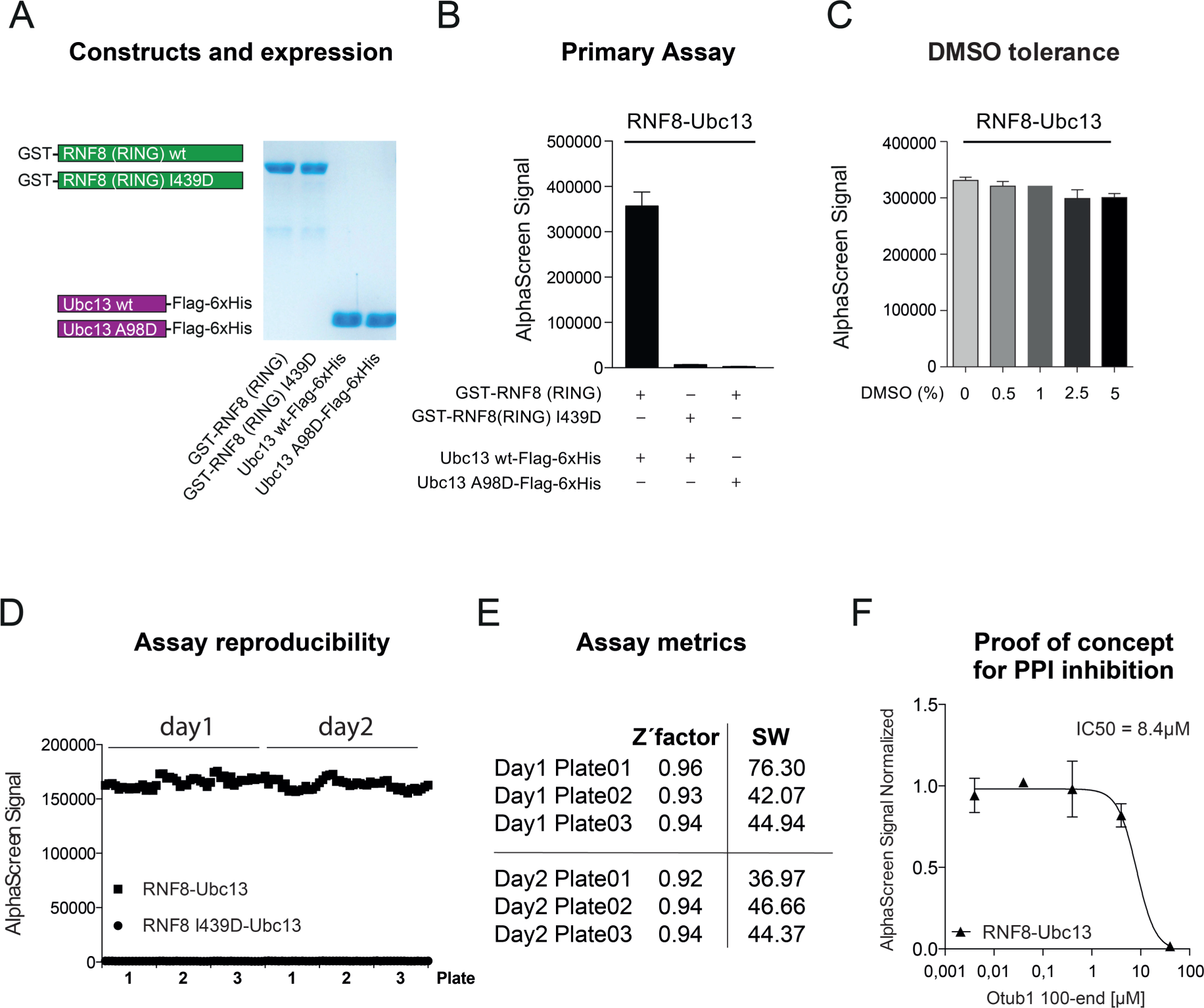
Primary screening assay. (
Once a number of hits have been obtained from a primary HTS campaign, a selection process is necessary to identify those compounds, which specifically act on the target. False-positive hits interfering with the screening technology (this involves compounds possessing inherent redox potential or direct oxygen transfer) or showing promiscuous behaviors on different protein target classes should be excluded using chemoinformatics analysis (e.g., filter for frequent AlphaScreen hitters) and counterassays.17–19 Ideally, confirmation assays should be based on technologies different from the primary screening assay and/or need to include appropriate control proteins or reagents. Therefore, we established, in addition to an AlphaScreen, a TR-FRET assay for the RNF8-Ubc13 PPI pair. Instead of GST-RNF8 (RING), we used a MBP-tagged RNF8 (RING) version to increase variations on the tags for TR-FRET detection. Again, this protein was purified in high yields and purity (data not shown). In this TR-FRET assay, the fusion proteins were detected via α-MBP and α-Flag antibodies rather than by the GSH/Ni-NTA matrices. Also, for this assay we could demonstrate excellent assay performance (Z′ = 0.97; SW = 112.44) and highly robust signals ( Fig. 2A ). We carefully checked both TR-FRET channels, that is 665 and 615 nm, and subsequently calculated their ratio representing the TR-FRET signal. The RNF8-Ubc13 PPI produced a specific TR-FRET signal that is reduced when using the Ubc13 A98D mutant rather than Ubc13 wild type. Importantly, the TR-FRET signal is specifically dependent on the 665 nM channel, whereas 615 nm is an internal reference and stays stable independent of the PPI pair used ( Fig. 2A ). Also, for the TR-FRET assay we checked the DMSO tolerance of the assay system ( Fig. 2B ) and the ability of OTUB1 to interfere with the RNF8-Ubc13 interaction ( Fig. 2C ). Both tests clearly certify that the TR-FRET assay represents a valid and specific secondary confirmation assay with high DMSO resistance and sensitivity for PPI inhibition.
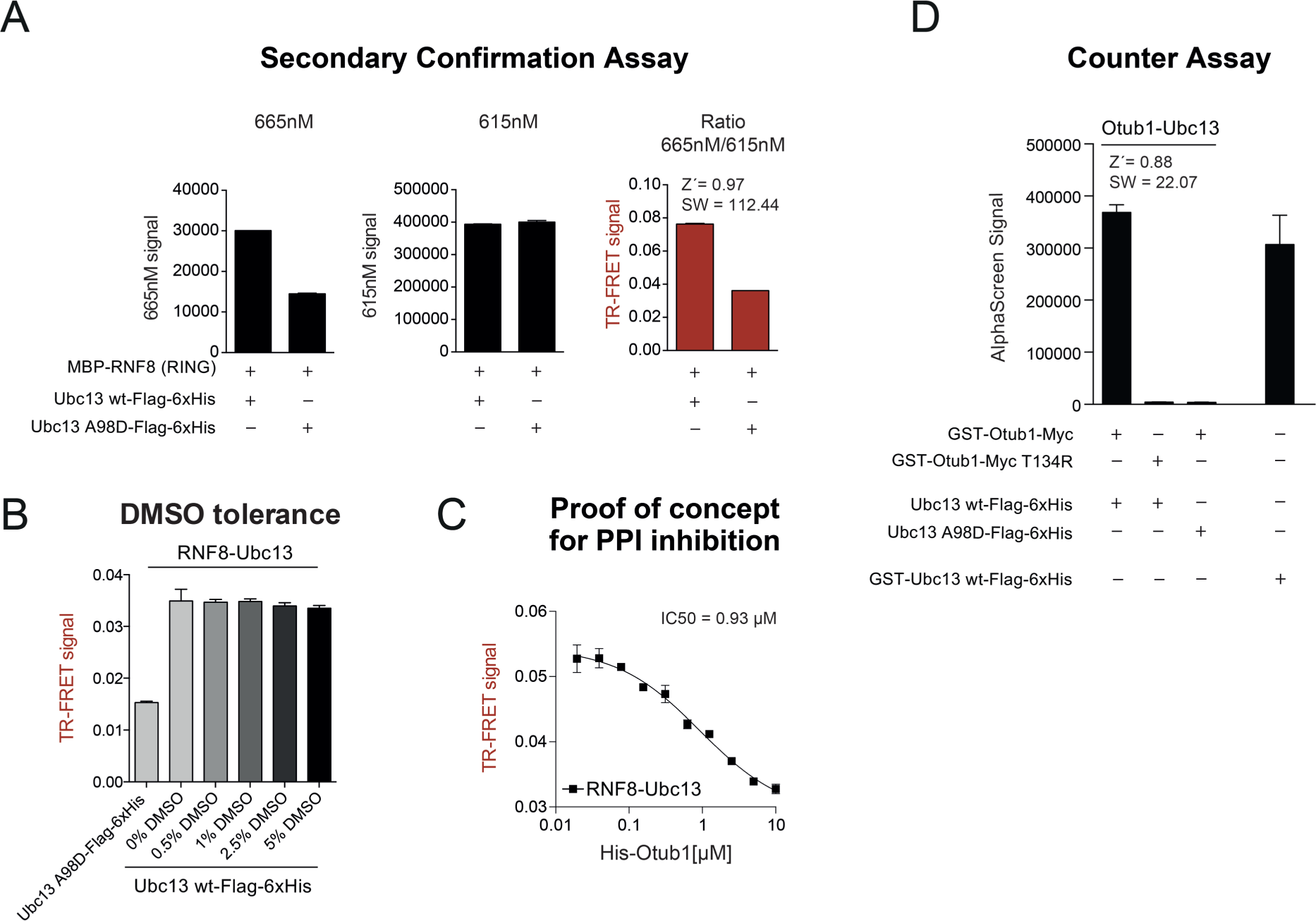
Secondary confirmation and counterassays for the identification of false-positive hits. (
Moreover, we established counterassays based on the AlphaScreen technology similar to the primary assay. Distinct AlphaScreen assays with different target proteins but identical protein tags and concentrations allow the identification of molecules that (1) interfere with the AlphaScreen technology, (2) bind to protein tags (GST/His), or (3) bind to the corresponding affinity matrices (GSH/Ni-NTA).17,18 We established an OTUB1-Ubc13 PPI assay to uncover unspecific Ubc13 binders. As already shown before, OTUB1 competes for binding of RNF8 to Ubc13 in the AlphaScreen system ( Fig. 1E ). Therefore, we purified GST-OTUB1 and a GST-OTUB1 T134R variant, which cannot bind to Ubc13. Also, for these proteins we could yield high purity (data not shown). We were able to establish an assay protocol for OTUB1-Ubc13 (Z = 0.88; SW = 22.07) with identical protein concentrations as for the RNF8-Ubc13 AlphaScreen PPI combination ( Fig. 2D ). As a second counterassay, the linear fusion of GST-Ubc13-Flag-His was introduced to have an AlphaScreen assay not relying on a PPI ( Fig. 2D ). An ideal hit candidate should result in reduction of the AlphaScreen signal only for the RNF8-Ubc13 PPI pair, but not for the OTUB1-Ubc13 pair or the Ubc13 linear fusion protein.
In the next step of the validation chain, small molecules that were selected from a biochemical screening campaign should be analyzed in cell-based systems to ensure their activity in a cellular environment. This secondary phenotypic confirmation assay will give an indication about the capacity at which a compound enters the cell and results in phenotypes that were also described by genetic modification (e.g., RNAi). For this purpose, we set up a high-content-analysis protocol to analyze known cellular DSB repair readouts. We used irradiation to induce DSBs, which led to massive recruitment of different DNA repair factors, as well as various posttranslational modifications at the site of the break, such as ubiquitination. U2OS cells were seeded in a 96-well format and treated with siRNAs targeting RNF8, Ubc13, and OTUB1 and subsequently irradiated with 2 Gy or left untreated. Afterward, cells were stained for ubiquitin (FK2) and imaged in the Operetta HCS system. Next, we implemented an automated analysis protocol, counting the cell number and the FK2 foci as a well-established indicator for the DNA damage response (DDR). We classified positive cells having more than five FK2 foci per nucleus. As previously described, siRNF8, as well as siUbc13, treatment reduced ubiquitin foci formation ( Fig. 3A , B ), whereas siOTUB1 showed opposing effects. Also, for this cell-based assay we were able to calculate a Z′ factor that was above 0.5, indicating an excellent assay. 20 Moreover, for all siRNAs we could demonstrate efficient downregulation of protein levels as shown in Western blot studies for RNF8, OTUB1, and Ubc13 ( Fig. 3C ). Treatment of cells with compounds gained from primary screen campaigns should result in reducing FK2 foci formation comparable to the siRNF8 treatment presented in this study.
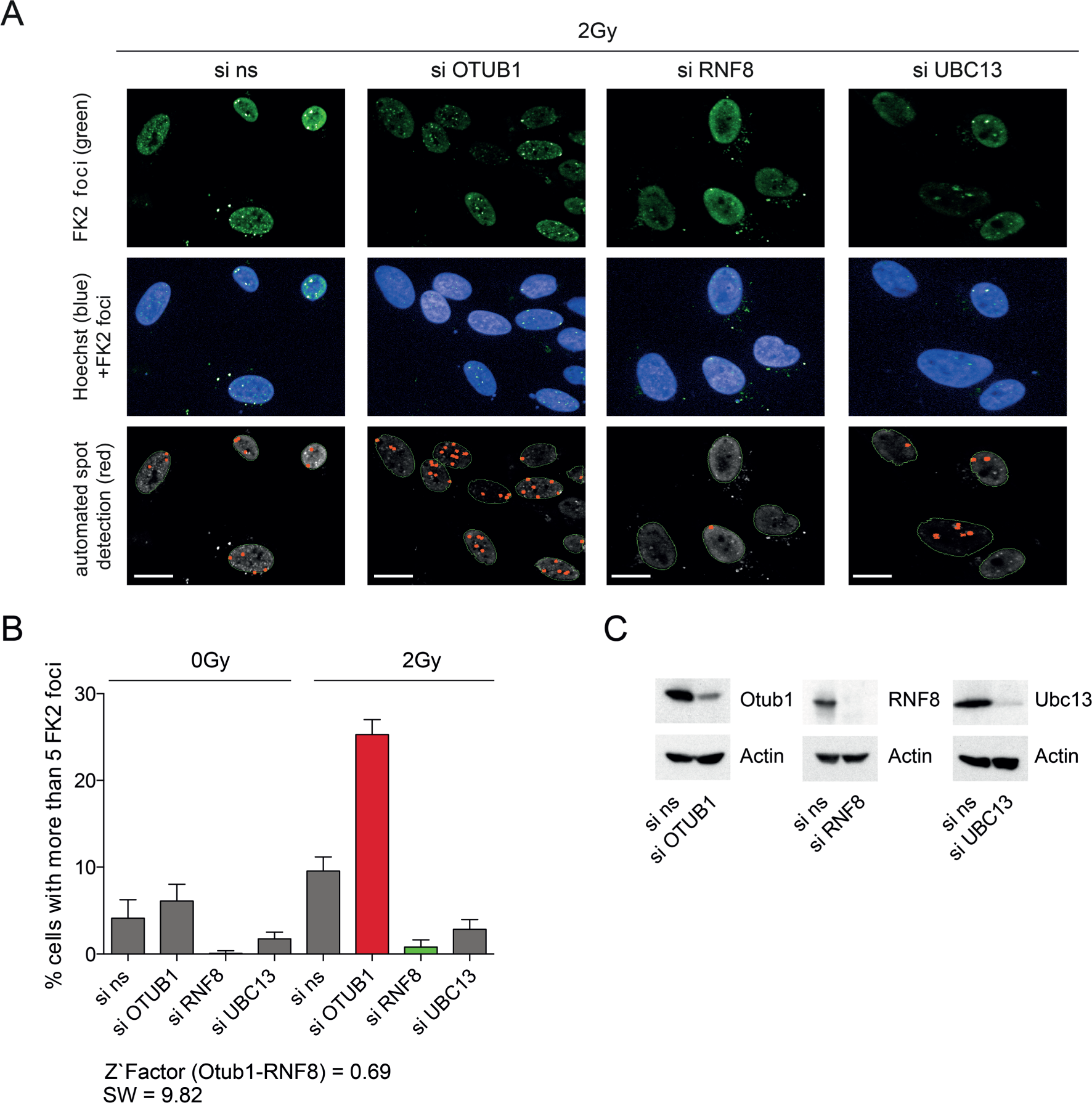
Cell-based phenotypic assay for further hit selection. (
In conclusion, we have developed and validated a sensitive and robust primary assay that can be used as a screening tool to identify RNF8-Ubc13 inhibitors. Additionally, we provide confirmation and counterassays, as well as a cell-based secondary assay, to be able to triage future primary hits from screening campaigns toward specific and selective RNF8 inhibitors. This multilevel strategy from hit identification until thorough hit validation is depicted in Figure 4 . Of note, the strategy could be easily reconfigured, using the TR-FRET assay as a primary assay and the AlphaScreen technology for the validation process as both cell-free assay types are suitable for fully automated HTS platforms. Finally, this multilevel strategy for the identification of RNF8-Ubc13 PPI inhibitors comprising primary as well as various secondary counter- and confirmation assays can be used as a basis to develop disrupters of other PPI target proteins.
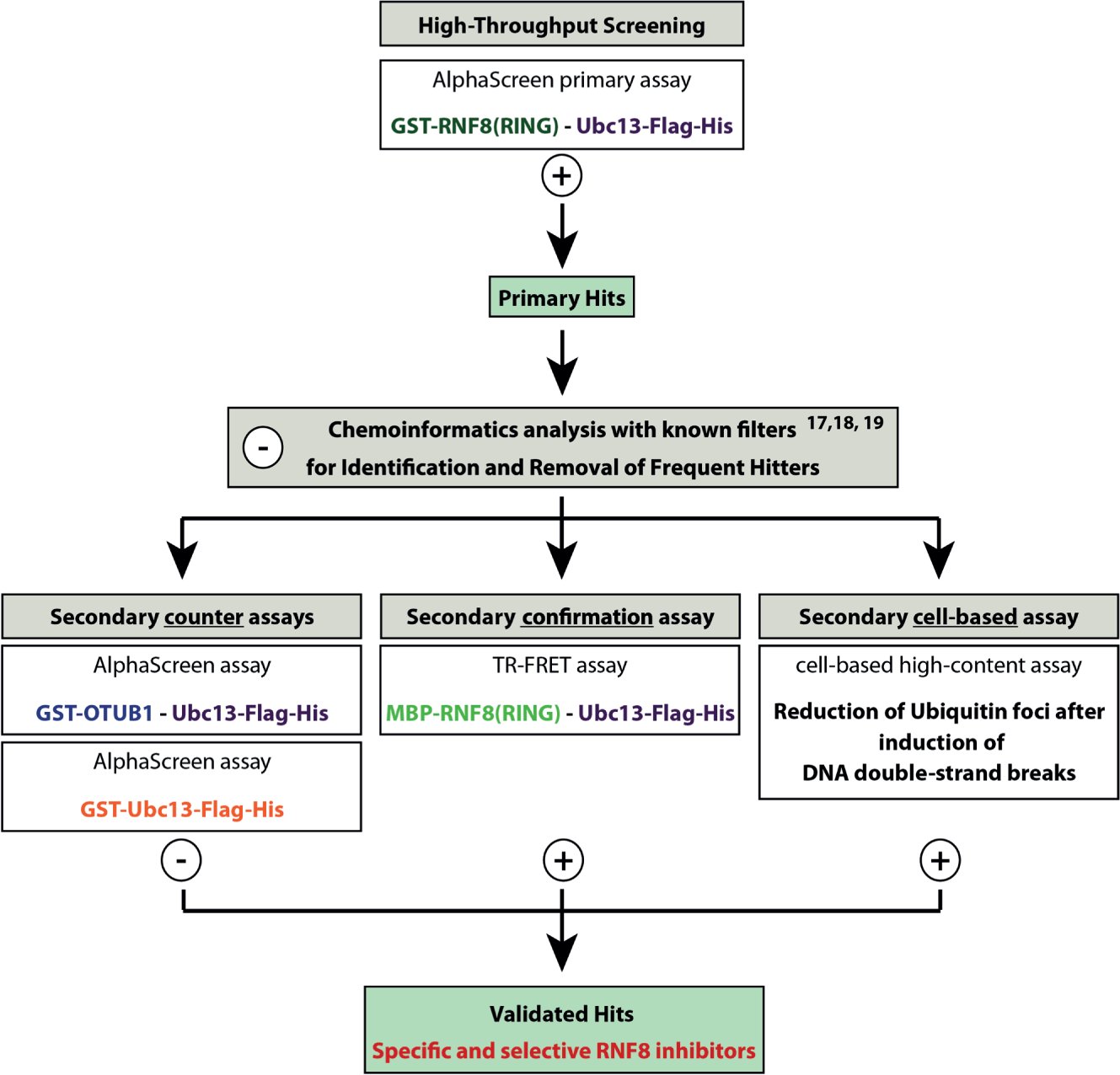
Overall RNF8 screening strategy. The screening campaign starts with a primary AlphaScreen assay for the RNF8-Ubc13 PPI. All small molecules that lead to a decrease in the AlphaScreen signal are classified as primary hits. With appropriate chemoinformatics analysis, secondary (counter, confirmation, and cell-based) assays false positives can be identified, and only those small molecules that show favored effects in all assay formats are designated as validated hits.
Footnotes
Supplementary material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.