
Abstract
The transfer of the small protein ubiquitin to a target protein is an intricately orchestrated process called ubiquitination that results in modulation of protein function or stability. Proper regulation of ubiquitination is essential, and dysregulation of this process is implicated in several human diseases. An example of a ubiquitination cascade that is a central signaling node in important disease-associated pathways is that of CBLB [a human homolog of a viral oncogene
Introduction
Protein homeostasis is a tightly regulated and essential component of cellular viability. In mammalian cells, this process is partially regulated by posttranslational modification of proteins by the addition of the small protein ubiquitin. The successful transfer of ubiquitin to a target protein is part of a complex poly-enzymatic process referred to as ubiquitination. Successful completion of this modification requires the presence of a ubiquitin-activating enzyme, commonly called an E1; a ubiquitin-conjugating enzyme (E2); a ubiquitin ligase (E3); the target protein; ubiquitin; and the cofactors adenosine triphosphate (ATP) and magnesium. While ubiquitination requires the presence of all components, it is the E3 that confers target specificity toward the final acceptor of ubiquitin.
1
This specificity makes the E3 an attractive therapeutic target. The discovery of an E3-specific inhibitor is well suited for creatively designed and robustly implemented high-throughput screening (HTS).
2
To this end, we developed, optimized, and implemented a HTS platform for the discovery of modulators of E3 activity. As a proof of principle, we have used this assay to discover inhibitors of the
The mammalian CBL family (CBL, CBLB, and CBLC) of enzymes plays an integral role in the regulation of receptor tyrosine kinase (RTK) signaling by coordinating the ubiquitination of activated RTKs, ultimately resulting in their internalization and lysosomal degradation. 4 Modulation of epidermal growth factor receptor (EGFR) signaling through CBL-mediated ubiquitination has been widely studied and well characterized.5,6 As members of the Really Interesting New Gene (RING) finger ubiquitin ligase superfamily, CBLs all contain the catalytic RING finger (RF) domain, a tyrosine kinase binding (TKB) domain, a proline-rich SH3-interacting domain, and, in CBL and CBLB, a ubiquitin-associated (UBA) domain. 4 Uniquely among the CBL family, CBLB negatively regulates adaptive (T cell) and innate (NK cell) antitumor immunity. In T cells, CBLB inhibits the costimulatory pathway by ubiquitinating the p85 subunit of PI3 kinase (PI3K) and preventing the recruitment of PI3K to the CD28 costimulatory receptor. 7 In NK cells, CBLB inhibits NK cell activity downstream of the inhibitory Tyro/Axl/MerTK (TAM) receptor activation.7,8 Genetic ablation of CBLB in mice results in increased CD8 T cell– and NK cell–mediated killing of transplanted and spontaneous tumors.7,9,10 Importantly, reconstitution experiments of either wild-type or RF mutant CBLB into CBLB knockout mice demonstrated that the loss of CBLB catalytic E3 activity is essential to tumor suppression in CBLB-null mice.8,11
Although we demonstrate that the platform we have developed can be applied broadly to other E3s, the potential translational applications led us to choose CBLB as our first target of interest for a 384-well formatted HTS for inhibitors of its ubiquitin ligase activity. From a screen of >70,000 small molecules, including natural products, we discovered a variety of pharmacophores, with a range of potencies, capable of inhibiting CBLB activity in in vitro biochemical assays. One structural class, natural product–derived alkaloids named methyl ellipticiniums (MEs), inhibited CBLB at low-micromolar concentrations. Herein, we describe the first CBLB-specific HTS and identify the chemical structure of an intracellularly active small-molecule inhibitor of CBLB. MEs represent a new pharmacophore for ubiquitin ligase inhibition with the potential to improve our understanding of the function of CBLB and therapeutic modulation of this target.
Materials and Methods
Recombinant Protein Production
Experimental procedures are described in greater detail in the Supplemental Material. Briefly, the UBA domains of CBLB (aa 914–982) and Cbl (aa 839–906) were cloned into pGEX 2TK or pGEX 5X-1 (GE Healthcare Biosciences, Pittsburgh, PA), respectively, as previously described.
12
The N-terminal regions containing the tyrosine-binding domain, linker, and RF of CBLB (aa 2–448) and Cbl (aa 2–469) were cloned into pGEX 6P-1 as previously described.4,13 The bacterial expression construct for Ube2d2, pETb-UbcH5b, was kindly provided by Dr. Allan Weissman. Recombinant GST proteins were produced as previously described.
13
Recombinant Ube2d2 was produced and purified as previously described.
14
Recombinant HRD1 (
Large-scale preparation of recombinant proteins was performed by the Protein Expression Laboratory, Frederick National Laboratory for Cancer Research, by modification of the above procedures.
UBA-Dependent Polyubiquitin Enrichment Experiments
This experimental design is a modification of previously published protocols and is described in greater detail in the Supplemental Material.12,15,16
Establishing a Plate-Based IVUA
This assay is a modification of a previously published assay, with minor changes. 15 Most notably, we have previously used radiolabeled ligand to follow ubiquitin transfer; however, we have modernized the assay to instead use biotinylated ubiquitin, whose presence can be detected through the use of avidin conjugated to horseradish peroxidase (HRP). Complete descriptions of methods are provided in the Supplemental Material.
IVUA HTS Development and Implementation
Following comprehensive assay optimization (Supplemental Materials), 384-well HTS was executed in the following manner. Black Fluotrac 600 high protein-binding plates (subsequently referred to as reaction plates; Greiner Bio-One, Monroe, NC) were coated overnight in a solution of 10 µg/mL UBA-GST in 1× phosphate-buffered saline (PBS) pH 7.4 supplemented with 1 mM ZnCl at a volume of 50 µL per well. On the following day, plates were emptied and manually filled with 100 µL of blocking solution [1× Tris-buffered saline with Tween 20 (TBST) supplemented with 1% bovine serum albumin (BSA)] using a Microfill device (BioTeK, Winooski, VT). Following a 1 h blocking period, plates were then washed thrice using an automated plate washer (ELx405 equipped with a plate stacker, BioTek) with 1× TBST and left filled with 1× PBS while the reaction solution was prepared. The reaction solution (1.2×) is composed of 1.2× CBLB reaction buffer [60 mM Tris pH 7.5, 0.12 mM dithiothreitol (DTT), 0.6 mM MgCl2, 0.012% Triton X-100, and 0.6 mg/mL bovine gelatin] with 12 nM E1, 60 nM E2, 90 nM E3, 600 nM unlabeled ubiquitin, and 60 nM biotinylated ubiquitin. The reaction solution is prepared and kept on ice immediately before use. Substances are diluted from stock plates into dilution plates containing a solution whose final concentration after compound addition is sixfold higher than the screening concentration. The composition of substance dilution buffer is 100 mM Tris pH 7.5, 600 µM ATP, 3 mM MgCl2, 2.4% DMSO, and 60 µM library substance. Substance dilution plates also contain low controls in column 1 (100 mM Tris pH 7.5 and 2.4% DMSO) and high controls in column 2 (100 mM Tris pH 7.5, 2.4% DMSO, 3 mM MgCl2, and 600 µM ATP). A Biomek FX-2 liquid handler (Beckman Coulter, Pasadena, CA) is used to assemble the complete substance dilution plates. UBA-coated, blocked, and washed reaction plates are emptied of 1× PBS and refilled with 25 µL per well of reaction solution. Filled reaction plates are then placed on the FX-2, and the reaction is initiated by the 384-well-head mediated transfer of 5 µL from the substance dilution well to its respective well of the reaction plate. This brings the final screening conditions to 30 µL at 1× concentration of all reaction components and initiates the in vitro ubiquitination reaction. Reactions are then allowed to proceed at room temperature for 1 h prior to quenching with the addition of 20 µL of 6 mM ZnCl in 100 mM Tris pH 7. Plates are then sealed and left on a plate rocker overnight. On the following day, plates are washed thrice with 1× TBST before the addition of 100 µL per well of a 1:10,000 dilution of Avidin-HRP in 1× TBST supplemented with 1% BSA. Incubation with Avidin-HRP proceeds for 1 h, during which time the working solution of QuantaBlu (QB) reagent is generated according to the manufacturer’s protocol and allowed to come to room temperature. Following another cycle of triple washing with 1× TBST, 50 µL of QB working solution is added to each well, and HRP-dependent fluorophore generation is allowed to occur. After 1 h, 50 µL of QB stop solution is added to each well, and plates are then shaken on a plate shaker, equilibrated, and read for fluorescence [excitation (ex): 320 nm; emission (em): 420 nm] on a Tecan M1000 Infinite plate reader (Tecan Group, Männedorf, Switzerland). Substance wells on each plate are normalized to their plate-based controls based on the following formula:
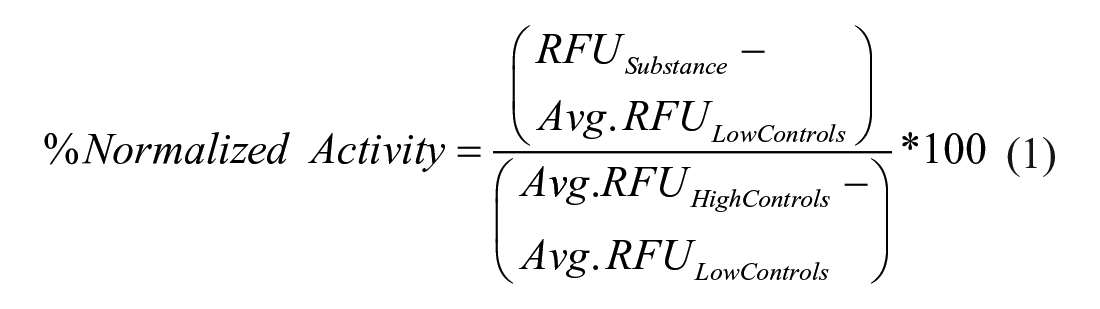
In addition, an assessment of data quality (Z-factor) for each plate was calculated using the following formula:
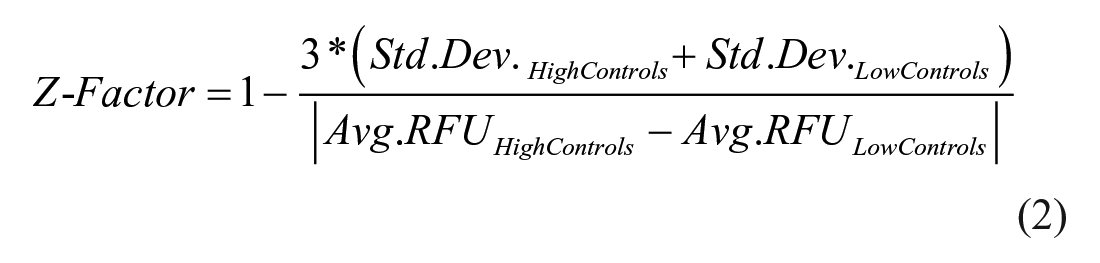
Quadruplicate reconfirmation and 5-point dose response were executed identically to the primary screening except that the substance dilution plates were assembled such that substances were formatted in quadruplicate (2×2 pattern) or 5-point duplicate [2 (duplicate) × 5 (doses) pattern across a row] dose response. Buffer composition was identical to that of primary screening. p38 kinase dose–response testing was carried out as previously published. 17
Primary lead (and derivatives) compound IC50 determination was carried out manually in a similar manner to that of primary screening. Dry resupplied compounds were resuspended in DMSO at a concentration of 100 mM. From this stock solution, a 6× high test concentration solution was prepared with the following composition: 600 µM substance, 600 µM ATP, 3 mM MgCl2, 3% DMSO, and 100 mM Tris pH 7.5. This solution was then used to generate a 10-point 6× dose–response solution set from 600 to 0.5 µM (varying on the substance concentration). Vehicle control (no substance) and “No ATP” control solutions were also assembled. A 1.2× reaction solution was assembled as described above. Using 96-well plates (Thomas Scientific, Swedesboro, NJ), 12 reactions (10 substance and 2 control) were set up and initiated by the addition of substance solutions to reaction solutions using a 12-channel pipettor; initiated reactions were then transferred to a UBA-coated, blocked, and washed reaction plate. All substance concentrations were screened in quadruplicate and executed as for the primary screen once the initiated reactions were transferred to the reaction plates. Following processing and normalization as above, IC50 values were calculated by fitting the normalized data to the following formula using Prism Software (GraphPad Software, San Diego, CA) using a variable slope and a least-squares fit:
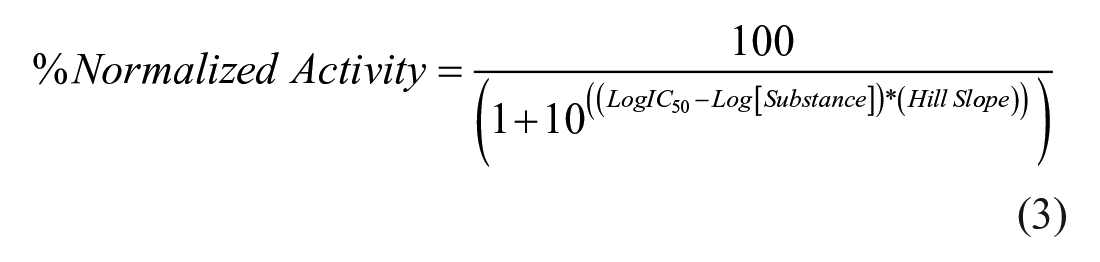
IC50 determination using E6AP was carried out as above for CBLB using the following final ubiquitination conditions: 30 nM E1, 50 nM E2 (UbcH7/UBE2L3, Boston Biochem, Cambridge, MA), 100 nM E3 (E6AP/UBE3A, Boston Biochem), 500 nM ubiquitin, and 50 nM biotinylated ubiquitin.
Methods for the assay optimization experiments shown in
Gel-Based Assay for E1/E2 Activity
A polyacrylamide gel electrophoresis (PAGE) assay was designed to assess the ubiquitination cascade upstream of the E3 ligase activity. Assay reactions, as described above for establishing the IVUA, were performed in Eppendorf tubes containing 28 µL of master mix containing 50 mM Tris-HCl pH 7.5, 0.5 mM MgCl2, 0.2 mM ATP, 0.1 mM DTT, 500 nM ubiquitin, and 85 nM biotinylated ubiquitin with or without 1 nM E1. To test the activity of compounds, 1 µL of 30× prepared dilutions of each stock compound to be tested was added to achieve the final desired concentrations of 0.1, 1, or 10 µM. As a negative control, 1nM E1 was used in the absence of ubiquitin and ATP. 0.5 mM MgCl2 without the other assay reagents (e.g., no ubiquitin or ATP). The reaction was initiated by the addition of 1 µL of E2 (0.016 µg) and incubated at RT for 2 h. The reaction was stopped with the addition of 8 µL of 4× Laemmli loading buffer, and the samples were immediately loaded onto Tris-Glycine4-20% polyacrylamide gradient precast gels (Bio-Rad, Hercules, CA) without boiling and without addition of reducing agent. The proteins were resolved by sodium dodecyl sulfate (SDS)-PAGE, transferred to nitrocellulose membranes using an iBlot2 transfer apparatus (Life Technologies, Waltham, MA), and immunoblotted with primary antibodies overnight. Goat anti-rabbit immunoglobulin G (IgG) HRP conjugate (cat. no. 1721019, BioRad, Hercules, CA) or goat anti-mouse IgG HRP conjugate antibodies (cat. no. 1721011, BioRad) were used with SuperSignal West Pico Plus detection reagent (Thermo Fisher Scientific, Waltham, MA) to visualize the blots on a Li-Cor Odyssey Imager (Li-Cor Biotechnology, Lincoln, NE).
Cell-Based Assay for CBL Family–Dependent EGFR Ubiquitination
HeLa cells (CCL2) were obtained from ATCC (American Type Culture Collection, Manassas, VA) and maintained in culture in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Small interfering RNA (siRNA) transfection was performed with Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific). 2×106 cells were plated in 10 cm dishes and allowed to adhere overnight. On the following day, cells were transfected with the following siRNAs: Cbl (cat. no. s2476), CBLB (cat. no. s2479), or a negative control (cat. no. 4390844) (all Thermo Fisher Scientific) according to the manufacturer’s protocol for 24 h, and then trypsinized, pooled, and replated into two 10 cm plates to make the transfections equal among sets. The cells were further incubated for 24–48 h and then starved in DMEM without serum for 3 h before stimulation with 25 ng/ml epidermal growth factor (EGF). Immunoblotting and immunoprecipitation are described below.
To develop HeLa cells with a genomic knockout of either CBL, oligos targeting CBL (sense: AAACGCATCTCCGTACTATCTTGTG; antisense: CACCGACAAGATAGTACGGAGATGC) were cloned into the PX459 vector. The HF-PX459 (V2) was a gift from Mike McGrew (Addgene plasmid no. 118632, RRID:Addgene_118632, Addgene, Watertown, MA, http://n2t.net/addgene:118632). 18 These vectors contained the Cas9 gene, a single guide RNA, and puromycin resistance. Cells were selected in puromycin, single-cell clones expanded, and knockout clones were identified by immunoblotting and confirmed by sequencing the genomic DNA. These stable knockouts were then used in subsequent experiments, in which they were pretreated with the indicated compounds for 18 to 48 h prior to a 10 min EGF (100 ng/mL) stimulation and lysate creation, as described below.
To harvest proteins, cells were washed twice in ice-cold Dulbecco’s phosphate-buffered saline (DPBS) and then lysed in ice-cold Nonidet P40 (NP40) cell lysis buffer [50 mM Tris pH 7.4, 250 mM NaCl, 5 mM ethylenediaminetetraacetic acid (EDTA), 50 mM NaF, 1 mM sodium orthovanadate (Na3VO4), 1% NP40, and 0.02% NaN3] supplemented with 1 mM Na3VO4 and protease inhibitors (cOmplete Protease Inhibitor Cocktail Tablets, Millipore Sigma, St. Louis, MO). The lysates were cleared of debris by centrifugation at 13,000 g for 10 min at 4 °C. The supernatants were collected, and protein concentrations were determined using a BioRad protein assay (BioRad). For immunoblotting, lysates were heated at 99 °C in 2× Laemmli sample buffer for 7 min. For immunoprecipitation, lysates containing 1 mg protein were incubated with anti-EGFR antibody 199.12 (cat. no. MA5-13319, Invitrogen, Rockford, IL) and Protein A/G+ agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA) overnight at 4 °C with tumbling. Immune complexes were collected by centrifugation and washed five times in cold lysis buffer, resuspended in 2× Laemmli sample buffer, and heated at 99 °C for 7 min. The proteins were resolved by SDS-PAGE, transferred to nitrocellulose membranes (Protran BA85, Whatman, Sanford, MA) or nitrocellulose iBlot2 membranes (Thermo Fisher Scientific), and incubated with primary antibody overnight at 4 °C. HRP-linked secondary antibodies donkey anti-rabbit IgG (cat. no. NA934V, GE Healthcare, Piscataway, NJ), HRP-linked donkey anti-mouse IgG (cat. no. NA931, GE Healthcare), goat anti-rabbit IgG HRP conjugate (cat. no. 1721019, BioRad), or goat anti-mouse IgG HRP conjugate (cat. no. 1721011, BioRad) were used with ECL SuperSignal substrates (Pierce Biotechnology, Rockford, IL) to visualize target proteins, using either a film processor or Li-Cor Odyssey Imager. Alternatively, the KwikQuant Western Blot Detection Kit (cat. no. R1004) and KwickQuant Imager (cat. no. D1001) from Kindle Biosciences (Greenwich, CT) were used to detect proteins on the nitrocellulose membrane. Each experiment was repeated at least three times. Densitometric analysis of immunoblot band intensities was performed using Adobe Photoshop software (version CC 2017, Adobe Systems, San Jose, CA) or Image Studio Software (Li-Cor Biotechnology).
Reagents and Chemicals
These are comprehensively described in the Supplemental Material.
Results
E3-Specific Product Detection as Proof of Principle toward High-Throughput Assay Development
Ubiquitination is an iterative cycle of isopeptide bond formation; E1 covalently accepts the ubiquitin before covalently transferring it to E2, RF E3s then orient the E2–ubiquitin complex to facilitate ubiquitin transfer to the target, and the process begins again with either a new non-ubiquitinated target or a mono-ubiquitinated target from the first cycle. 1 As such, this process generates a distribution of ubiquitinated products. Each step of this process can be visualized by low-throughput techniques, like Western blots, or by using chemically tagged ubiquitin (e.g., biotinylation) and simply monitoring molecular weight changes as ubiquitination cycles proceed. 15 To establish an E3-specific assay, however, we needed to develop a way to capture only those products that are directly derived from the activity of the CBLB ligase, not from upstream E1 and E2 activity. To do this, we took advantage of two features of CBLB biochemistry: CBLB autoubiquitination and the ability of the UBA domain of CBLB to specifically bind polyubiquitinated products. CBLB autoubiquitination is believed to be a means of negative feedback to regulate the intracellular activity of CBLB.6,12,16 Incorporating this feature of CBLB into the assay allows us to reduce the total number of proteins in the assay while maintaining an appreciable signal-to-noise ratio. More importantly, we have previously shown that the UBA domain of CBLB (UBAb) preferentially binds to polyubiquitin chains and only weakly to monoubiquitin.12,16 In contrast, the UBA domain of CBL (UBAc) does not bind to either polyubiquitin chains or monoubiquitin.
As shown in
Figure 1A
, by precoating a high-protein-binding 96-well plate with recombinantly expressed CBLB UBA domain protein, we have developed a capture plate capable of enriching polyubiquitinated proteins over monoubiquitinated ones. Plates coated with the UBAb bound tetraubiquitin but not monoubiquitin (
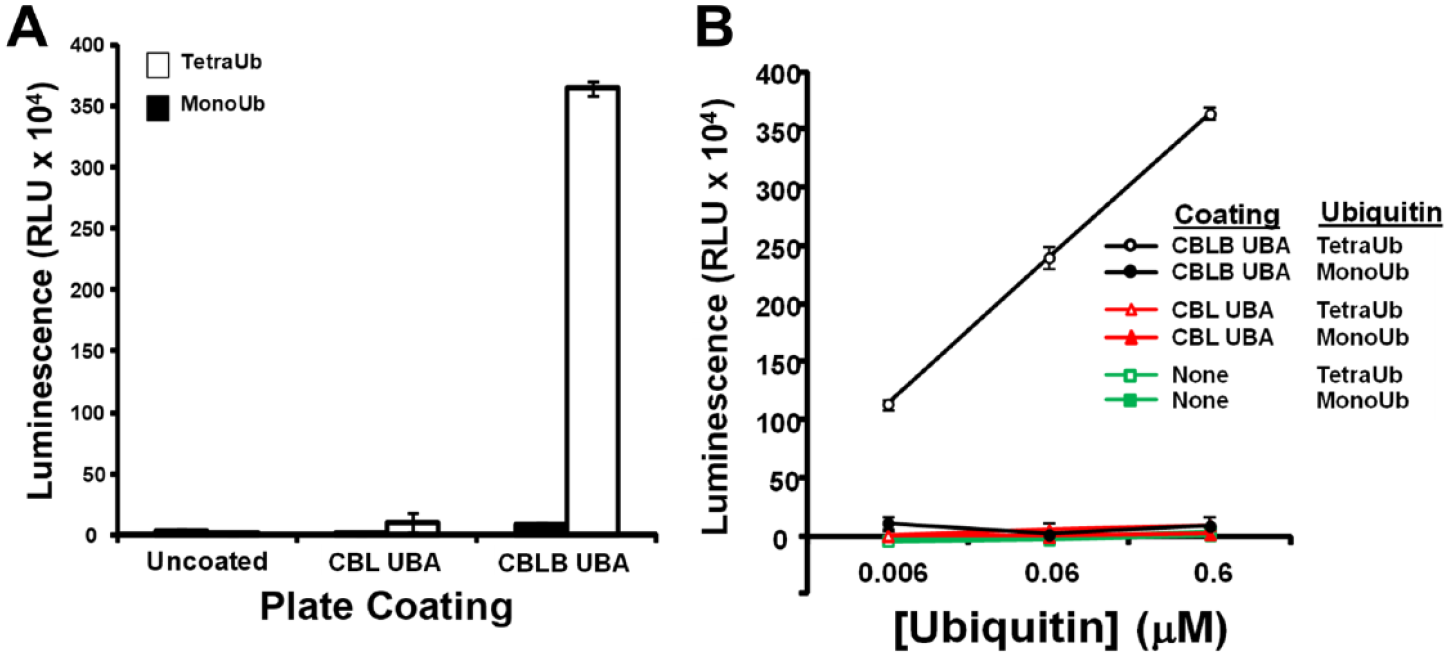
Recombinantly expressed ubiquitin-associated (UBA) domain of
After establishing a possible means for capturing polyubiquitin chains from a complex mixture, we then set about applying this technique to an IVUA in a 384-well format (
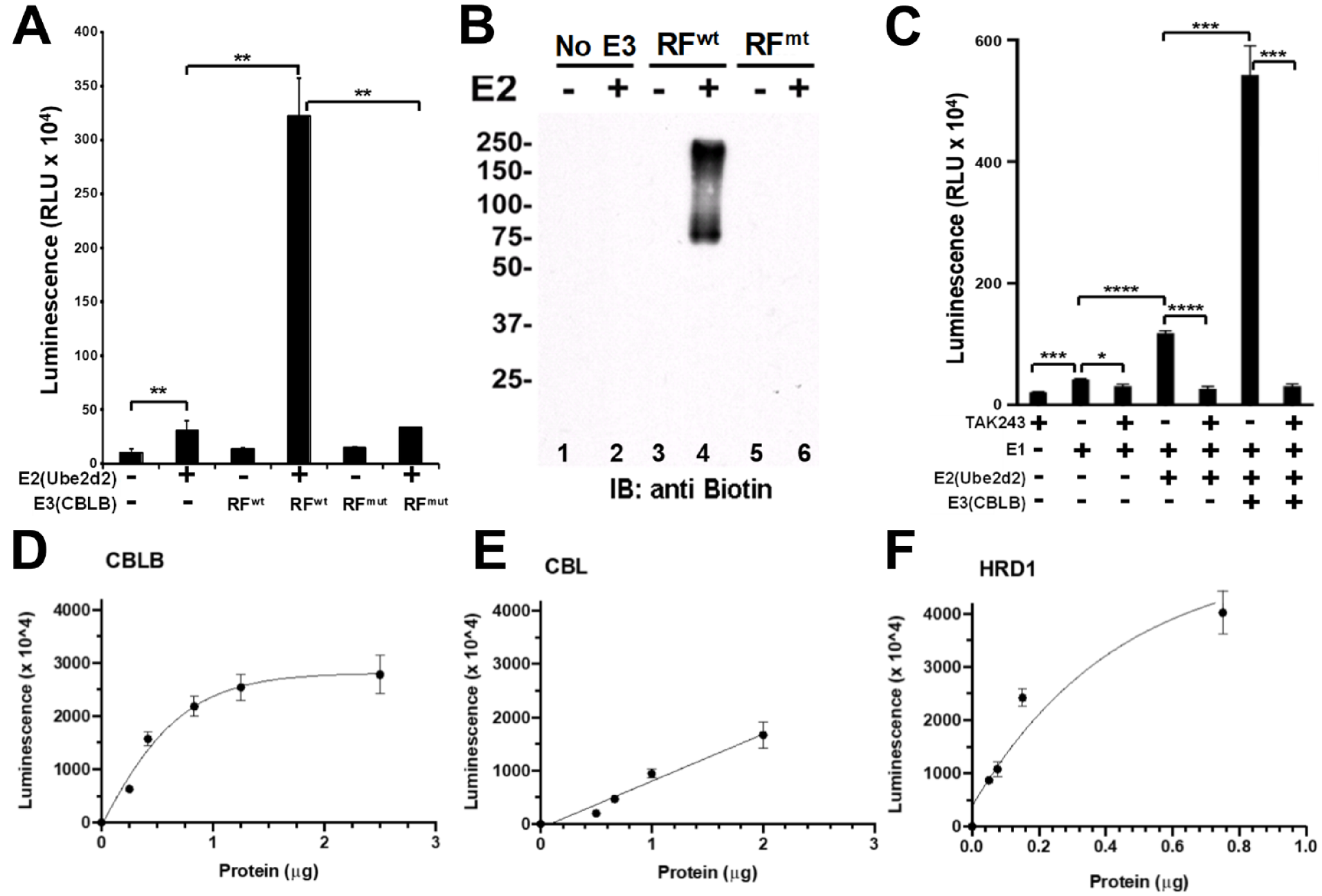
In vitro plate-based E3 assay specifically detects
Sensitivity to Ubiquitination Inhibition and CBLB Specificity Controls
To ensure that the assay we are using is aligned with the central objective of inhibitor discovery, it was necessary to demonstrate that we could detect the presence of a ubiquitination cascade inhibitor. Using the well-described E1 inhibitor TAK243,
Figure 2C
clearly demonstrates that we are able to detect inhibition of the ubiquitination cascade.
21
The E3-dependent signal is significantly reduced in the presence of the E1 inhibitor (
In addition to being able to determine where along the ubiquitination cascade inhibition may occur [either by plate-based methods (
HTS Optimization
After establishing that our UBA capture strategy was adaptable to a 384-well format, it was then necessary to fully optimize the assay for cost-effective reagent use and automated liquid handling. The assay relies on the UBA domain–dependent capture of CBLB polyubiquitinated reaction products, and as such, a titration response curve was experimentally generated to establish the relationship between the observed signal and the concentration of UBA domain used to precoat assay plates (
The drug discovery literature is filled with reports of compounds whose discovery initially appeared to be target specific yet on further experimentation proved to be active in a variety of assays of widely divergent formats.24,25 These compounds have come to be colloquially known as pan-assay interfering substances (PAINS).
24
While there is no universal way to eliminate their discovery during screening, there are several ways to diminish the likelihood of their appearance. A common mechanism of action for PAINS is nonspecific protein binding, often resulting in localized denaturation and thus apparent enzyme inhibition.
26
One way to reduce the appearance of this behavior is through the inclusion of an excess amount of excipient protein to essentially act as a “molecular sponge,” thus reducing nonspecific denaturation of the targeted enzyme.
25
Because different assays have different tolerances for both the concentration of excipient protein and its identity, we tested the inclusion of both casein and bovine gelatin for the effect on the assay over a range of concentrations (
Our IVUA can be thought of as a modified sandwich ELISA (enzyme-linked immunosorbent assay) in that the UBA domain–coated plates capture polyubiquitinated products that contain modified biotinylated ubiquitin. These captured products are then probed for the presence of biotin by using a HRP-linked avidin (HRP-avidin) molecule. This provides two additional vectors on which the assay must be optimized, biotinylated ubiquitin concentration as well as the HRP-avidin concentration. Both of these variables were examined in a concentration-dependent manner (
HTS Outcomes
Following complete assay optimization, we executed an HTS of the Molecular Targets Program [Center for Cancer Research, National Cancer Institute (NCI), Bethesda, MD] pure compound library. This screen encompassed 71,201 discrete substances distributed among 236 384-well plates. A common HTS measure of robustness is the Z-factor, a weighted ratio of the difference in standard deviations to the difference between average readings for high and low controls, with a theoretical maximum value of 1 and a minimum acceptable threshold of 0.5.
27
Using this metric, our assay performed well, with an average Z-factor of 0.73 and only six of 236 plates failing to meet the threshold cutoff of 0.5 (
Primary-Screening Lead Evaluation
The 80 confirmed primary-screening actives were then subjected to 5-point duplicate dose–response screening over a concentration range of 2.5 to 40 µM in the CBLB screening assay. To further ensure that these compounds were CBLB-specific inhibitors, these confirmed screening leads were also screened in tandem against the serine–threonine kinase p38 in a previously published HTS format (data not shown).
17
This tandem screening and availability of resupply further reduced our confirmed primary-screening actives to a pool of eight distinct chemical scaffolds capable of reducing the CBLB-dependent screening signal by at least 50% at 40 µM while not reducing the p38 signal by more than 10% (

Verified screening leads. Structures, identifiers, and screening assay IC50 values of lead molecules discovered through high-throughput screening (HTS) for inhibitors of
As shown in
Figure 3
, these compounds represent several different chemotypes and have a range of measured potencies; complete IC50 curves are provided as
While the primary leads do not have novel chemical composition of matter, the IVUA platform that we have developed is unique from other published CBL family–oriented HTS campaigns in that our platform is built on enzymatic activity rather than fluorescence anisotropy changes upon protein–protein interactions.36,37 In addition, to the best of our knowledge, our campaign is the first published inhibitor screen specifically targeting CBLB. Of the previously published CBL-directed screens, three compounds were identified as disrupting the interaction of CBL and a peptide derived from an E2 binding partner, catapol, methylprotodioscin, and leonuride.
37
These three were tested in our IVUA screening platform and were found to be inactive (
After obtaining sufficient resupply of verified primary-screening leads, their activity was first assessed in an E3 specificity assay similar to that used to generate
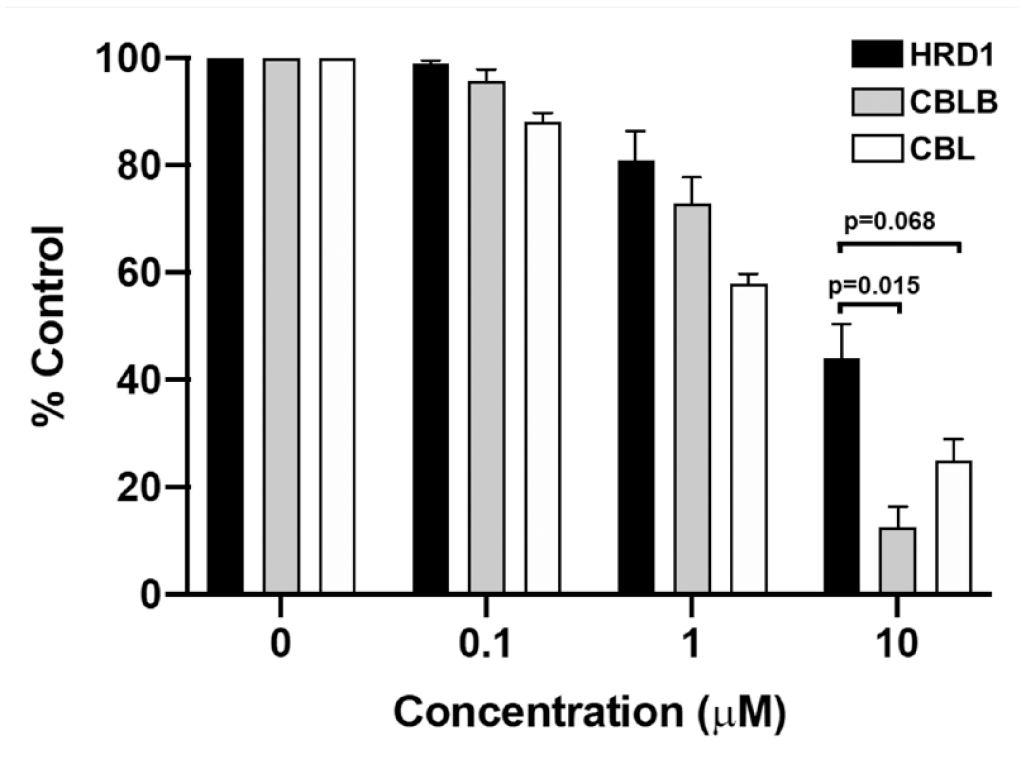
Compound 1 exhibits dose-dependent inhibition and relative specificity for
Although the lack of specificity for CBLB over CBL is disappointing, it is perhaps not surprising because the CBLBN1/2 enzyme used in our assays shares 76% sequence identity to the homologous portion of the CBL protein and the catalytic RF domains share 95% identity. 38 Encouraged by the specificity of compound 1 for the CBL family relative to HRD1, we prioritized our efforts on further characterization of the specificity and potency of compound 1 and related ME analogs.
Although the CBL family E3 specificity assay suggests that compound 1 is functioning at the E3 stage of the ubiquitination cascade (otherwise, all three E3 conditions would be equally inhibited since they share common E1 and E2 elements), it was necessary to definitively demonstrate this selectivity. To determine if compound 1 is interfering with either the E1 ubiquitin activation step or the E2 ubiquitin conjugation step, we carried out IVUAs using only reagents necessary for each step (E1 or E1/E2) and determined what impact, if any, the presence of compound 1 had on these processes. As shown in Figure 5 , compound 1 did not inhibit the monoubiquitination of E1 (as indicated by the molecular weight shift seen in lanes 2–5 compared to lane 1 when ATP and ubiquitin are omitted from the reaction).
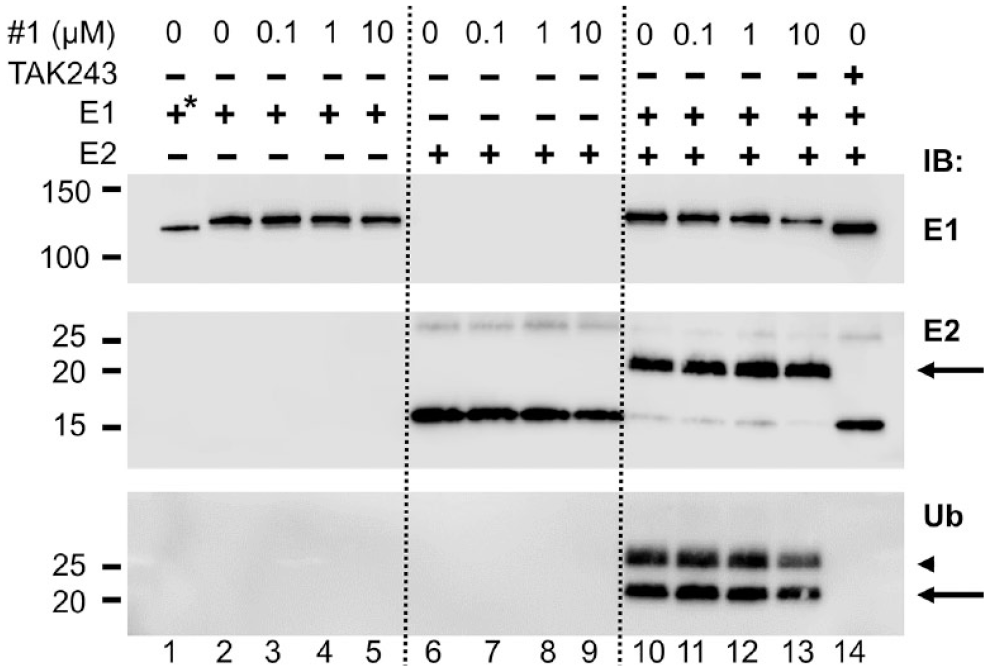
Compound 1 does not inhibit E1 activation of E2 ubiquitin conjugation. Assays were performed in tubes, as in
Compound 1 also did not inhibit the monoubiquitination of the E2, as measured by both size shift and ubiquitin immunoblotting when E1 is present (
Although compound 1 did show modest specificity for the CBL family of E3 ligases over HRD1, it unfortunately also demonstrated cytotoxicity toward the HeLa cervical cancer cell line (

Methyl ellipticinium (ME) derivatives with improved
This toxicity-to-potency ratio provides insight into which compounds may be best suited to further characterization. In addition, several of the tested derivatives also had increased CBLB E3 specificity, using a ratio of the IC50s for HRD1 to CBLB. The ratio for compound 1 was 3.47, while the ratio for some of the tested derivatives exceeded 10 to 1 (
To extend the in vitro observations obtained using our IVUA platform, it was necessary to generate a cell-based model in which to evaluate the activity of our discovered inhibitors against single E3s. Due to the high homology between CBL proteins, it is possible for CBL family members to show substrate redundancy when coexpressed.4,7,13 For example, in HeLa cells, which normally express CBL and CBLB but not CBLC, we have previously shown that the siRNA-mediated reduction of CBL or CBLB leads to decreased ubiquitination of activated EGFR, and the simultaneous siRNA-mediated knockdown of CBL and CBLB leads to almost a complete loss of ubiquitination of the activated EGFR (Ref.
13
and
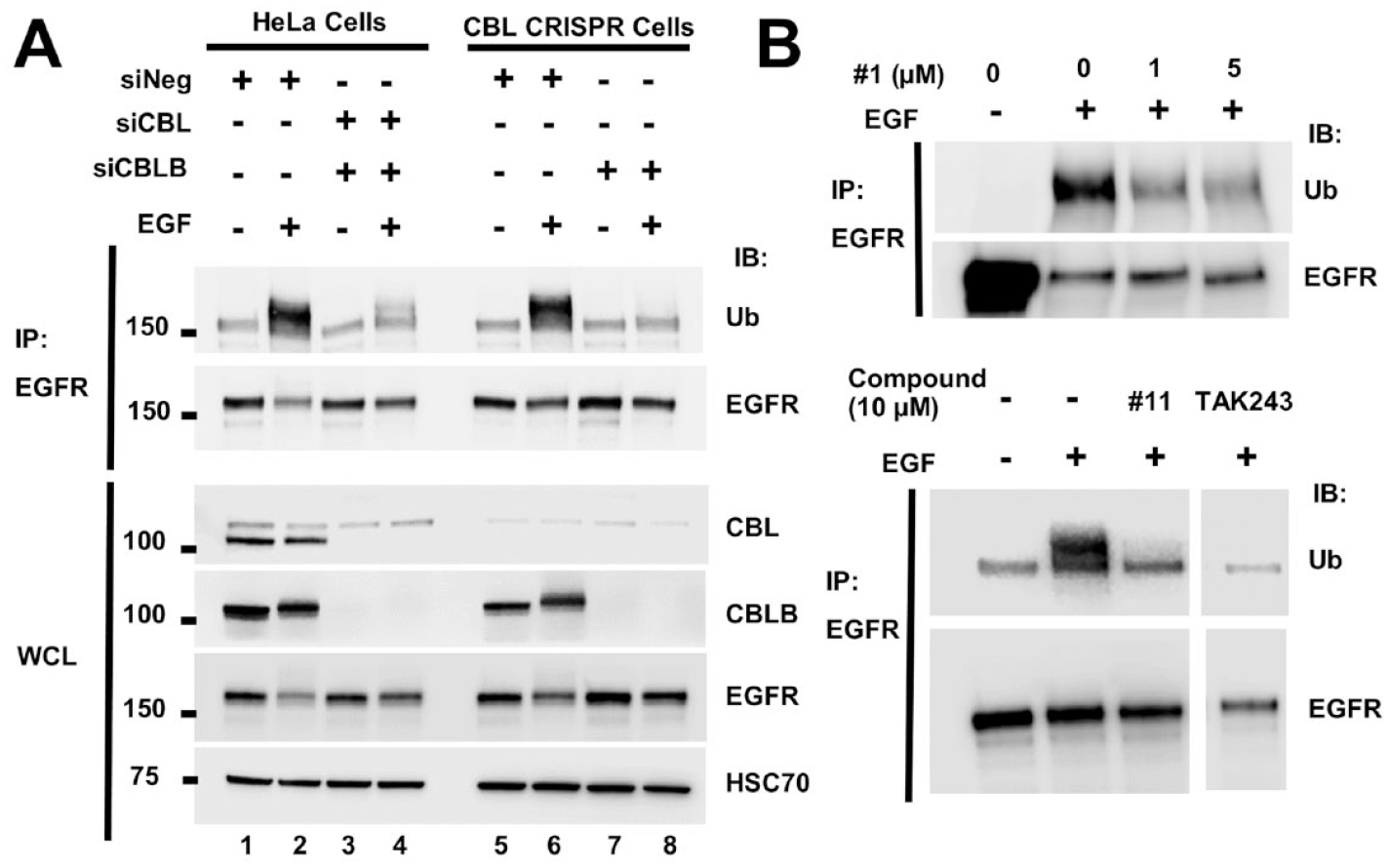
Discussion
The importance of ubiquitination is apparent when one considers how many disease processes are associated with its dysregulation.1,7,41–43 Despite this fact, there are few published screens looking for modulators of this process and no clinically approved drugs targeted at these pathways. While there has been clinical success inhibiting proteasomal degradation of ubiquitinated products (i.e., bortezomib and carfilzomib) and new excitement at the prospect of redirecting ubiquitin machinery through the use of proteolysis-targeting chimeras (PROTACs), clinical development of ubiquitination inhibitors has remained largely restricted to inhibitors of either E1 enzymes or the E3 MDM2 (www.clinicaltrials.gov). One challenge to progress in drug discovery and development is that the multienzyme (E1/E2/E3) and multi-cofactor (ATP/magnesium/ubiquitin) ubiquitination process itself is very difficult to incorporate into a robust HTS. To address the challenge of E3-targeted drug discovery, we have established a ubiquitination screening platform that we have demonstrated can be applied to both RF and HECT family ubiquitin ligases.
Using this IVUA platform, we have determined that MEs appear to broadly inhibit E3-dependent ubiquitin ligation. While the exploration of ME analogs does not represent a systematic attempt at a structure–activity relationship study, the data support several conclusions. The structural diversity tested largely consisted of modifications of the substituents of the quaternary nitrogen of the A ring and changing the identity but not the location of the polar substituent in the D ring. Our experimental evidence (
As a RF ubiquitin ligase, the biological significance of CBLB enzymatic activity is a rapidly evolving area of research, with the discovery of its role in immunomodulation of the innate immune system published in 2014.7,8,45–49 This research activity highlights the need for small-molecule modulators of CBLB activity that can be used to better understand both the biology of this target in a research setting as well as those that can be translated into a clinical setting to improve disease outcomes. The experiments described in this article are the first published cell-free screen for modulators of CBLB enzymatic activity. The assay that we have developed offers several potential advantages over previously published ubiquitin ligation screening assays. 50 First, our assay does not rely on proprietary reagents for either the ubiquitin capture or the signal development, increasing its broad potential use. Second, the use of a non-homogenous assay allows us to build in analyte wash steps prior to signal development, which, depending on the screening library composition, may be important to avoid spectrophotometric interference due to the presence of photometrically active library components. Finally, we have shown that this assay can be readily adapted to other ubiquitin ligases, including both RF and HECT E3s, broadening its appeal to the ubiquitin-related research community. We have used our robust screening assay to identify and characterize several classes of chemical compounds that can be used to advance the understanding of both CBLB activity and ubiquitination more broadly.
Prior to this work, several groups have described inhibitors of CBL or CBLB activity. Two groups have reported peptidomimetic compounds that bind to the TKB domain of either CBL or CBLB, and block interaction with a specific substrate.36,48,51–53 These compounds are not catalytic inhibitors of E3 activity but rather allosteric agents that prevent specific substrates from being targeted by CBL or CBLB. Similarly to substrate peptidomimetics, Wu et al. developed modified peptides based on the structure of the CBL RF interface with the E2 UbcH7.37,49 They showed that intraperitoneal (IP) injection of the peptides could protect mice against high-fat diet-induced obesity and insulin resistance, phenocopying the effects of CBL gene knockouts.
49
The same group also identified several small molecules that compete for UbcH7 binding to CBL at this site.
37
In our autoubiquitination assay, these compounds do not inhibit CBLB E3 activity (
Supplemental Material
sj-pdf-1-jbx-10.1177_24725552211000675 – Supplemental material for In Vitro Ubiquitination Platform Identifies Methyl Ellipticiniums as Ubiquitin Ligase Inhibitors
Supplemental material, sj-pdf-1-jbx-10.1177_24725552211000675 for In Vitro Ubiquitination Platform Identifies Methyl Ellipticiniums as Ubiquitin Ligase Inhibitors by Brice A. P. Wilson, Donna Voeller, Emily A. Smith, Antony Wamiru, Gang Liu, Stanley Lipkowitz and Barry R. O’Keefe in SLAS Discovery
Footnotes
Acknowledgements
The authors would like to acknowledge the efforts of Lauren Procter, Morgan Pagonis, and Jane Jones of the Protein Expression Laboratory, Frederick National Laboratory for Cancer Research, for their work scaling up the production of various recombinant proteins.
Supplemental material is available online with this article.
Authors’ Note
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government.
Author Contributions
B.A.P.W., B.R.O., and S.L. planned the research objectives; D.V. established pre-HTS assays and executed post-HTS lead evaluation, including all cell-based workups; B.A.P.W. established and optimized HTS parameters; E.A.S. and A.W. executed HTS; E.I.G. performed data analysis, and B.A.P.W., D.V., B.R.O., and S.L. composed the manuscript. All authors reviewed and edited the manuscript. B.A.P.W. and D.V. contributed equally to this manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research has been supported by the Intramural Research Program of the National Institutes of Health (NIH), National Cancer Institute, Center for Cancer Research. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.