
Abstract
Although there has been substantial success in the development of specific inhibitors for protein kinases, little progress has been made in the identification of specific inhibitors for their protein phosphatase counterparts. Inhibitors of PP1 and PP5 are desired as probes for research and to test their potential for drug development. We developed and miniaturized (1536-well plate format) nearly identical homogeneous, fluorescence intensity (FLINT) enzymatic assays to detect inhibitors of PP1 or PP5. The assays were used in an ultra-high-throughput screening (uHTS) campaign, testing >315,000 small-molecule compounds. Both assays demonstrated robust performance, with a Z′ of 0.92 ± 0.03 and 0.95 ± 0.01 for the PP1 and PP5 assays, respectively. Screening the same library with both assays aided the identification of class inhibitors and assay artifacts. Confirmation screening and hit prioritization assays used [32P/33P]-radiolabel protein substrates, revealing excellent agreement between the FLINT and radiolabel assays. This screening campaign led to the discovery of four novel unrelated small-molecule inhibitors of PP1 and ~30 related small-molecule inhibitors of PP5. The results suggest that this uHTS approach is suitable for identifying selective chemical probes that inhibit PP1 or PP5 activity, and it is likely that similar assays can be developed for other PPP-family phosphatases.
Introduction
In eukaryotic cells, reversible phosphorylation alters the biological actions of many proteins, acting to regulate the catalytic activities of enzymes, affect the ligand affinities of receptors, alter protein stability, control intracellular localization, and alter protein-protein interactions.1,2 Protein phosphorylation occurs predominately at serine (ser), threonine (thr), or tyrosine (tyr) residues, consisting of the transfer of a gamma phosphate from adenosine triphosphate (ATP) to the side chain hydroxyl. Such reactions are targeted in a sequence-specific manner and are catalyzed by one or more of the >500 known ser/thr- or tyr-protein kinases. The removal of phosphate is accomplished by one or more of the >150 ser/thr-, tyr-, or dual specificity phosphatases, which are also dynamic and highly regulated enzymes.1–4 Because the phosphorylation state of a particular protein at any instant in time reflects the combined activity of both protein kinases and protein phosphatases, the development of inhibitors targeting specific protein kinases or protein phosphatases is greatly desired. Indeed, to date, a tremendous effort has been devoted to the development of compounds that control the actions of protein kinases, producing many specific or selective inhibitors that are useful to probe phosphorylation-regulated processes. In addition, >20 inhibitors of protein kinases (e.g., STI-571/Gleevec, ceritinib, dabrafenib, gefitinib) have been developed into Food and Drug Administration–approved drugs, most for the medical management of human cancers. In contrast, little progress has been made in the development of specific inhibitors of protein phosphatases.
The screening campaign described here was designed to identify highly selective inhibitors of two PPP-family serine/threonine protein phosphatases, PP1C and PP5C (often designated in the literature as PP1 and PP5; encoded by genes PPP1CA and PPP5C, respectively). The PPP family of phosphatases contains five additional subfamilies designated as PP2AC (PPP2CA), calcineurin/PP2B (PPP3CA), PP4C (PPP4C), PP6C (PPP6C), and PP7 (PPP7C/PPEF1). All PPP family members share a common catalytic mechanism, and several natural toxins target this family, including okadaic acid, microcystin, nodularin, tautomycetin, and calyculin A.2–5 Most of the natural product inhibitors are quite potent (IC50; sub nM to low nM range) against PP1C/PP2AC/PP4C/ PP5C/PP6C but demonstrate no or modest selectivity.2,4 These broad-spectrum compounds have some utility as research reagents, but the combined inhibition of PP1C-PP6C is toxic to most, if not all, eukaryotic cells.
Concerns related to systemic toxicity have hampered the development of PPP family inhibitors, and many investigators considered PPP phosphatases as undruggable for years. However, selective inhibitors of calcineurin/PP2B 5 (i.e., cyclosporine A and FK506 [tacrolimus]) have been developed into immunosuppressive drugs with a global market exceeding one billion U.S. dollars per year. In addition, fostriecin, a natural product produced by Streptomyces pulveraceus, that acts as a highly selective inhibitor of PP2AC/PP4C (IC50 <2 nM) and a weak inhibitor of PP1C and PP5C (IC50 >70 µM)4,6 demonstrated marked antitumor activity in animals and entered phase I human clinical trials.3,7 LB100, 8 an inhibitor of PP2AC, has also recently entered phase I trials, suggesting that concerns related to systemic toxicity can be overcome by the development of highly selective inhibitors. Still, to date, there are no reports of small-molecule inhibitors that can be used to easily distinguishing the activity of PP1C versus PP5C, and compounds that act on both generally demonstrate class inhibition. Thus, there are advantages to conducting PP1C and PP5C screening campaigns in parallel.
There are compelling data suggesting that specific inhibitors of PP5C may be useful for the medical management of cancer. First, the expression of PP5C is responsive to hypoxia-inducible factor-1 9 and estrogen, 10 both of which have been linked to the progression of human breast cancer. Second, a positive correlation exists between elevated PP5C expression and some breast cancers, notably in patients diagnosed with invasive ductal carcinoma having metastatic lesions at the time of diagnosis (p < 0.001). 11 Third, in mouse models of tumor progression, constitutive overexpression of PP5C promotes tumor growth in a high-estrogen environment. 12 In addition, the suppression of PP5C expression with small interfering RNA/antisense oligonucleotides in cell culture studies prolongs several stress-induced signaling cascades that favor apoptosis and sensitize cancer cells to stress- or drug-induced apoptosis.9,13 Cell culture–based investigations to determine the actions of PP5C have revealed it plays important and complex roles in the regulation of the phosphorylation states and functions of many cell-signaling proteins, including mitogen-activated protein kinases, p53, glucocorticoid receptors, DNA-damage activated protein kinase, apoptosis signal-regulating kinase, and Raf-1 proto-oncogene serine/threonine kinase.2,9,13,14 These studies suggest that PP5C is a potentially important regulator of signaling networks affecting cellular responses to growth factors and stress and that dysregulation of PP5C activity may be an important factor in the development of some cancers. Finally, PP5C-knockout mice are viable, fertile, and demonstrate only a modest phenotype, the inability to gain weight when placed on a high-fat diet. 15 This suggests highly selective PP5C inhibitors will have low systemic toxicity. Together, these studies suggest PP5C represents an attractive target for antitumor drug development.
PP1C activity has been linked to many ailments,16–19 and a specific inhibitor of PP1C is highly desired as a chemical probe. However, the heterogeneous actions of PP1C in vivo (e.g., affecting smooth-muscle contraction in addition to cell cycle progression) make it unclear if a catalytic inhibitor of PP1C will be useful to treat human disease.
To facilitate the identification of novel inhibitors of PPP phosphatases, we developed homogeneous, fluorescence intensity (FLINT) biochemical assays that are useful to detect inhibitors of PP1C or PP5C. 20 Here we describe miniaturized versions of these robust FLINT assays conforming to a 1536-well format for ultra-high-throughput screening (uHTS) of large libraries containing small molecules. To increase the chances of identifying specific inhibitors, we developed assays for both PP1C and PP5C, using nearly identical assay conditions, and then screened compounds from the NIH’s Molecular Libraries Small Molecule Repository (MLSMR; a ~315,000 small-molecule collection) 21 for inhibitors of PP1C and PP5C.
Materials and Methods
Vector Construction
Expression constructs for human PP1C (PPP1CA; Human genome nomenclature committee [HGNC] ID: HGNC:9281) and PP5C (PPP5C; HGNC:9322) were created using standard cloning methods as previously described.6,20,22,23 Expression and purification of PP5C were performed essentially as described previously.22,23 For PP1C, the commercial vector pMal-c2E (New England Biolabs, Ipswich, MA) was used to express the N-terminal maltose binding protein (MBP) fusion to the PPP1CA ORF in BL21(DE3) Tuner pLacI cells (Novagen, Madison, WI). The MBP-PP1C fusion protein was purified using heparin-sepharose affinity chromatography. Following digestion with enterokinase (New England Biolabs), PP1C was separated from MBP by anion exchange chromatography on Q-sepharose HP (GE Life Sciences, Pittsburgh, PA). Phosphatase preparations were divided into aliquots and stored at −80 °C (PP1C storage buffer: 20 mM tris pH 7.4, 0.01 mM EDTA, 0.01% CHAPS, 1 mM MnCl2; PP5C storage buffer: 40 mM tris pH 7.5, 1 mM tris-carboxyethyl phosphine, 1 mM sodium azide). Working stocks of enzymes for low-throughput confirmation assays were stored at −20 °C in tris buffer containing 50% glycerol, 2 mM DTT, 0.1 mg/mL bovine serum albumin (BSA), and 2 mM MnCl2.
PP1C and PP5C uHTS Assays
Assays were performed in a buffer/substrate system that we previously developed for a low-throughput 96-well format endpoint assay. 20 A stepwise protocol is presented in Table 1 . Briefly, PP1C inhibition assays were performed by dispensing 3 µL of enzyme solution containing 670 pM PP1C in 1.3× assay buffer (40 mM HEPES, pH 7.0; 0.13 mg/mL BSA; 0.013% Triton-X100; 1.3 mM DTT; 1.3 mM sodium ascorbate; 1.3 mM MnCl2) into each well of 1536-well plates. The plates next received 27 nL per well of test compounds from the MLSMR, 100% inhibition control (15 mM cantharidin, yielding a final assay concentration of 0.1 mM) or DMSO (0% inhibition control; 0.7% final DMSO concentration) using a Pin Tool transfer unit (GNF/Kalypsys). The plates were incubated at room temperature for 10 min before receiving 1 µL of substrate (3-O-methylfluorescein phosphate [OMFP]; 400 µM in 3 mM HCl) to start the reaction. The HCl had only a minor effect on the assay buffer pH, reducing it by ~0.05 units (data not shown). OMFP is a fluorogenic substrate with favorable kinetic parameters (low Km, robust rate of hydrolysis) for both PP1C and PP5C. 20 The miniaturized assays, as well as the low-throughput versions they were derived from, are endpoint assays with a reaction time chosen to accommodate efficient workflows on the automated assay platform. After 30 min of incubation at room temperature, 2 µL of 300 mM potassium phosphate at pH 10.0 were added to each well to stop the assay reaction. OMFP hydrolysis yields O-methylfluorescein (OMF), which was measured with a PerkinElmer Viewlux (Waltham, MA) using fluorescein filters (480 nM excitation/540 nm emission: bandwidth of 20 nm). PP5C inhibition assays were conducted in a similar manner, using 310 pM PP5C in 1.3× assay buffer (40 mM HEPES, pH 7.0; 0.13 mg/mL BSA; 0.013% Triton-X100; 1.3 mM DTT; 1.3 mM sodium ascorbate; 0.13 mM MnCl2) as the enzyme solution and a 200 µM OMFP substrate solution. As reported previously, 20 a higher concentration of MnCl2 in the PP1C assay buffer is necessary to maintain stable PP1C activity at room temperature over the multihour time frames necessary for storage of the diluted enzyme solution in the liquid-handling robot during uHTS runs. PP5C was found to be much less susceptible to activity loss, and 0.13 mM MnCl2 was sufficient to maintain activity of the diluted enzyme for at least 24 h at room temperature. 20
PP1C and PP5C inhibition assay protocol in 1536-well plate format.
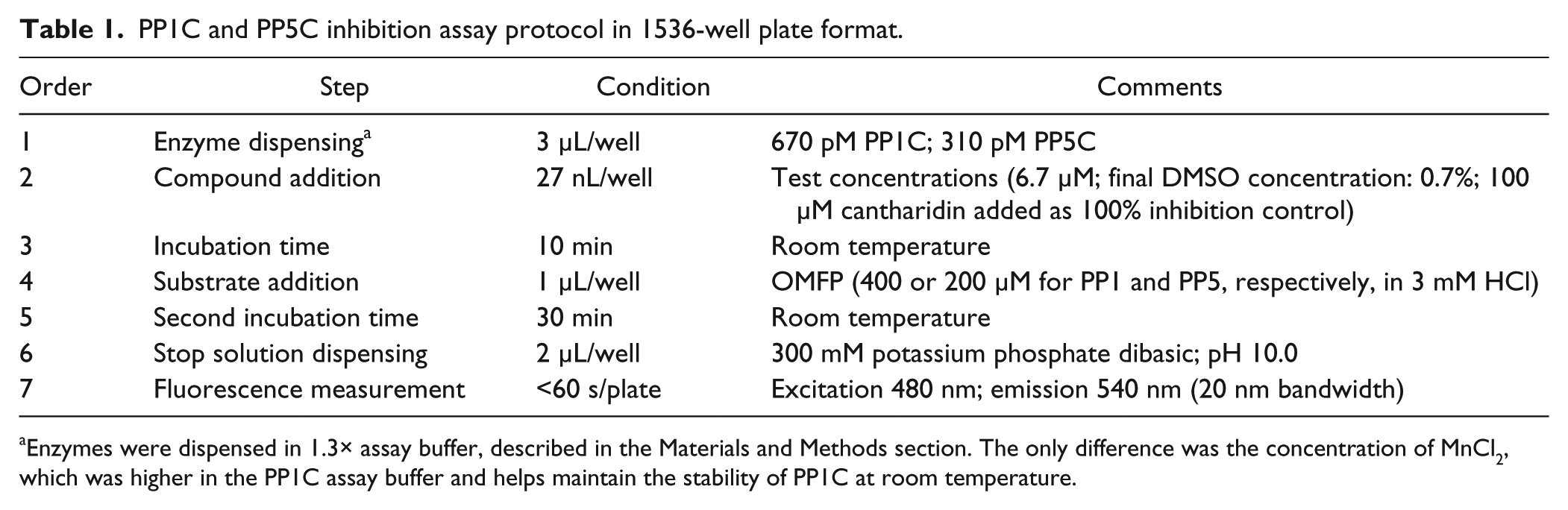
Enzymes were dispensed in 1.3× assay buffer, described in the Materials and Methods section. The only difference was the concentration of MnCl2, which was higher in the PP1C assay buffer and helps maintain the stability of PP1C at room temperature.
uHTS Screening Assays
A summary relative to each step of the PP1C and PP5C uHTS campaigns is shown in Figure 1 , and details are provided in Tables 2 through 4 . During the primary screen, MLSMR compounds were tested in singlicate at a final nominal test concentration of 6.7 µM (final DMSO concentration of 0.7%) using the automated GNF/Kalypsys robotic platform at the Scripps Research Institute Molecular Screening Center (The Scripps Research Institute, Jupiter, FL). The percentage inhibition of each test compound was calculated on a per-plate basis as described in the next section. Nominally inhibiting compounds in the primary screens were determined using a mathematical algorithm described previously. 20 Briefly, the hit cutoff used to qualify active compounds was calculated as the sum of the average percentage inhibition of all compounds tested plus three times their standard deviation (µ + 3SD). The confirmation screen was performed with the same conditions as the primary uHTS, except that putative hits were assessed in triplicate and the results for each compound were reported as the average percentage inhibition of the three measurements, plus or minus the associated SD. Any compound exhibiting an average percentage inhibition greater than the hit cutoff calculated for the primary screen was declared active. For titration experiments, assay protocols were identical to those described above, with the exception that each compound was prepared, using an automated liquid handler (Beckman Coulter, Brea, CA), as a set of 10-point, 1:3 serial dilutions in DMSO starting from a nominal 10 mM stock (highest test concentration in assay = 67 µM) and assessed in triplicate.
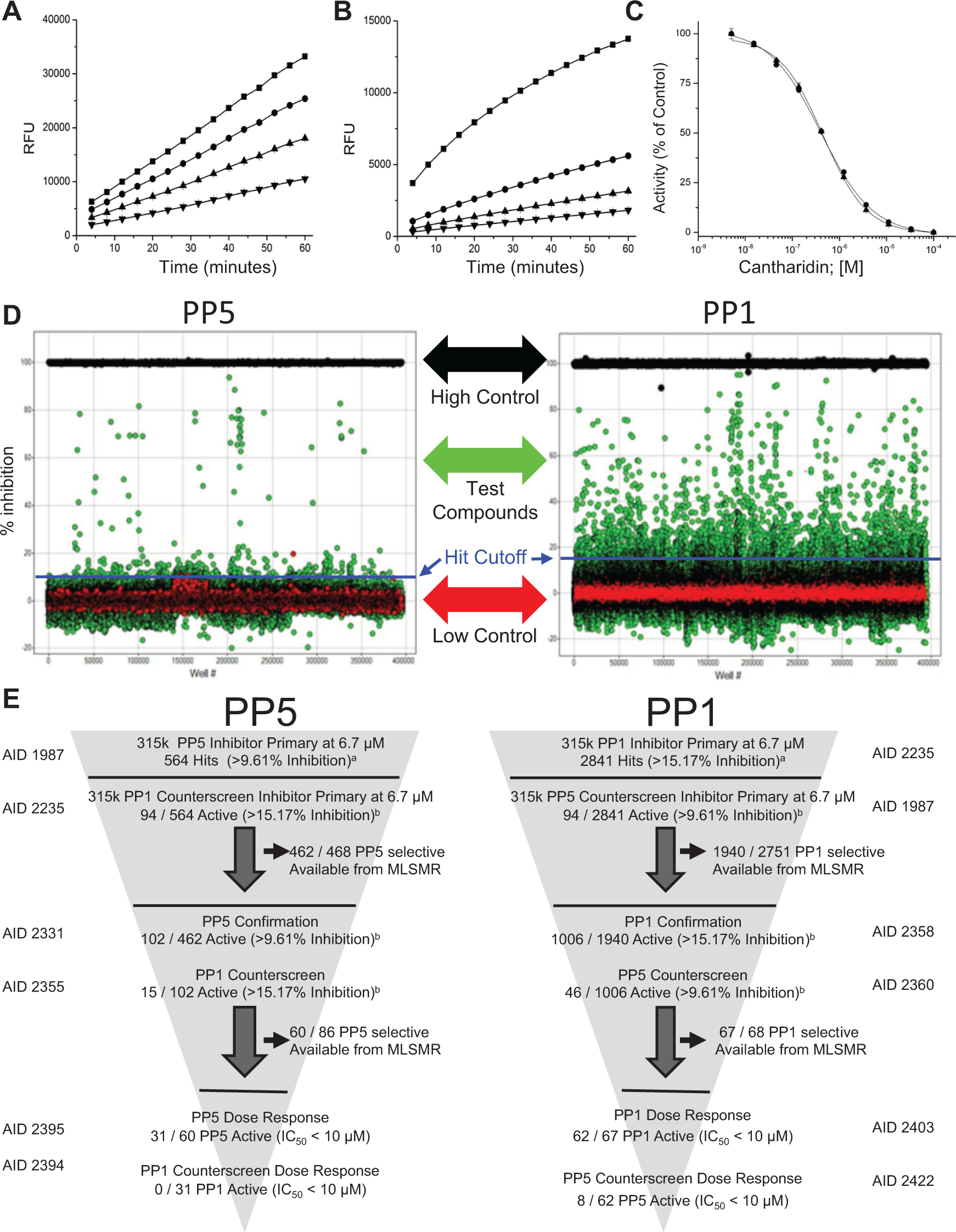
PP1C and PP5C primary ultra-high-throughput screening (uHTS) campaign performance. (
Run statistics for the PP1C and PP5C primary/selectivity screens.
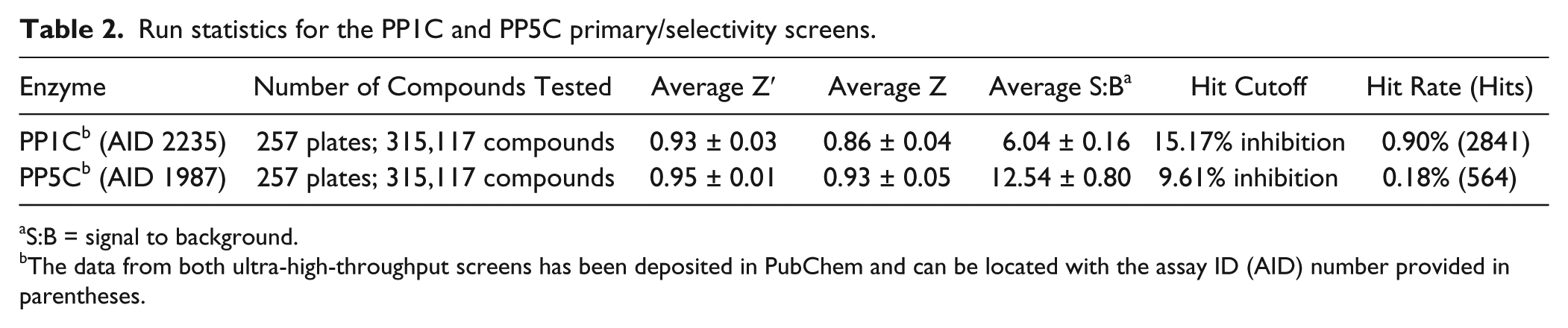
S:B = signal to background.
The data from both ultra-high-throughput screens has been deposited in PubChem and can be located with the assay ID (AID) number provided in parentheses.
PP1C ultra-high-throughput screening campaign summary and results.
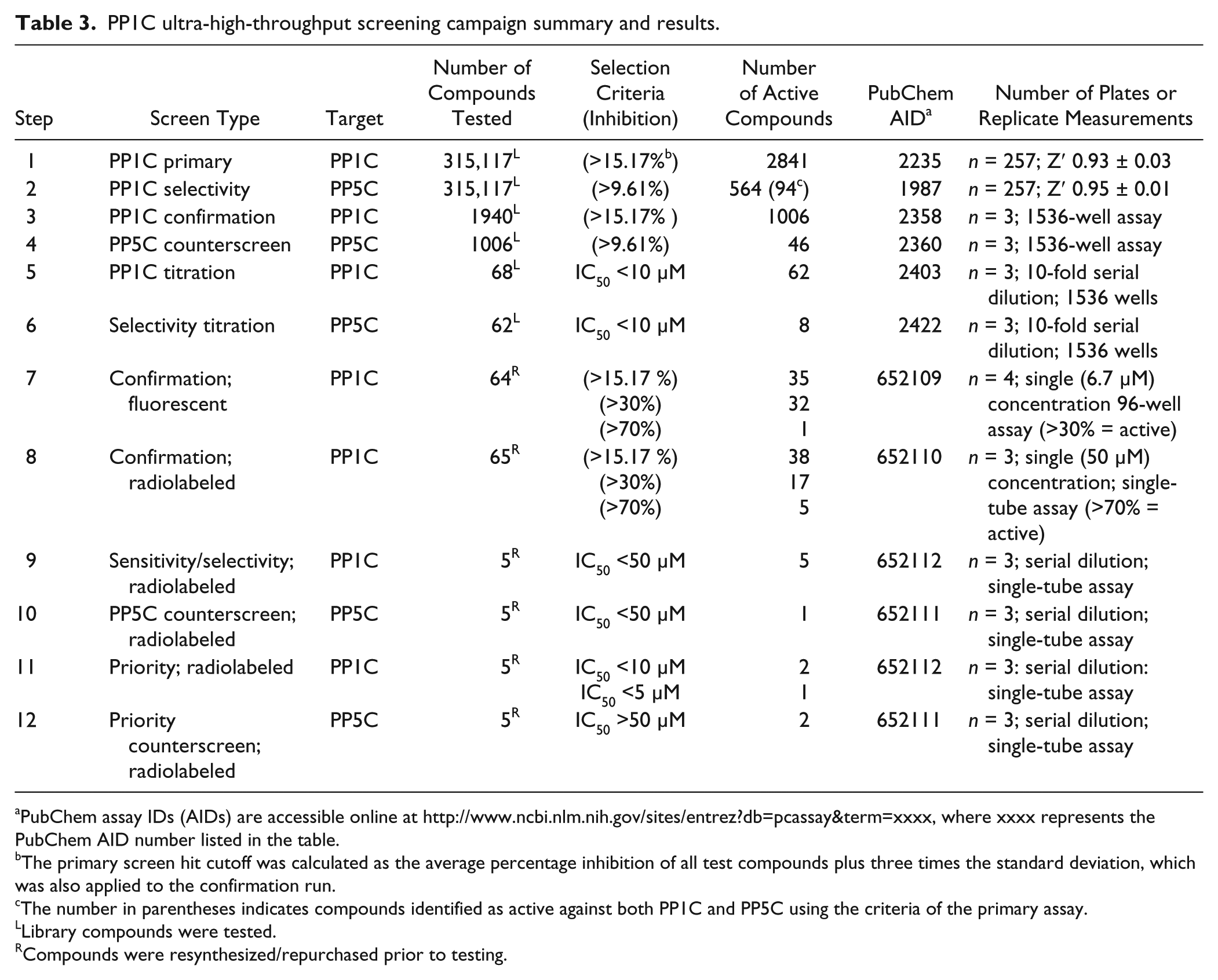
PubChem assay IDs (AIDs) are accessible online at http://www.ncbi.nlm.nih.gov/sites/entrez?db=pcassay&term=xxxx, where xxxx represents the PubChem AID number listed in the table.
The primary screen hit cutoff was calculated as the average percentage inhibition of all test compounds plus three times the standard deviation, which was also applied to the confirmation run.
The number in parentheses indicates compounds identified as active against both PP1C and PP5C using the criteria of the primary assay.
Library compounds were tested.
Compounds were resynthesized/repurchased prior to testing.
PP5C ultra-high-throughput screening campaign summary and results.
PubChem assay IDs (AIDs) are accessible online at http://www.ncbi.nlm.nih.gov/sites/entrez?db=pcassay&term=xxxx, where xxxx represents the PubChem AID number listed in the table.
The primary screen hit cutoff was calculated as the average percentage inhibition of all test compounds plus three times the standard deviation (SD), which was also applied to the confirmation run.
The number in parentheses indicates compounds identified as active against both PP1C and PP5C using the criteria of the primary assay.
Library compounds were tested.
Compounds were resynthesized/repurchased prior to testing.
Screening Data Acquisition, Normalization, Representation, and Analysis
Raw fluorescence data from the ViewLux plate reader were uploaded into an HTS database (MDL Information Systems, San Ramon, CA). Activity of each well was normalized on a per-plate basis, and the percentage inhibition for each compound was calculated as follows:
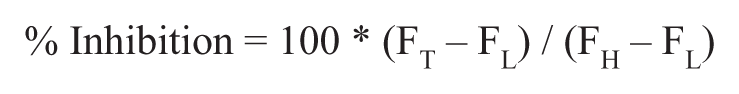
where FT represents the measured fluorescence intensity signal of the well containing test compound and FH is the median fluorescence intensity of the fully inhibited (100 µM cantharidin) controls (n = 24). FL is the median fluorescence intensity of the low-inhibition control wells containing 0.7% DMSO vehicle (n = 24). A Z′ value greater than 0.5 was required for plate validation during the quality control process. 24 For uHTS dose-response experiments, triplicate percentage inhibition values were plotted against compound concentration, and a four-parameter equation describing a sigmoidal dose-response curve was then fitted with an adjustable baseline using Assay Explorer software (MDL Information Systems). Data were also represented using Prism version 4.03 (GraphPad Software, San Diego, CA), with fitting of dose-response curves and IC50 determinations performed using the variable slope (four-parameter) sigmoidal curve analysis tool of Prism. The data produced from screening, confirmation, and orthogonal assays presented in this article have been deposited in the National Institutes of Health’s (NIH’s) PubChem database and can be accessed using the AID numbers provided in Figure 1E (http://pubchem.ncbi.nlm.nih.gov/).
DiFMUP Confirmation Assays
DiFMUP (6,8-difluoro-4-methylumbelliferyl phosphate) is another fluorogenic substrate that is dephosphorylated by both PP1C and PP5C and can be used in FLINT-based assays. 22 DiFMUP-based assays were conducted in a 96-well format using 100 µM DiFMUP (final assay concentration) in the same buffers described above for the OMFP assays.
Radiolabeled Confirmation Assays
To eliminate false-positives from uHTS hits, [32P]- or [33P]-labeled phosphohistone or glycogen phosphorylase substrates were produced. [32P/33P]-phosphohistone was prepared by the phosphorylation of bovine brain histone (type-IIAS) with cyclic adenosine monophosphate (cAMP)–dependent protein kinase (PKA) from rabbit muscle in the presence of cAMP and [32P/ 33P]ATP as described previously.4,6 [32P/33P]-labeled phosphorylase was prepared by phosphorylation of rabbit muscle glycogen phosphorylase b with rabbit muscle phosphorylase kinase in the presence of [32P]- or [33P]ATP essentially as described previously. 25 Protein phosphatase assays with phosphoprotein substrates were performed in the same buffer system as used for the FLINT-based assays. Phosphatase activity was measured by the quantification of [32/33P]-labeled orthophosphate liberated from substrates using the assay buffers described above and established protocols.6,26
Results
Development, Validations, and Miniaturization of the In Vitro HTS Assay to Detect Inhibitors of PP1C and PP5C
The primary screening assay was designed to measure the effect of test compounds on the activity of PP5C. To facilitate the identification of specific inhibitors versus inhibitors of the class, a nearly identical assay to detect inhibitors of PP1C was simultaneously developed. Both assays used OMFP as a substrate. Upon dephosphorylation, the product, OMF, produces a strong fluorescent signal. 20 Previous optimization for a 96-well format revealed that both the PP1C- and PP5C-OMFP–based assays were reliable, robust, and sensitive. 20 To reduce cost per well and allow higher throughput, the assay was miniaturized to a 1536-well plate format in a final volume of 6 µL/well (i.e., 4 µL reaction volume plus 2 µL stop reagent). Miniaturization of the assay was straightforward and accomplished essentially by scaling down the reaction volume by a factor of 37.5 while keeping final buffer and cofactor concentrations the same. A reduction in the BSA concentration improved the consistency of automated solution dispensing, and slight adjustments were made to enzyme concentrations for optimization of assay linearity with the uHTS equipment. In addition, the OMFP substrate solutions were prepared in dilute HCl rather than water in order to suppress background substrate hydrolysis during the long screening runs and the relative proportions of enzyme. Substrate solutions were adjusted from 2:1 in the 96-well format to 3:1 in the 1536-well format. Volumes of test compounds, cantharidin controls, and DMSO controls were reduced by more than 37.5-fold to accommodate Pin Tool dispensing. This kept the DMSO concentration in the reactions below 1%, a level previously confirmed to have no effect on PP1C or PP5C activity. 20 Because specific inhibitors of PP1C are also greatly desired, both the PP1C and PP5C assays were miniaturized, maintaining similar assay conditions whenever possible. Critical variables of the assay, such as enzyme and substrate concentrations, incubation times, enzyme stability, and DMSO tolerance were systematically assessed and optimized both before and during the miniaturization phase to provide the best compromise between assay performance, reagent consumption, and protocol compliance with the robotic platform.
Assay linearity at different enzyme concentrations plotted versus run times are shown in Figure 1A and B . Both assays proved linear and robust over a wide range of enzyme concentrations and time; 670 pM PP1C and 310 pM PP5C (enzyme solution concentrations) were chosen for use in the uHTS. The stepwise protocol resulting from the optimization of each assay variable is detailed in Table 1 . Under these conditions, cantharidin, a known inhibitor of both PP1C and PP5C, produces a concentration-dependent inhibition, acting on PP1C and PP5C with an IC50 of 0.47 ± 0.04 and 0.52 ± 0.06 µM (n = 10), respectively ( Fig. 1C ). Thus, cantharidin (100 µM final assay concentration) was used as a high-inhibition control for both PP1C and PP5C.
PP1C and PP5C uHTS Assay Performance
The scatter diagrams of percentage inhibition data for the PP1C and PP5C uHTS campaigns ( Fig. 1D ) illustrate excellent segregation between high and low controls and the absence of positional effects that can be associated with assay artifacts (e.g., evaporation issues, liquid dispensing errors). In the uHTS, every plate contained both low controls (0% inhibition; n = 16) and high controls (100% inhibition; n = 16) to (1) ensure responsiveness to inhibition, (2) monitor quality control through Z′ and signal/basal calculations, (3) verify proper compound dispensing, and (4) normalize data on a per-plate basis. For both assays, the percentage inhibition values of all test compounds exhibited an approximately normal distribution. Hit cutoffs (indicated by blue lines in Fig. 1D ) were 9.61% and 15.17% (mean + 3SD) for PP5C and PP1C, respectively, with a majority of compounds showing little or no activity (hit rates were 0.18% and 0.9% for PP5C and PP1C, respectively). As demonstrated by a Z′ that was >> 0.5, both uHTS assays proved to be highly robust and sensitive ( Table 2 ).
PP1C and PP5C Screening Campaign Results
The uHTS funnels used to identify inhibitors of PP1C or PP5C are illustrated in Figure 1E , and a step-by-step description of the inhibitor development paths are present in Tables 3 and 4 . Both the PP1C and PP5C OMFP-based FLINT assays were used as co-first pass to screen a total of 315,117 distinct chemical entities from the NIH’s MLSMR 17 at a final concentration of 6.7 µM. A set of 2841 compounds, which exhibited percentage inhibition of greater than that calculated by a nominal cutoff algorithm (15.17%; see the Materials and Methods section for details) was designated as primary hits for PP1C. For PP5C, the calculated nominal cutoff algorithm was 9.61% inhibition, and a set of 564 compounds was designated as primary hits. Because the same compound library was screened for inhibitors of both PP1C and PP5C, the primary counterscreen for class inhibitors could be derived from a comparison of the two uHTS data sets. Using these criteria, 94 compounds were identified as inhibitors of both PP1C and PP5C, suggesting class inhibition or assay artifacts.
The first confirmation screens tested 1940 of the primary hits identified for PP1C and 462 of the primary hits for PP5C, with compound selection based on availability from the MLSMR. The assays employed the same conditions as the primary uHTS, with each compound assessed in triplicate. From triplicate screening results, 1006 had confirmed (>15.17% inhibition at 6.7 µM) activity against PP1C, and 102 had confirmed (>9.61% inhibition at 6.7 µM) activity against PP5C. The confirmed hits were then counterscreened in triplicate for inhibitory activity against PP1C or PP5C, respectively. Forty-six of 1006 PP1C-selective compounds were eliminated because of activity (>9.61% inhibition) against PP5C, and 12 of 102 PP5C-selective compounds were eliminated because of activity (>15.17% inhibition) against PP1C. The remaining PP1C or PP5C selective compounds were ranked based on percentage inhibition at 6.7 µM. Sixty-seven compounds demonstrating greatest inhibition for PP1C and 60 of the best inhibitors of PP5C were selected for titration assays (10-point, 1:3 serial dilutions starting at 67 µM and also assessed in triplicate) against both PP5C and PP1C. Comparison of the individual concentration-response curves produced by testing against both PP1C and PP5C facilitated the triage of class selective inhibitors.
Of the 67 compounds tested against PP1C, titration data analysis identified 62 compounds that had an IC50 <10 µM, 8 of which were eliminated because they also demonstrate IC50 <10 µM against PP5C. For PP5C, 31 of 60 produced IC50 <10 µM, none of which demonstrated IC50 <10 µM against PP1C. Before proceeding, all compounds of interest were resynthesized or repurchased, analyzed for purity and compound identity, and then retested for inhibition of PP1C/PP5C activity with the OMFP FLINT-assay in a 96-well format (volumes scaled up 50-fold from uHTS assays), testing in quadruplicate at a single test concentration (6.7 µM). Only compounds having similar inhibitory activity to test compounds in the MLSMR library were chosen for further development.
Radiolabeled Biochemical Confirmation Assays
Although OMFP is an excellent substrate for both PP1C and PP5C, it is a small molecule, and compounds that can displace OMFP may not disrupt the more complex interactions occurring between PP1C/PP5C and physiological phosphoprotein substrates. Therefore, radiolabeled [32P; 33P]-phosphoprotein assays were employed as additional confirmation assays. For radiolabeled assays, bovine histone phosphorylated by cAMP-dependent PKA or glycogen phosphorylase phosphorylated by phosphorylase kinase were used, both being established phosphoprotein substrates for PP5C and PP1C.6,27–29 The top 60 to 65 compounds were retested with [32/33P]-labeled phosphoprotein substrates alongside FLINT assays using OMFP and/or DiFMUP (a second previously validated FLINT assay 22 ). When tested in quadruplicate at a single concentration of 50 µM for the inhibition of PP1C, 38 of the 65 resynthesized/validated compounds demonstrated >15% inhibition, 17 demonstrated >30% inhibition, and 5 demonstrated >70% in the [32/33P]-based assays. Comparison of the inhibition data produced by FLINT and [32/33P]-protein based assays revealed that nearly all of the chemically validated compounds produced similar results with both substrates (steps 7 and 8 in Tables 3 and 4 ; for additional details, see Pubchem AIDs 463223, 463224, 463225, 463226 and 652109, 6521010, 652111, 652112). The majority of compounds eliminated in this round failed to produce sufficient inhibitory activity with both substrates upon resynthesis/chemical validation.
To further prioritize compounds for development, serial dilutions were tested to assess the strength of inhibition against both PP1C and PP5C, first using the FLINT assays. Serial dilutions of the five strongest inhibitors identified in each campaign were then retested using radiolabeled assays. All 10 of the strongest inhibitors identified using the FLINT assays were confirmed with the [32P/33P]-labeled phosphoprotein substrates. One of the five strongest inhibitors of PP1C was eliminated from further consideration based on having an IC50 <50 µM for PP5C using the radiolabel protein substrates. For PP1C prioritization, three compounds with IC50 <10 µM were identified, two of which produced IC50 for PP5C inhibition >50 µM, representing the greatest selectivity for any compounds identified in this screening effort. A dose-response curve for the most selective inhibitors of PP1C identified in this campaign is shown in Figure 2A .
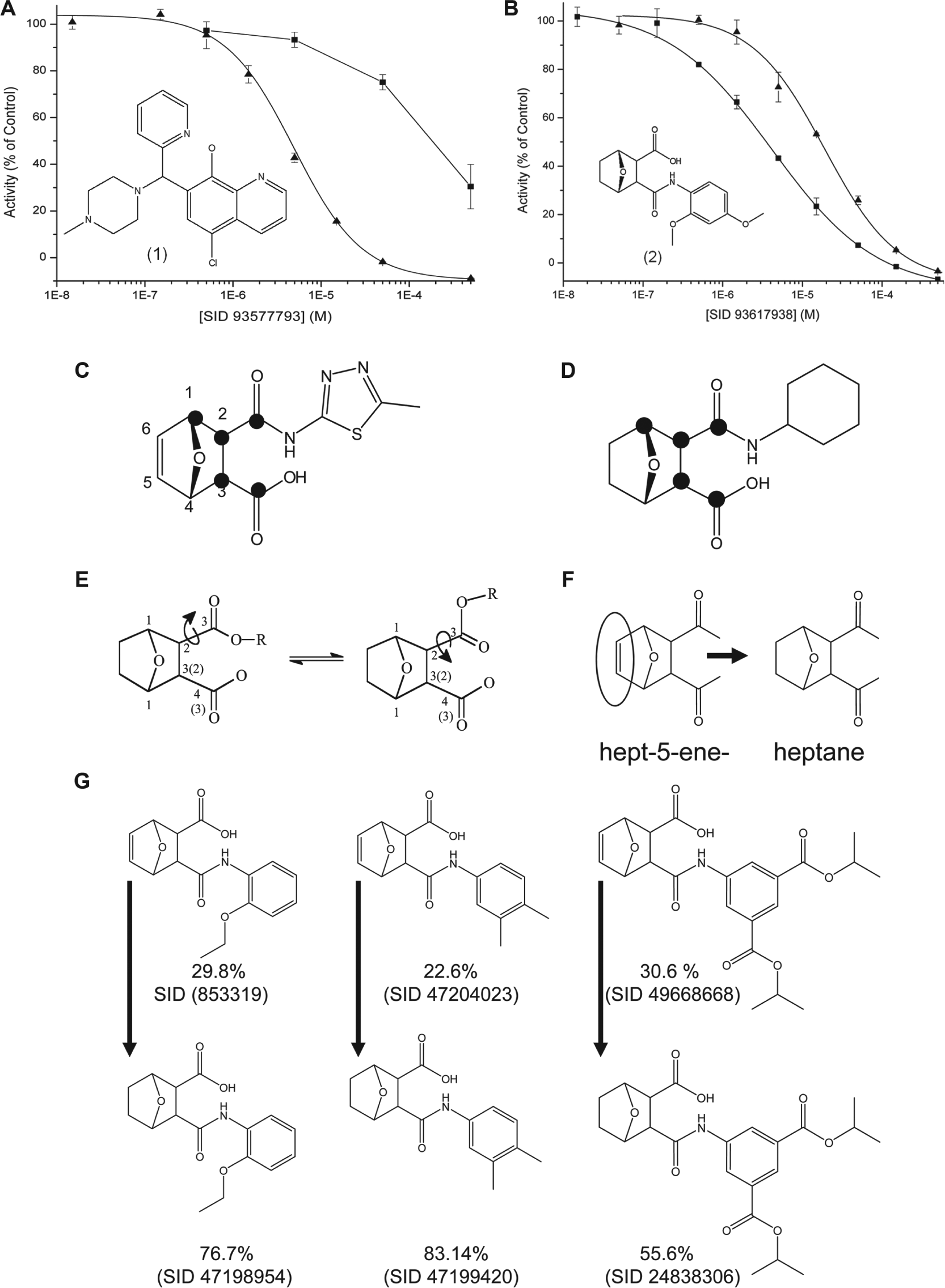
Concentration-response curves for radiolabeled phosphoprotein substrate confirmation assays. (
When ranked based on relative potency and selectivity for PP5C, 3-{[(2,4-dimethoxyphenyl)amino]carbonyl}-7-oxabicyclo[2.2.1]heptane-2-carboxylic acid [SID 93617938, henceforth referred to as (2)] was identified as one of the best inhibitors identified in the uHTS. Structure activity relationship analysis revealed that 30 of the top 35 confirmed hits for PP5C shared a 1,3 or 1,4-dioxy functionality within a common 7-oxybicyclo[2.2.1] heptane/heptene scaffold ( Fig. 2D–G ). For all of the tested inhibitors with this functionality, conversion from hept-5-ene to heptane increased the strength of inhibition but produced no obvious advantage related to selectivity (as illustrated in Fig. 2 ). A complete list of all compounds tested and all assay data results for each step described above has been deposited in PubChem and can be accessed using the AID numbers provided in the figures and tables.
Discussion
Here we report the development of a sensitive, rapid, and robust screening protocol for identifying small-molecule inhibitors of PP1C or PP5C. PPP family phosphatases share a common catalytic mechanism, and several natural compounds that potently inhibit the class (i.e., PP1C, PP2AC, PP4C, PP5C4,6,26,29–31) demonstrate marked toxicity to eukaryotic cells. Therefore, to be useful as research probes or as lead compounds for drug development, inhibitors of PP1C or PP5C need to be highly selective. To facilitate the identification of specific inhibitors, we developed nearly identical OMFP-based assays for both PP1C and PP5C. The assays measure enzyme-catalyzed hydrolysis of a synthetic fluorogenic phosphomonoester substrate, OMFP. Upon dephosphorylation, OMFP produces a robust fluorescence signal, and OMFP proved to be an excellent substrate for both enzymes.
With Z′ values >0.9 and a throughput of ~18,000 compounds per hour, both the PP1C- and PP5C-OMFP 1536-well format assays were easily implemented in the context of automated uHTS, demonstrating excellent assay performance over the course of screening compounds from the NIH’s MLSMR (a small-molecule collection encompassing >315,000 distinct chemical entities). During assay optimization, we found that preparing OMFP in 3 mM HCl greatly reduced background fluorescence, because of nonenzymatic loss of phosphate occurring over time in substrate solutions used during screening runs. This increased assay data consistency. The conditions and composition of the final assay buffers used for the PP1C and PP5C uHTS were nearly identical, and it is likely that only minor modifications of the procedures described here will be needed to develop similar assays to detect inhibitors of other PPP family phosphatases.
As expected from screening of a nonbiased compound collection that covers a large chemical space, the percentage inhibition of all test compounds displayed a distribution approximating a normal distribution, and the hit rates of the primary uHTS assays were low (0.9% and 0.18%, PP1C and PP5C, respectively). Screening the same library for inhibitors of both PP1C and PP5C helped to identify compounds that were assay artifacts or acted as inhibitors of the class. Still, it should be noted that some of the class inhibitors eliminated in this PP1C-PP5C campaign may still have residual value, as they can be tested to determine if they demonstrate selectivity against other PPP-family members (i.e., PP2AC, PP4C, or PP6C).
Analysis of the PP5C screening data revealed a common feature (7-oxybicyclo[2.2.1] heptane/heptene scaffold) in 30 of the 35 strongest semiselective inhibitors of PP5C. For all 7-oxybicyclo[2.2.1] heptene/heptane–based compounds tested, reduction of the C5-C6 double bond (heptene to heptane; illustrated in Fig. 2E–G ) produced an increase in potency but failed to provide an advantage related to selectivity. The typical orientation of the 7-oxybicyclo[2.2.1] heptane core scaffold within the active site of PP5C has been reported in the high-resolution co-crystal structures of PP5C in complex with several cantharidin and norcantharidin derivatives.32,33 Inspection of these structures (PDB IDs 4zx2 and 4zvz) suggests that the lower affinity of the heptene is likely due to the trigonal planar geometry of the heptene C5 projecting the C5 hydrogen toward the amide hydrogen of Val429 in PP5C, thereby creating an unfavorable steric interaction that could distort the high-affinity binding mode observed for the heptane scaffold. If the binding mode is conserved in PP1C, then a similarly unfavorable interaction with the equivalent valine residue (Val250 in PP1C) would be expected. The heptane configuration of the core is also shared with cantharidin, the weakest natural product inhibitor that targets PP1C, PP2AC, PP5C, and PP6C. 30
Analysis of the PP1C screening data revealed more structural diversity among the PP1C-selective inhibitors. SID-93577793 ( Fig. 2 ) represents the most selective inhibitor identified in this campaign, and studies to produce the co-crystal structures of PP1C and PP5C in complex with several of the hits identified in this screen are in progress. Such structures should prove valuable in the development of compounds with greater selectivity and potency.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Abraham Mitchell Cancer Research Fund (to R.E.H. and to A.W.), Mayer Mitchell Annual Award for Excellence in Cancer Research (to R.E.H.). National Institute of Health Molecular Libraries Probe Production Centers Network (NIH/MLPCN) grant 5U54MH084512 (to Scripps Research Institute Molecular Screening Center), grants from NIH (R01CA60750, RO3MH05702, R21NS071553 to R.E.H.) and U54HG005031 (to the KU Specialized Chemistry Center). We thank Pierre Baillargeon, Lina DeLuca, and Louis Scampavia (Lead Identification Division, Scripps Florida) for compound management.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.