
Abstract
Histone deacetylases (HDACs) are important epigenetic factors regulating a variety of vital cellular functions such as cell cycle progression, differentiation, cell migration, and apoptosis. Consequently, HDACs have emerged as promising targets for cancer therapy. The drugability of HDACs has been shown by the discovery of several structural classes of inhibitors (HDACis), particularly by the recent approval of two HDACis, vorinostat (ZOLINZA) and romidepsin (Istodax), for the treatment of cutaneous T-cell lymphoma by the US Food and Drug Administration. The outstanding potential of HDACis, with a defined isoform selectivity profile as drugs against a plurality of diseases, vindicates increased effort in developing high-throughput capable assays for screening campaigns. In this study, a dual-competition assay exploiting changes in fluorescence anisotropy and lifetime was used to screen the LOPAC (Sigma-Aldrich, St Louis, MO) library against the bacterial histone deacetylase homologue HDAH from Bordetella, which shares 35% identity with the second deacetylase domain of HDAC6. The binding assay proved to be highly suitable for high-throughput screening campaigns. Several LOPAC compounds have been identified to inhibit HDAH in the lower micromolar range. Most interestingly, some of the hit compounds turned out to be weak but selective inhibitors of human class IIa and IIb HDACs.
Introduction
T
The outstanding potential of HDACis with defined isoform selectivity profiles as drugs against a plurality of diseases vindicates an increased effort in the development of high-throughput capable assays and screening campaigns. Besides classical enzyme activity assays with chromogenic, fluorogenic, or luminogenic substrates, 12 indirect reporter gene assays 15 and binding assays on the basis of fluorescence quenching, 16 fluorescence anisotropy 17 or fluorescence resonance energy transfer, 18 and even surface plasmon resonance 19 have been developed. Very recently, a dual-parameter binding assay based on fluorescence anisotropy and fluorescence lifetime (FLT) was developed featuring extraordinary robustness with respect to autofluorescence of test compounds. 20
In this study, the dual-parameter binding assay was used to screen the LOPAC library against HDAH. To our best knowledge, this study reports the first screening data of a binding assay for an HDAC homologue. The data were compared with the outcome of a conventional fluorogenic enzyme activity assay. Several LOPAC compounds that are pharmacologically active against other targets than HDACs and similar chemical structures included in the confirmative retesting have been identified to inhibit HDAH in the micromolar range. Selected hit compounds were also tested on representatives of HDAC classes I, IIa, and IIb to determine their selectivity profile. Finally, it was shown that the fluorescent probe could also be used as a ligand for recombinant human HDAC8.
Materials and Methods
Reagents
Methanol was purchased from Roth (Karlsruhe, Germany); trypsin (bovine pancreas), DMSO, and Pluronic F-68 were from Sigma-Aldrich (St. Louis, MO); and the two acetylated (Ac) and trifluoroacetylated (TFA) substrates, Boc-Lys(Ac)-AMC (I-1875) and Boc-Lys(TFA)-AMC (I-1985), respectively, were from Bachem (Bubendorf, Switzerland). All other chemicals were purchased from Roth, Merck, or AppliChem (Darmstadt, Germany). Recombinant His-tagged HDAH from Bordetella/Alcaligenes strain FB188 was produced according to Hildmann et al. 9 The expression of HDAH was induced in Escherichia coli XL-1 blue in LB medium using isopropyl-β-D-thiogalactopyranosid (IPTG; AppliChem) and purified to homogeneity by Zn2+-IMAC sepharose column chromatography (GE Healthcare, Piscataway, NJ), applying a linear concentration gradient from 40 to 500 mM imidazol (Fluka, Buchs, Switzerland) using an ÄktaPrime system (GE Healthcare). Recombinant human HDAC8 was prepared similarly according to Riester et al. 21 Human HDAC1, HDAC6, and HDAC7 were purchased from BPS Bioscience (San Diego, CA). The Atto-hydroxamate ligand was synthesized as described elsewhere. 20
Binding of Atto-hydroxamate ligand to HDAH and HDAC8
In total, 50 nM Atto-hydroxamate ligand was mixed with increasing concentrations of HDAH of HDAC8 enzymes in FB188 buffer (15 mM Tris-HCl [pH 8.0], 50 mM KH2PO4/K2HPO4, 250 mM NaCl, 250 µM EDTA, and 0.001% Pluronic F-68) at 21 ± 2 °C. In the case of HDAH, both fluorescence parameters, anisotropy and FLT, changed upon binding to the enzyme. In contrast, addition of HDAC8 to the fluorescent ligand probe caused only the change of fluorescence anisotropy. Therefore, for HDAH, both fluorescence parameters were used to calculate the dissociation constant (Kd), whereas with HDAC8, only the fluorescence anisotropy could be exploited. The Kd values of HDAH (E) and Atto700-HA (L) were obtained from fitting the data to a simple bimolecular binding model using the progam Origin 7 (OriginLab Corporation, Northampton, MA):
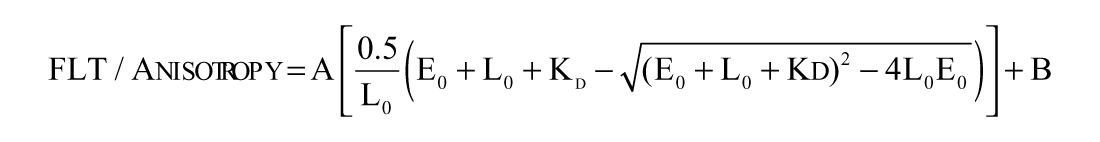
where A is amplitude, B is background, Kd is the dissociation constant, E0 is the total concentration of enzyme, and L0 is the total concentration of the Atto-hydroxamate ligand.
Library screening
The LOPAC compound library was obtained from Sigma as 10-mM stock solutions in DMSO. In total, 1 µL of each compound solution was diluted 1:10 with DMSO in deep-well plates (HJ Bioanalytik, Mönchengladbach, Germany), 20-fold diluted with methanol, and subsequently distributed into eight 96-well half-area replica plates (Greiner Bio-One, Monroe, NC). After complete evaporation of methanol, the plates were sealed with silver seal (Greiner Bio-One) and stored at −20 °C. Screening of the compound library for inhibitors of the target enzyme HDAH was performed using a conventional one-step fluorogenic assay 12 as well as the dual-parameter competition assay 20 at a total volume of 40 µL and a final compound concentration of 31 µM. In short, the fluorogenic enzyme activity is based on the deacetylation of an ε-acetylated lysine substrate and the subsequent cleavage of the deacetylated substrate by trypsin, resulting in a release of fluorescent 7-amino-4-methylcoumarin. The substrate Boc-Lys(Ac)-AMC and HDAH were applied at final concentrations of 10 and 2 µM, respectively. The corresponding increase in fluorescence was measured using a POLARstar fluorescence microplate reader (BMG Labtech, Ortenberg, Germany) equipped with a 340/10-nm excitation filter and a 460/10-nm emission filter. The dual-parameter competition assay exploits the concurrent change of fluorescence anisotropy and FLT upon the reversible binding of the fluorescent Atto700-hydroxamate ligand to the enzyme. The presence of inhibitors was indicated by a reversal of the fluorescence properties caused by an effective displacement of the ligand. The principle of the binding assay is shown in Figure 1 . All assay steps were performed in FB188 buffer at 21 ± 2 °C using final concentrations of 50 nM Atto-hydroxamate ligand and 2 µM HDAH. Fluorescence anisotropy and FLT were measured in an LF502 NanoScan FLT multimode fluorescence microplate reader from Berthold Detection Systems (Pforzheim, Germany). The samples were excited by short light pulses at 635 nm, and the emitted polarized light was collected after passing through 740/30-nm band pass filters. The performance of both screens was monitored by inserting control plates with 48 negative and 48 positive controls between compound plates to calculate the Z′ factor throughout the screening campaign. In addition, there was one row with negative (DMSO) controls on each compound plate.
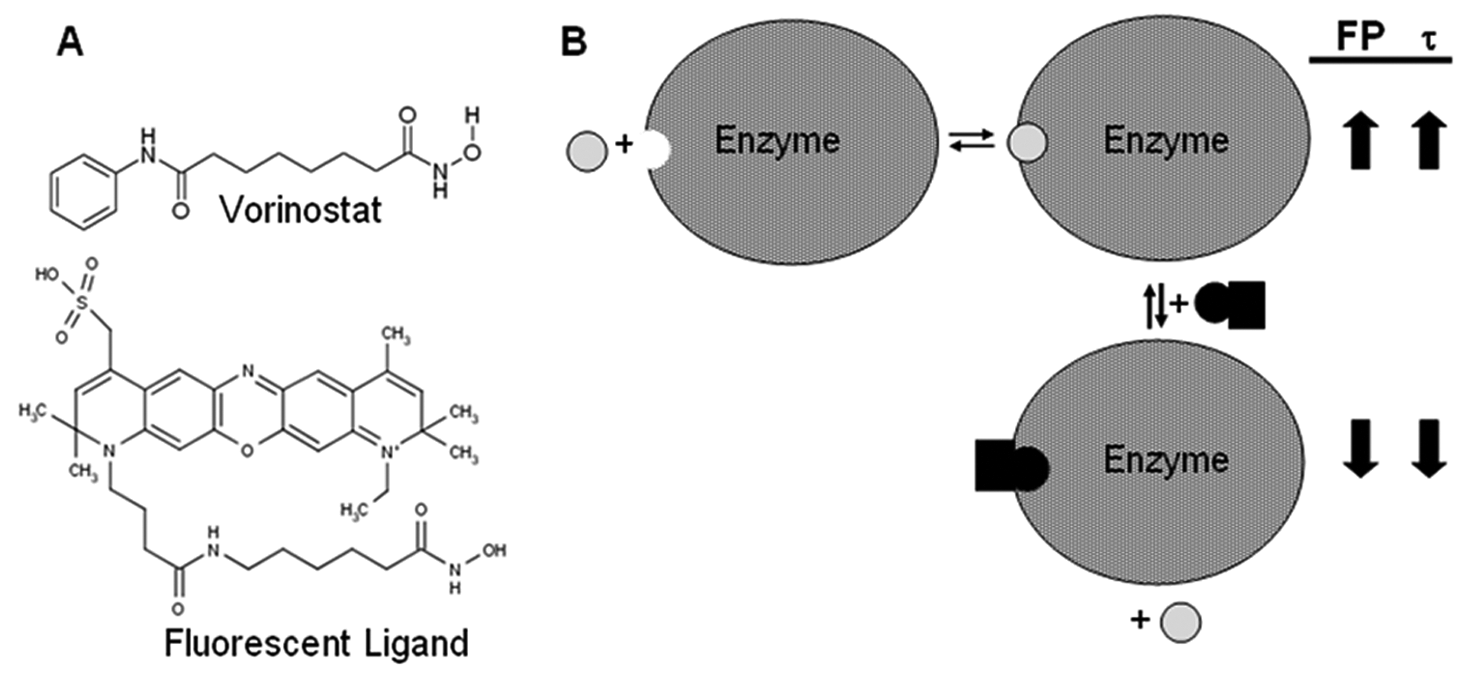
Scheme of the dual-parameter competition assay. (
Dose–response curves
Dose–response experiments with HDAH protein were carried out for hit compounds resulting from the screen using the one-step enzyme activity assay or dual-parameter competition assay at different compound concentrations. The highest final concentration in each concentration series was 100 µM. In the case of the human HDAC isoforms, trypsin cannot be added to the deacetylation reaction because HDACs are prone to proteolytic hydrolysis by trypsin. Therefore, with human HDACs, a two-step enzyme activity assay was used that decouples the deacetylase reaction and the fluorogenic detection reaction with trypsin. 22 In HDAH, HDAC1, and HDAC6 assays, Boc-Lys(Ac)-AMC was used as substrate at 50, 80, and 100 µM, respectively. Boc-Lys(TFA)-AMC was used for HDAC7 at a concentration of 60 µM. The concentrations comply with Km values. The final concentrations of HDAC1, 6, and 7 were 4.5, 2.8, and 0.3 nM, respectively. The dose–response curves were fitted to the four-parameter logistic function 23 using the program Origin (OriginLab Corp., Northampton, MA).
The displacement of the Atto-hydroxamate ligand from its complex with human recombinant HDAC8 was measured as follows: The complex was preformed by mixing 3.9 µM HDAC8 and 50 nM of the fluorescent ligand in FB188 buffer. Then different concentrations of inhibitor (e.g., suberoylanilide hydroxamic acid [SAHA]) were added, resulting in a dose-dependent change of fluorescence anisotropy.
Calculation of protonation status
Chemical structures were drawn and their protonation status was calculated using Marvin Sketch 5.4.0.1. from ChemAxon Ltd. (Budapest, Hungary).
Calculation of similarity between chemical structures
The Tanimoto coefficient, a commonly used measure for the similarity between chemical structures, 24 was calculated on the basis of chemical hashed fingerprints used by JChem (ChemAxon Ltd.). The Tanimoto coefficient between two chemical hashed fingerprints a and b with k dimensions is defined as
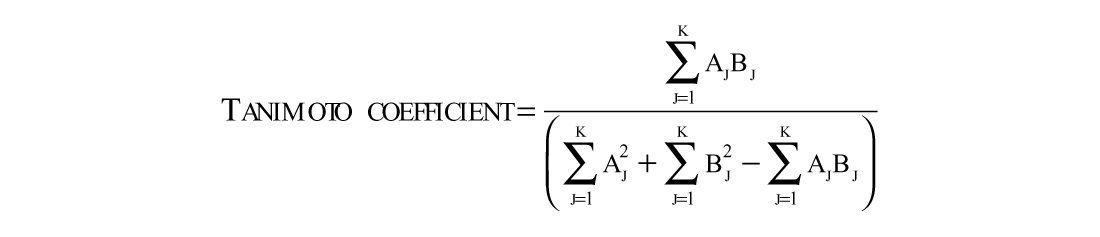
The Tanimoto similarity can only be applied to a binary variable. The highest similarity equates a Tanimoto coefficient of 1.0.
Results
Performance of the dual-parameter competition assay
Fluorogenic assays that generate free amino-coumarin dyes as a measure of enzyme activity are prone to autofluorescent and quenching test compounds. In many cases, amino-functional rhodamine dyes yield better performances because they are excited in the visible range. Binding assays may provide complementary information about the direct interaction of target and test compounds. Our aim was the development of a robust binding assay for HDAH and human HDACs and to demonstrate its suitability for high-throughput screening (HTS). To reduce potential optical interferences with test compounds, we chose a largely red-shifted dye (Atto 700) as the probe. To further increase the robustness of the optical readout, we combined fluorescence anisotropy and fluorescence lifetime. The resulting dual-parameter competition assay and its extraordinary robustness against up to 50 µM 7-amino-4-methyl-coumarin have been described recently. 20 The assay is based on simultaneous changes in fluorescence anisotropy and FLT upon reversible binding of a fluorescent ligand to an HDAC homolog (see Fig. 1 ). In this study, the dual-parameter competition assay was used to screen the LOPAC library to obtain data under realistic screening conditions. Fluorescence anisotropy and FLT have been measured consecutively from the same samples, whereas the HDAH screen was carried out in two runs on different days. The mean fluorescence lifetimes of the free ligand and its complex with HDAH were 1.56 and 1.98 ns, respectively. The HDAH concentration used throughout the screening campaign was determined from a titration curve of Atto-hydroxamate-ligand with HDAH to be 2 µM (see Fig. 2 ). The performance of the second run after compound number 880 was considerably better than that of the first run. The corresponding Z′ factors were calculated to be 0.59 (anisotropy) and 0.62 (FLT) for the first run and achieved an excellent 0.85 (anisotropy) and 0.87 (FLT) throughout the second run. This obvious improvement was mainly accomplished by a thorough readjustment of the pipetting robot, including replacement of old pipetting needles and optimizing the settings for solvent class within the robots software.
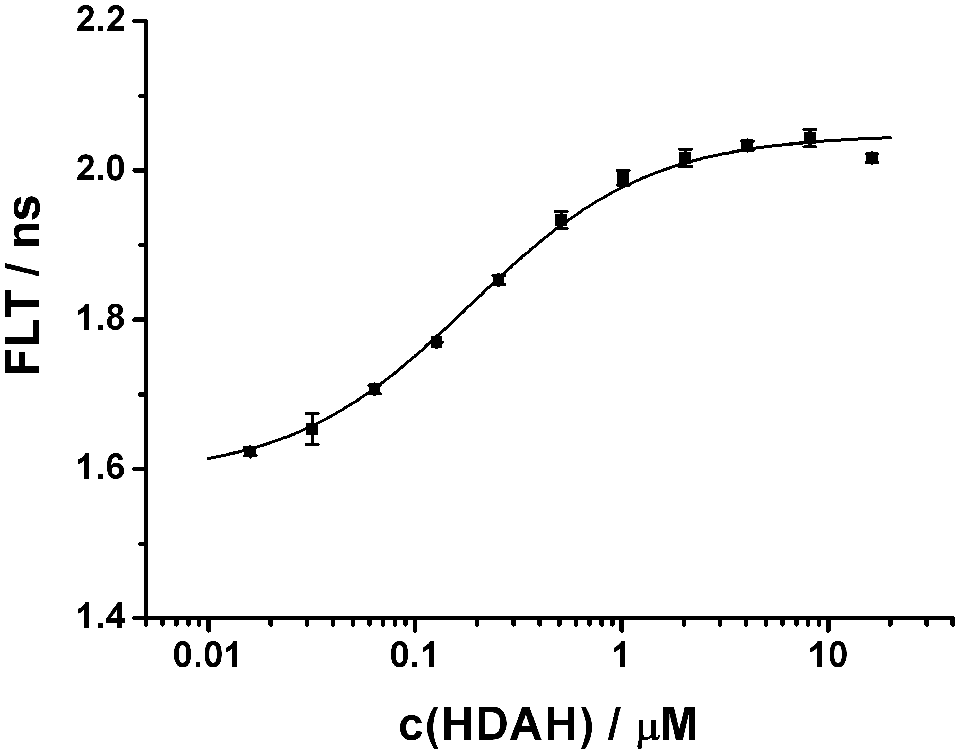
Binding of HDAH to the Atto-hydroxamate ligand. In total, 50 nM Atto-hydroxamate ligand is titrated with increasing concentrations of HDAH in FB188 buffer at 21 ± 2°C. The fluorescence lifetime (FLT) is plotted versus the concentration of HDAH. The data points represent means of three measurements. The dissociation constant Kd of 0.2 ± 0.02 µM was obtained from fitting the data to a simple bimolecular binding model (smooth line).
Performance of the enzyme activity assay
In addition, the LOPAC library was screened separately using a standard fluorogenic enzyme activity assay. 12 The final concentration of Boc-Lys(Ac)-AMC substrate, 10 µM, was well below the Michaelis constant of 90 µM, ensuring high sensitivity of the assay, and the concentration of HDAH was chosen to obtain a Z′ factor as high as 0.82. Although the nominal Z′ factor between samples in the presence and absence of HDAH suggested excellent performance of the enzyme activity assay, the data of the enzyme activity screen showed significantly more variation than the anisotropy and FLT data of the binding assay screen, which had about the same Z′ factor ( Fig. 3 ). One reason for this observation is the extraordinary low standard deviation of the negative control samples of the enzyme activity assay (see Fig. 3C ).
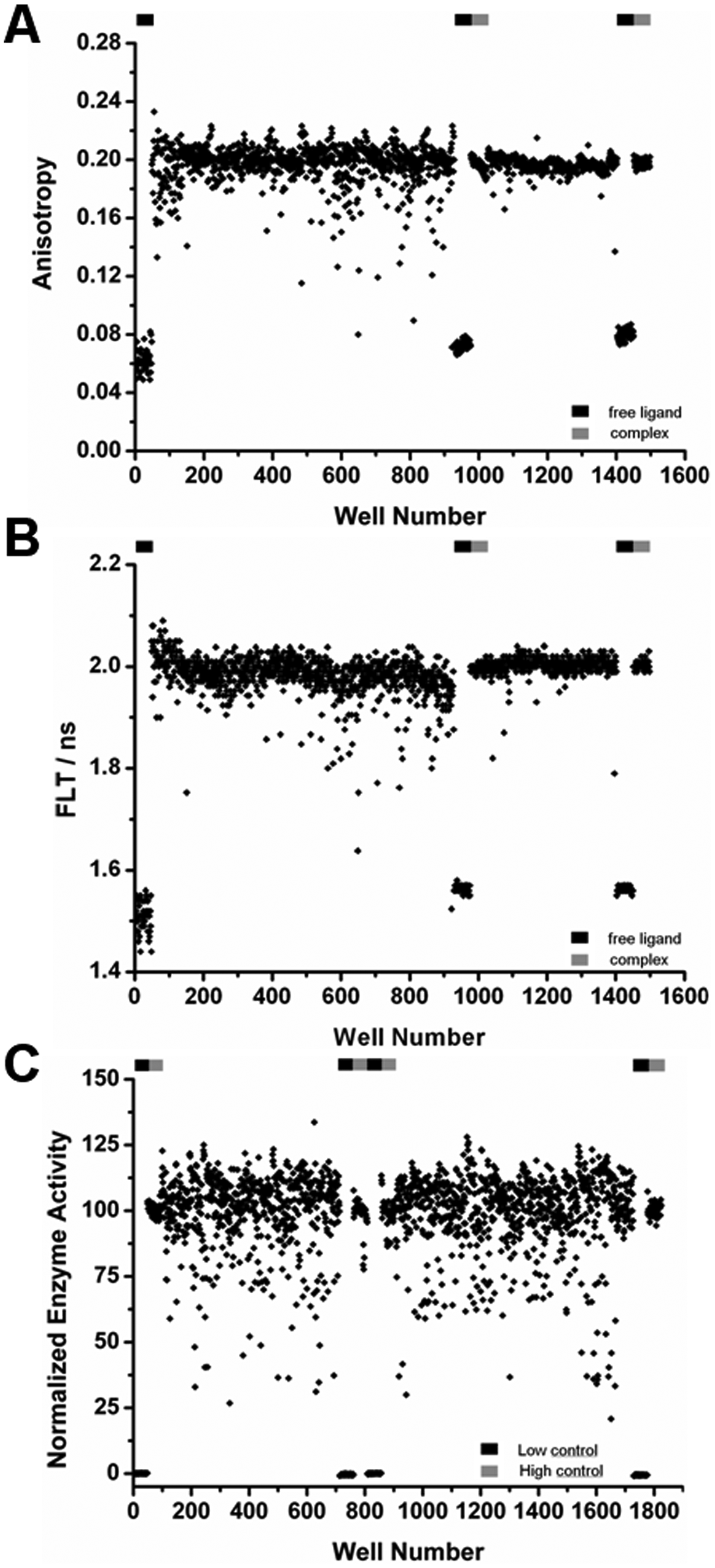
HDAH screen. The LOPAC library was screened against HDAH using the dual-parameter competition assay (
Screening results
The screening data of both types of assays, the dual-parameter competition assay and the enzyme activity assay, yielded three data sets. Two data sets, fluorescence anisotropy and lifetime, resulted from the dual-parameter competition assay and one data set from the enzyme activity assay. All three data sets are shown in Figure 3 . A comparison of the assay performance and robustness of the dual-parameter competition and the enzyme activity assay with respect to all three readouts showed comparable Z′ factors of 0.82 to 0.87 (second run of binding assay only) and confirmation rates of about 60% to 70%. But in contrast to fluorescence anisotropy and FLT, the threshold of the enzyme activity assay was set much lower at a normalized activity of 40 due to the variance of the data. Fluorescence anisotropy and FLT showed a strong correlation and only few hits (0.6%), deviating significantly from the mean values of neutral controls at 100. Compounds with a normalized anisotropy of less than 70 and FLT less than 92.5 were defined to be hits and marked by the shaded rectangle in Figure 4 . The most potent hit compounds were oleic acid (O 1008) and PPARδ agonist L-165,041 (L 2167). The compound identification in brackets refers to the catalog number of Sigma. These two compounds caused a strong decrease in FLT, fluorescence anisotropy, and enzyme activity. Both compounds carry a carboxylic acid group that may interact with the zinc ion inside the active site of HDAH. Weaker hits comprise compounds I 0404, I-117, M 6383, M-166, and D-142 (see Fig. 4 ). Furthermore, A 9561 and F-114 were added to the hit list because of a normalized FLT-value below 92 and a normalized enzyme activity less than 70.
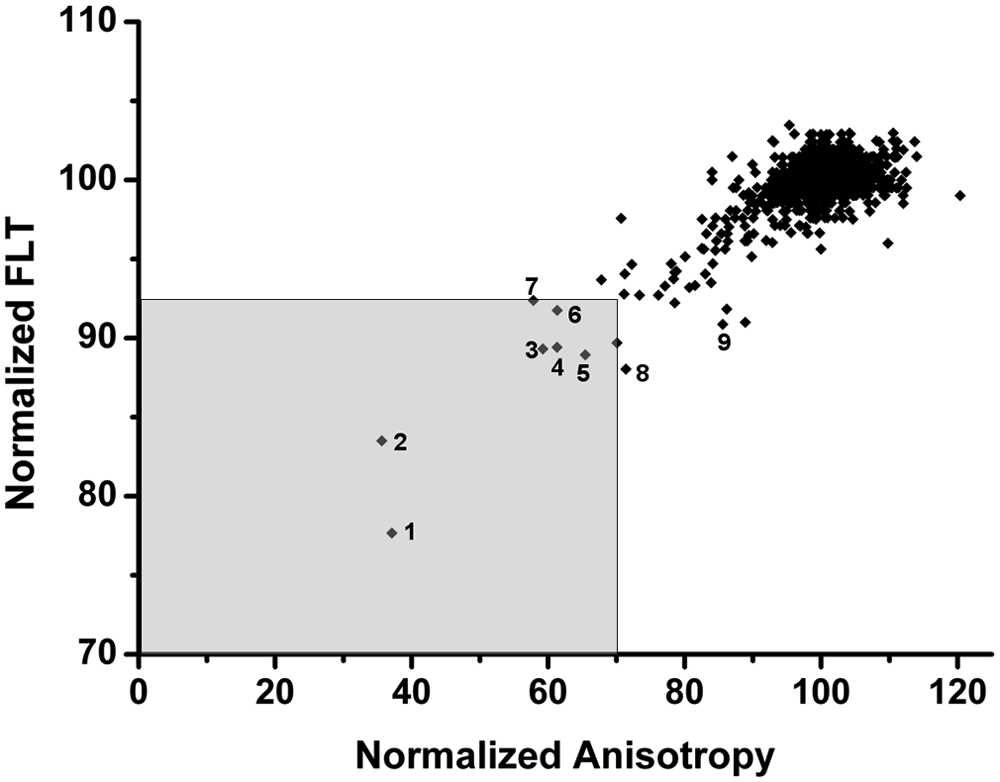
Hit criteria for the dual-parameter competition assay. The normalized fluorescence lifetime (FLT) of all LOPAC compounds is plotted versus the corresponding fluorescence anisotropy. Hits are defined by the shaded rectangle in the lower left corner. Hit compounds (catalog numbers of LOPAC library are given in brackets) comprise 1 (O 1008), 2 (L 2167), 3 (I 0404), 4 (I-117), 5 (M 6383), 6 (M-166), and 7 (D-142). Compounds 8 (A 9561) and 9 (F-114) have been added to the hit list because of normalized FLT <92 and normalized enzyme activity <70.
In a direct comparison of both assay formats, there were also compounds such as O 1008, I 0404, I-117, and M-166 emerging as hits only from the binding assay-based screen and showing more than 80 of normalized enzyme activity (see Fig. 5 ). On the other hand, compounds such as C 0253, A 7410, F 2927, W 108, and U 5882 turned out to be negative in the binding assay-based screen (FLT >95) and strongly positive in the enzyme activity-based screen (enzyme activity <40).
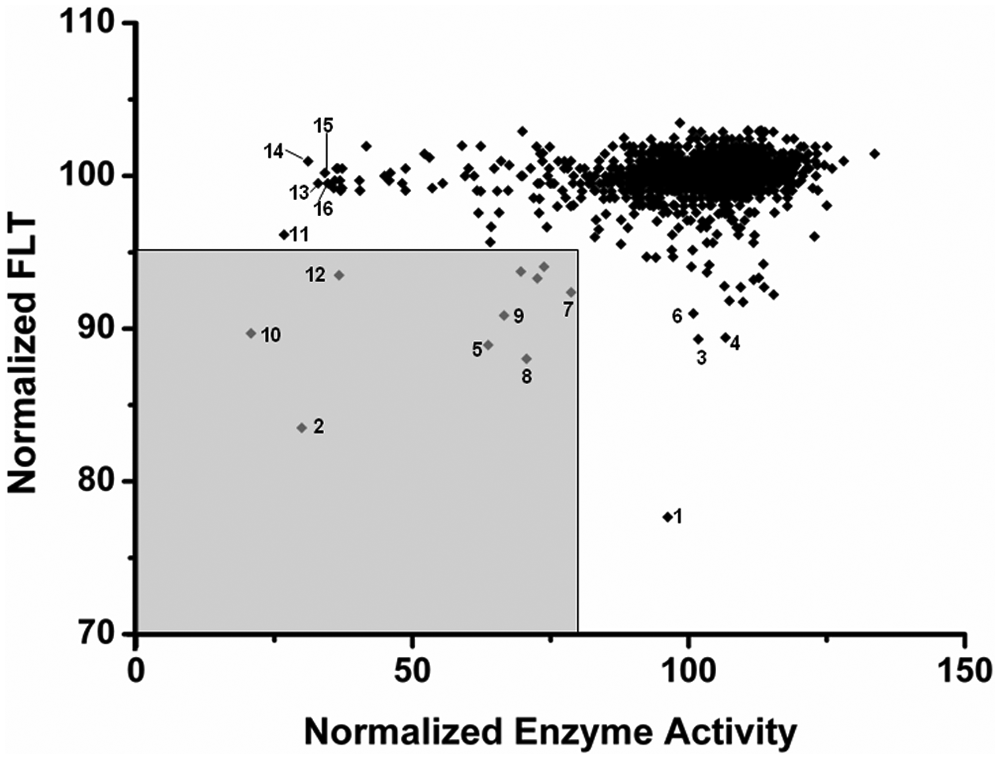
Comparison of dual-parameter competition and enzyme activity assay. The normalized fluorescence lifetime (FLT) of all LOPAC compounds tested versus HDAH is plotted versus the corresponding enzyme activity. Compounds marked by the shaded rectangle in the lower left corner are supposed to be the most promising hits. Numbering of data points corresponds to Figure 3 plus compounds 10 (T 9177), 11 (C 0253), 12 (P 4509), 13 (A 7410), 14 (F 2927), 15 (W 108), and 16 (U 5882).
To reconfirm the results, all hit compounds were retested using both assay formats. The corresponding IC50 values are summarized in Table 1 . Eight of nine hit compounds identified by the dual-parameter competition assay were confirmed in both assay formats with IC50 values of less than 60 µM. The best two compounds (O 1008, I 0404) showed IC50 values under 10 µM. Eight of 13 compounds appearing as very potent hits only in the enzyme activity screen (enzyme activity <40 and FLT >95) were confirmed. On the other hand, four of five tested exclusive FLT-based hits (FLT <92.5 and enzyme activity >70) could be confirmed. Taking a broad intersection of FLT and activity-based hits (FLT <95 and enzyme activity <80) defined 10 mutual hits (see Fig. 5 ); 7 of them were retested and 6 confirmed (see Table 1 ).
Selectivity Profile of HDAH Hit Compounds
Furthermore, to see differences between the bacterial HDAH and the human HDAC isoforms, the most potent HDAH inhibitors were profiled on HDAC1, 6, and 7 using the two-step enzyme activity assay. Similar structures of the confirmed hit P 4509 were included in the analysis as well. The dose–response curves of L-165,041 (L 2167) tested in HDAH and HDAC enzyme activity assays are shown in Figure 6A . None of the tested compounds turned out to be a strong inhibitor of any HDAC isoform in the lower micromolar range. However, oleic acid, L 2167, N-lauroyl-(L)-phenylalanine, N-lauroyl-(D/L)-phenylglycine, A 9561, and the methylketone 45E09, which was discovered earlier by Wegener et al. 12 in an enzyme activity-based HDAH screen of natural products, were able to inhibit class II HDACs at IC50 values of less than 50 µM (see Table 1 ). These compounds also showed a moderate isoform selectivity for class II HDACs because class I representative HDAC1 was not or only very weakly affected (IC50 >100 µM). Although compound L 2167 was active against both class IIa HDAC7 and class IIb HDAC6, the N-lauroyl amino acids turned out to be rather selective for class IIa HDAC7 (see Table 1 and Fig. 6B ). In contrast, compounds O 1008 and A 9561 were shown to be weak but rather selective inhibitors of class IIb HDAC6.
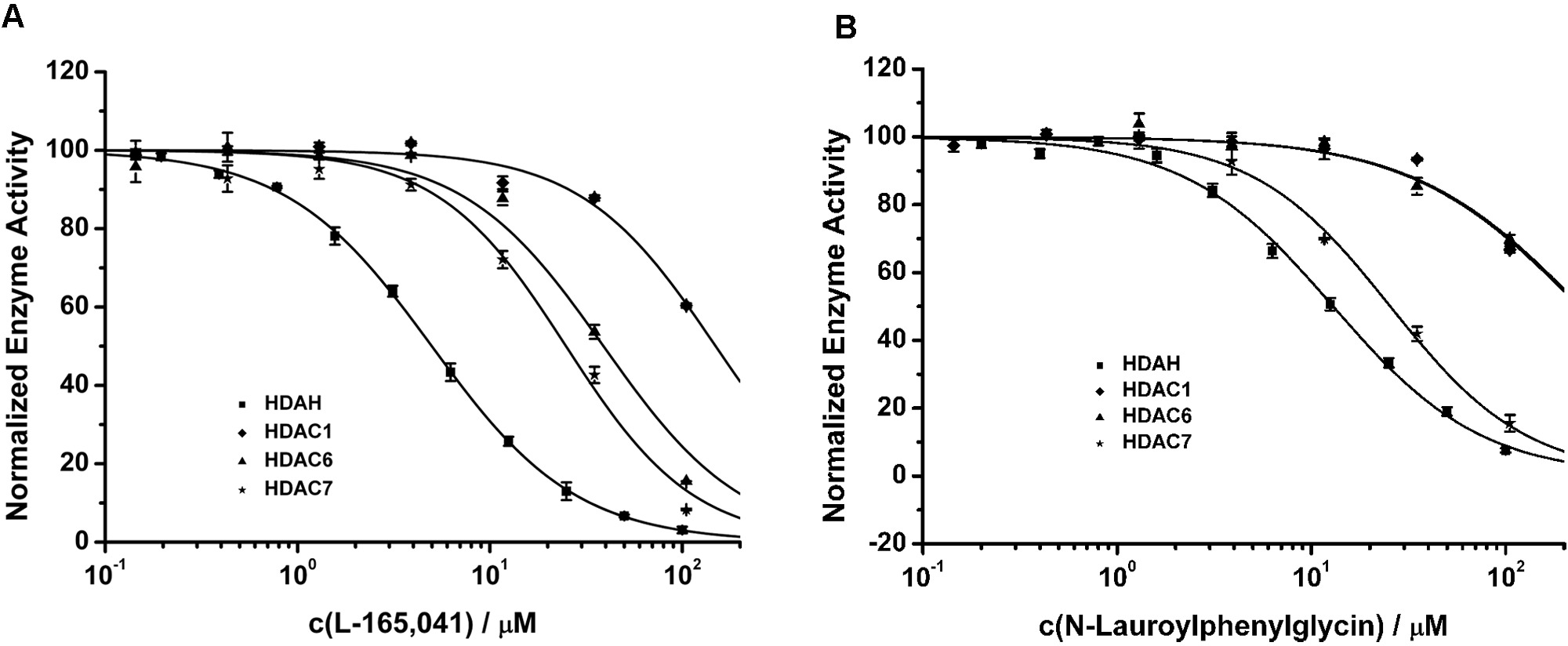
HDAC selectivity profiles of L-165,041 (
Competitive HDAC8 binding assay
To demonstrate the multiple usability of the Atto-hydroxamate ligand for HDAC assays, its binding to recombinant human HDAC8 was investigated. It could be shown that addition of HDAC8 to the fluorescent ligand leads to an increase in anisotropy, whereas FLT was not influenced. No saturated binding could be achieved up to 3.9 µM HDAC8. The binding degree at this concentration was estimated to be about 70% by fitting and extrapolating the data. Specific binding of the Atto-hydroxamate ligand to the active site of HDAC8 was demonstrated by the displacement of the Atto conjugate using increasing concentrations of SAHA ( Fig. 7 ). Fitting of the competitive binding curve to a simple one-site binding model 20 revealed that the fraction of binding active HDAC8 was clearly smaller than 100%. Depending on the fraction of functional HDAC8, a Kd value of about 0.1 to 0.5 µM could be deduced for SAHA.
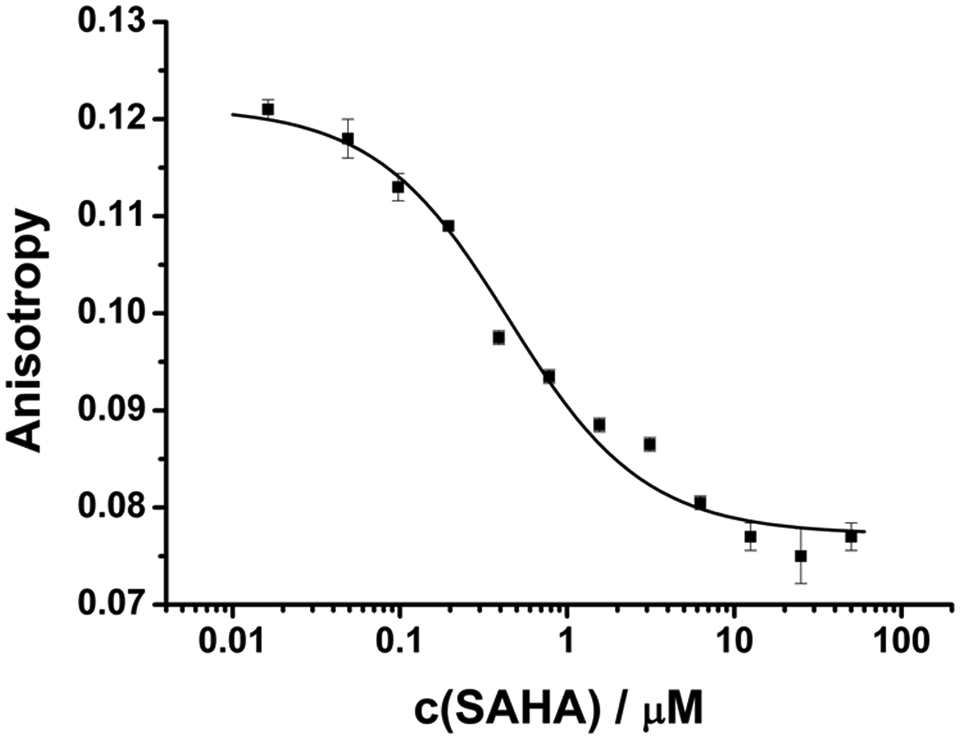
Dose–response curve of suberoylanilide hydroxamic acid (SAHA) binding to recombinant human HDAC8. The experiments were performed under standard assay conditions using 3.9 µM HDAC8 and 50 nM Atto-hydroxamate ligand. The Kd value of SAHA was estimated to be 0.1 to 0.5 µM. Each data point represents five measurements.
Discussion
The LOPAC compound library was screened to validate the high-throughput suitability of the recently developed dual-parameter competition assay based on fluorescence anisotropy and FLT. 20 The FLT readout of the assay was shown to be robust against up to 50 µM 7-amino-4-methylcoumarin previously. 20 To further validate the assay with respect to HTS suitability, the LOPAC library was tested and the resulting data were compared to a conventional fluorogenic assay. The binding assay screen was subdivided into two runs (see Fig. 3A , B ). The first run (880 compounds) showed just a moderate performance with a Z′ factor of 0.59 for fluorescence anisotropy and 0.62 for FLT. These Z′ factors were comparable to our first description of the assay with 0.66 for anisotropy and 0.62 for FLT. 20 Further optimizing the liquid handling of the pipetting robot improved the assay performance significantly to very good Z′ factors of 0.85 for anisotropy and 0.87 for FLT that could be achieved in the second run. Remarkably, the excellent Z′ factor for FLT results from a 0.5-ns change from 1.56 to 1.98 ns upon binding of HDAH to the Atto-hydroxamate ligand. Most organic dyes that emit fluorescence in the far red range are relatively large conjugated structures featuring comparably short FLTs. This also implies just small absolute changes in FLTs, if the chemical microenvironment is changed upon binding. On the other hand, the perturbance of autofluorescent compounds, assay ingredients, or cellular components is greatly diminished at long wavelengths. Our dual-parameter competition assay demonstrates that changes as small as 0.5 ns are sufficient to achieve excellent assay performance and robustness if the dye can be excited at 635 nm or above. When comparing the assay performance of the dual-parameter competition with the enzyme activity assay, the Z′ factors regarding FLT, anisotropy, and enzyme activity data turned out to be quite similar (Z′ = 0.82–0.87). But the data of the enzyme activity screen appeared to be much noisier than the corresponding anisotropy or FLT data. Looking for the reason for this seemingly contradictory result revealed that the low control of the enzyme activity assay in the absence of HDAH exhibited essentially no variation. Therefore, it appeared to be useful to compare the signals of DMSO controls, which are complemented reaction mixtures in the absence of screening compounds, for all readout parameters. Because the variation of the FLT and anisotropy signal of preformed HDAH and Atto-hydroxamate ligand was clearly less than the variation of the normalized enzyme activity of uninhibited HDAH, the hit thresholds for FLT and anisotropy could be set closer to the DMSO controls than for enzyme activity.
The Atto-hydroxamate ligand probe bound to HDAH as well as HDAC8. In both cases, inhibitors such as SAHA were able to compete with the fluorescent ligand for binding to the corresponding enzyme, proving the potential of the ligand probe to serve as a multiple-use ligand for human HDACs. The reversible binding of the fluorescent Atto-hydroxamate ligand to HDAC8 is in agreement with the pan-inhibitor properties of SAHA with respect to human HDACs, which guided the synthesis of the ligand. The estimated Kd value for SAHA and HDAC8 of about 0.1 to 0.5 µM is consistent with reported IC50 values between 0.2 and 2 µM.25,26 In contrast to the HDAH assay, only the fluorescence anisotropy changed upon binding of the fluorescent ligand to HDAC8. Obviously, the Atto dye experienced a different chemical microenvironment at the surface of HDAH than in the complex with HDAC8. The channel to the active site of HDAH is longer (1ZZ1, 11.5 Å) than in HDAC8 (1T69, 7.8 Å). Therefore, the Atto dye may poke more out of the channel and have fewer or no contacts to the surface of HDAC8 as compared with HDAH. Consequently, no or less changes in FLT would be expected. On the other hand, changes of fluorescence anisotropy will still be measurable if the chemical microenvironment remains unvaried. This finding emphasizes that the chemical microenvironment of sensing dyes and with it changes in FLT upon binding is hard to predict. If both fluorescence parameters change upon binding, hits can be identified more reliably and with lower false-positive rates. In the HDAH binding assay, FLT and fluorescence anisotropy were highly correlated, as shown in Figure 4 . This reflects the high robustness of FLT and anisotropy as readout parameters but is also due to the fact that the same molecular event, competitive binding, is measured in the same well at the same time.
The correlation between binding assay (FLT) and enzyme activity based on screening data was much less obvious ( Fig. 5 ). Since these types of assays measured different events, it was not expected beforehand that the data of both assay formats would correlate completely. Allosteric inhibitors of enzyme activity, for instance, would not necessarily act as a competitor of the fluorescent ligand probe. However, compounds that displace the fluorescent ligand from the active site are believed to inhibit enzyme activity as well. The relationship between binding and inhibition was also blurred due to the variance of the enzyme activity assay. More precise activity data have been obtained from dose–response curves of hit compounds. The corresponding IC50 values of confirmed hits showed good agreement between all three readout parameters ( Table 1 ). Eight of nine hits of the dual-parameter competition assay-based screen could be confirmed, underlining the very good performance of this assay format. Three compounds that appeared as clear hits in the dual-competition binding assay—O 1008, I 0404, and I-117—could be confirmed in both assay formats, although they were false negative in the enzyme activity screen probably due to its broad variance ( Fig. 5 ). On the other hand, hits such as C 0253, A 7410, and F 2927 were found in the enzyme activity-based screen and confirmed in both assay formats but were initially overlooked in the primary binding assay screen ( Fig. 5 ). Although exclusive hits for just one assay method would be potentially interesting in terms of the mode of interaction between enzyme and compound, all hits could be confirmed in both assay formats (see Table 1 ). The most potent hit found in both assays was L-165,041 (L 2167). The inhibition of HDAH by L-165,041 in the lower micromolar range is somewhat surprising because the compound is known as a potent and selective peroxisome proliferator activator receptor δ (PPARδ) agonist. 27 L-165,041 was not only confirmed in both assay formats but also found to inhibit class IIb HDAC6 and class IIa HDAC7, showing IC50 values of 39 ± 1.5 and 25 ± 1.9 µM, respectively.
The PPARδ agonist affected class I HDAC1 significantly less (see Fig. 6A ). L-165,041 may interact with the catalytic Zn2+ of HDAH via its carboxylate group. Besides a Zn2+ chelating group, long alkyl chains appear to be an important factor for potent binding to HDAH. So one of the most potent HDAH inhibitors that has been found so far is the dodecanone derivative 45E09. 12 In fact, the most potent hit in this screening effort was oleic acid. In addition, palmitoyl-(D/L)-carnitine chloride was confirmed as a potent HDAH binding molecule. Two other long-chain acyl amino acids, N-lauroyl-(L)-phenylalanine and N-lauroyl-(D/L)-phenylglycine, which were included in the confirmative assays, were also found to bind to and inhibit HDAH. In contrast to oleic acid and palmitoyl-(D/L)-carnitine chloride, the N-lauroyl amino acids were selective for class IIa HDAC7. It may be instructive to consider the structurally related phenylalanine-containing hydroxamic acids reported by Schafer et al. 14 to be HDAC class IIb selective inhibitors. In these compounds, the acyl chain of N-lauroyl-(L)-phenylalanine is replaced by a C6-acyl chain that carries a hydroxamate moiety at its far-out end that is supposed to chelate the catalytic Zn2+. The methylketone 45E09 was shown to display 13% inhibition of rat liver extract HDACs, which is supposed to contain mainly HDAC1, 2, and 3, at 50 µM. 12 This is in agreement with our finding that 45E09 is rather ineffective on HDAC1. However, we could show that the compound is a selective HDAC class II inhibitor exhibiting IC50 values of 19 ± 5.4 µM for HDAC7 and 59 ± 10 µM for HDAC6. The amiloride A 9561, known as an inhibitor of Na+/Ca2+ and Na+/H+ exchanger, 28 was the only compound with selectivity for class IIb HDAC6. There were three analogues of A 9561 with Tanimoto coefficients higher than or equal to 0.8 within the LOPAC library, but only analogue A 7410 turned out to be effective against HDAH, as well. However, A 7410 did not inhibit human recombinant HDACs. The analogues of A 9561 differ only at position 5 of the pyrazine ring. At buffer conditions (pH 8.0), compound A 9561 was calculated to be deprotonated and neutral and may interact through its guanidinium group with the Zn2+ ion inside the catalytic site. 29 Although the potency of A 9561 against HDAC6 is rather low, the finding that A 9561 shows selectivity for HDAC6 but not for class IIa HDAC7 may be of certain interest and supplement the knowledge for rational design of selective HDAC6 inhibitors.
In summary, we have demonstrated that the dual-parameter competition assay is suitable for HTS and that a difference as small as 0.5 ns in FLT between positive and negative controls of a long-wavelength fluorescent probe is sufficient for the development of a high-performing screening assay. The employed Atto-hydroxamate ligand can also be used to assay HDAC8 and presumably other HDAC isoforms. A series of HDAH inhibitors was identified with the most potent compounds having IC50 values in the low micromolar range. Six compounds were also shown to be moderately effective and selective against recombinant human class II HDACs. Despite their low potency, the structural features of these compounds may inspire the development of more potent class II selective HDAC inhibitors.
Footnotes
Acknowledgements
This work was supported by the Center of Research (ZFE) of the University of Applied Sciences Darmstadt. The encouragement of Thomas Leiser and Jan Brod during screening is gratefully acknowledged.