
Abstract
Fluorescence lifetime (FLT)–based assays have developed to become highly attractive tools in drug discovery. All recently published examples of FLT-based assays essentially describe their use for monitoring enzyme-mediated peptide modifications, such as proteolytic cleavage or phosphorylation/dephosphorylation. Here we report the development of competitive binding assays as novel, inhibitor-centric assays, principally employing the FLT of the acridone dye Puretime 14 (PT14) as the readout parameter. Exemplified with two case studies on human serine proteases, the details of the rationale for both the design and synthesis of probes (i.e., active site–directed low-molecular-weight inhibitors conjugated to PT14) are provided. Data obtained from testing inhibitors with the novel assay format match those obtained with alternative formats such as FLT-based protease activity and time-resolved fluorescence resonance energy transfer–based competitive binding assays.
Introduction
The use of fluorescence lifetime (FLT) as the readout parameter for in vitro assays increases biochemical assay robustness, reducing artifacts stemming from well-to-well volume variations, turbidity by precipitating particles, and compound autofluorescence. This makes the readout superior to other optical readout formats. 1 In addition, while providing high data quality, FLT-based assays are homogenous and scalable and therefore compatible with medium- and high-throughput experiments. With a predictable size of the assay window based on rationally derived rules for substrate design and the ease of substrate synthesis, this assay format offers a convenient and cost-efficient alternative to other established formats. 2 Assays based on monitoring enzyme-mediated peptide modifications that directly affect the FLT of a reporter dye have been successfully introduced for routine inhibitor profiling in automated processes within the hit-to-lead and lead optimization phases for enzyme classes such as proteases, kinases, and phosphatases.3–7
The assay principle presented here employs changes in FLT, enabling determination of binding potency of compounds to the active site of an enzyme through the displacement of a conjugated binding probe. Using such an approach, compound screening in cases in which enzymes exhibit only low activity with known substrates is possible. Furthermore, this new assay format can be used as an orthogonal assay format for, for example, validating binding of hits found by high-throughput screening (HTS) campaigns employing enzymatic activity assays.
However, the development of competitive binding assays for proteases as outlined here is an inhibitor-centric approach. It requires the availability of active site–directed binders, that is, low-molecular-weight (LMW) inhibitors with potency ideally in the double-digit nanomolar range and structural features that allow for (1) site-directed conjugation of an FLT dye and (2) a molecular moiety able to quench the fluorescence of the FLT dye intramolecularly.
To illustrate this approach, we present details of the assay development for two human serine proteases. One of these is β-tryptase, which is considered to be a potential target for immune diseases (e.g., rheumatoid arthritis and ulcerative colitis).8,9
For the rational design of the FLT reporter probes, the co-crystal structures of the proteases with known LMW inhibitors revealed the binding mode of the inhibitors in the complexes and their molecular interactions. Based on this information, the Puretime 14 (PT14) dye was attached to the solvent-exposed sites of the inhibitors. This dye has been previously used in the enzymatic activity assays based on the FLT readout. The enzyme-buried part of the inhibitor includes a nitrogen-containing, heterocyclic system, structurally similar to the indole moiety of tryptophan. In previously established FLT-based enzymatic activity assays, tryptophan effectively quenched the dye’s fluorescence. Thus, it was hypothesized that for the free inhibitor conjugate, the dye’s fluorescence could be quenched by interaction with the heterocyclic system. In contrast, in the protein-bound conjugate, the heterocyclic moiety should not be able to interact with the fluorophore; therefore, the emission signal of the solvent-exposed dye would remain unaffected.
This concept was experimentally proven. In both assays, a remarkable difference in the dye’s FLT of greater than 5 ns, between the bound and unbound state of the probe, was observed. Importantly, compared with the parent inhibitors, the affinity of the probes for their targets appeared almost unchanged upon conjugation to the dye, as predicted by the rational design. An excellent assay signal window enabled the subsequent development of the FLT-based displacement assays. The assays were validated with a selection of known inhibitors. The half-maximal effective concentration (EC50) data obtained with the novel displacement assay compare well with those obtained by previously established FLT and time-resolved fluorescence resonance energy transfer (TR-FRET)–based assays.
Materials and Methods
FLT Probes
The FLT dye PT14, required for the synthesis of both probes, was prepared in two steps by coupling 6-(9-oxoacridin-10(9H)-yl)hexanoic acid (as described in WO 2005/012901) with the commercially available tert-butyl (4-aminobutyl)carbamate, followed by preparative high-performance liquid chromatography (HPLC) purification and Boc deprotection.
The FLT probe 1 for β-tryptase ( Fig. 1 ; UPLC purity 100%) was prepared in house by coupling N-(4-aminobutyl)-6-(9-oxoacridin-10(9H)-yl)hexanamide (PT14) to the carboxylic acid function of the N-Boc precursor 1-((3S,5S)-1-(4-(((tert-butoxycarbonyl)amino)methyl)benzoyl)-5-((2-methyl-1H-indol-5-yl)carbamoyl)pyrrolidin-3-yl)-1H-1,2,3-triazole-4-carboxylic acid, followed by preparative HPLC purification and Boc deprotection.
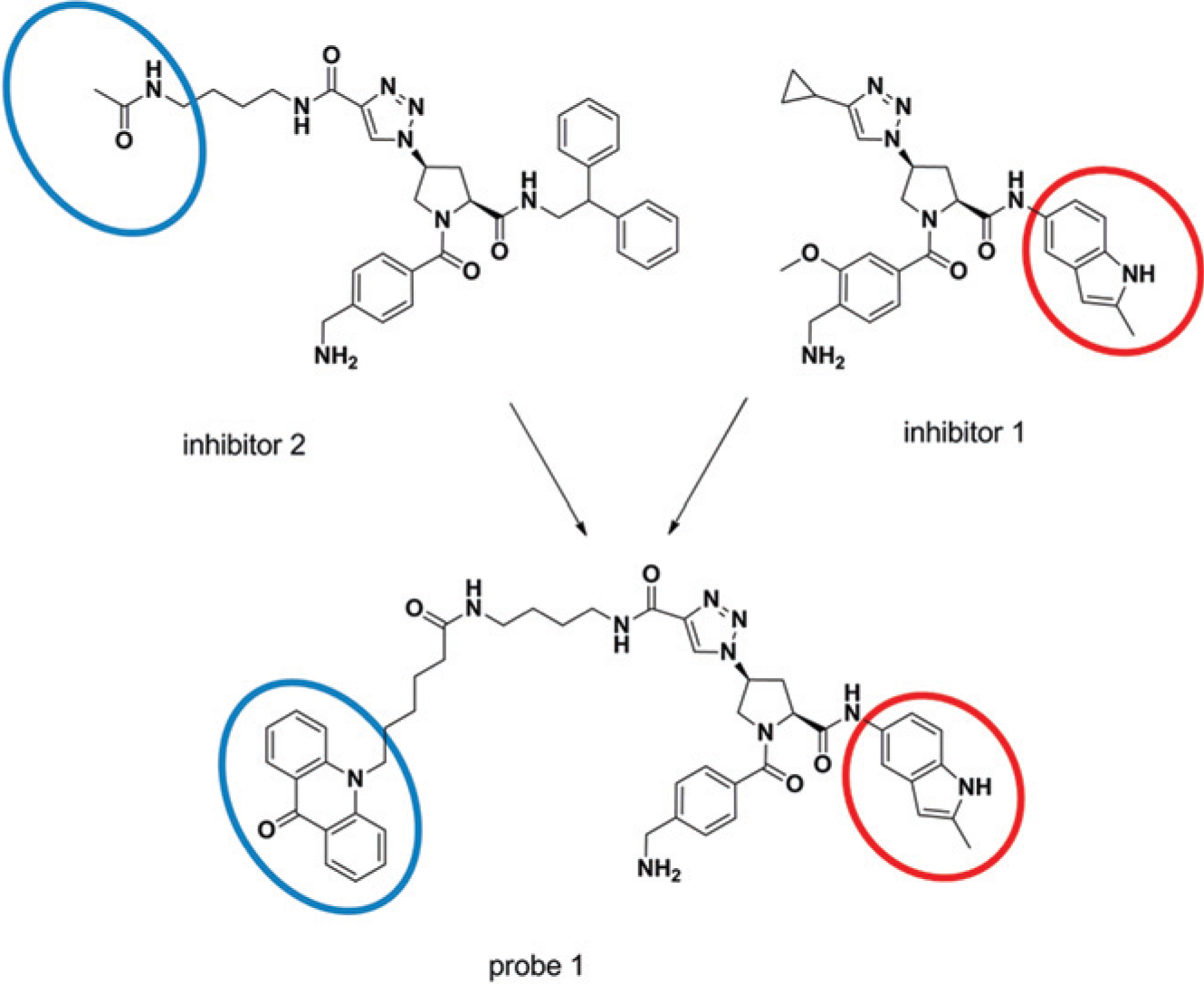
Schematic representation for illustrating the synthesis of the fluorescence lifetime (FLT) probe 1 for the β-tryptase competitive binding assay. Both inhibitor 1 and inhibitor 2 are active-site–directed inhibitors of β-tryptase characterized by IC50 values in the activity assay of 36 and 40 nM, respectively, determined with the enzyme activity assay. The inhibitor structures were merged, and the product was conjugated to the dye PT14 to yield the FLT probe 1. The blue and the red circle highlight the FLT dye PT14 and the molecular moiety putatively quenching PT14’s fluorescence, respectively.
The FLT probe 2 for the second serine protease (structure not shown) was prepared as described above for probe 1.
The concentrations of the FLT probes in solution were determined by absorption measurements using the molar extinction coefficient of ϵ = 7580 M−1cm−1 (at 394 nm in aqueous solution) for PT14.
Enzymes
Recombinant human lung tryptase was purchased from Promega (G563A, Lot 26023802; E.C. 3.4.21.59; Promega BioSystems, Sunnyvale, CA). The second human serine protease was recombinantly produced in house in its active form and stored as a 38.48 µM (1 mg/mL) stock solution in 50 mM Tris/HCl buffer at pH 8.0, containing 100 mM NaCl at −80 °C.
Buffer Conditions and Liquid Handling
The assay for β-tryptase was conducted in assay buffer comprising 50 mM Tris/HCl at pH 7.3, 50 mM NaCl, 0.05 % (w/v) CHAPS, and 50 mg/L Heparin Liquemin (Drossapharm, Basel, Switzerland).
The assay for the second protease was conducted in assay buffer comprising 50 mM HEPES/NaOH at pH 7.4, 2.5 mM MgCl2, 0.05 % (w/v) CHAPS.
All protein and probe containing solutions were handled in Maxymum Recovery tubes (Axygen Scientific Inc., Union City, CA). Compound, enzyme, and the substrate solutions were transferred to 384-well plates (black microtiter 384 plate, round well, Cat. No. 95040020; Thermo Electron Oy, Finland) by means of a CyBi-Well 96-channel pipettor (CyBio AG, Jena, Germany).
The reference inhibitors were dissolved in 90% (v/v) DMSO/water and kept at a stock concentration of 10 mM at 4 °C.
Instrumentation for Fluorescence Measurements
Fluorescence measurements were conducted using an Ultra Evolution FLT reader (TECAN, Maennedorf, Switzerland) or on the Fluospec FL plate reader (Assaymetrics, Cardiff, UK).
For TR-FRET measurements, the Tecan Ultra Evolution reader was equipped with bandpass filters, 340 nm (35 nm bandwidth) and 670 nm (25 nm bandwidth) for fluorescence excitation and emission acquisition, respectively. The measurement of fluorescence emission at 670 nm was time gated (70 µs) to reduce short-lived fluorescent background. The fluorescence intensity of each well was averaged over 30 flashes per measurement.
For lifetime measurements, the excitation light source was a semiconductor laser at 405 nm, producing picosecond light pulses with a selected repetition frequency of 10 and 5 MHz for the Tecan Ultra and the Assaymetrics Fluospec FL, respectively. The emission was collected through a 450 nm bandpass filter with 25 nm bandwidth (Tecan Ultra) and a 438 nm bandpass filter with 24 nm bandwidth (Fluospec), respectively. The reading time per well was typically 1 s, yielding approximately 1000 counts in the peak channel. The instrument control software carried out the initial curve fitting automatically, providing FLTs characterizing the recorded fluorescence intensity decay curve by a bi-exponential decay function. 10
The second, longer FLT, τ2, obtained from a bi-exponential fit of the fluorescence intensity decay curves, was used as an assay readout for the parameter for the FLT-based competitive binding assays of both proteases.
Determination of the Dissociation Constant for the Probe Binding to the Enzyme
The equilibrium constant for dissociation, KD, characterizing the affinity of the probe to the enzyme, was determined by titration of the protein (low pM to mid µM range) at constant probe concentration (20 nM for tryptase, 25 nM for second serine protease). The second, longer FLT resulting from the fit of a bi-exponential decay model to the fluorescence intensity decay data (raw data) was used as the readout parameter for subsequent calculations.
Calculations were made under the assumption of an equimolar binding interaction of the probe and the receptor. The bound and unbound state of the probe, characterized by the distinct FLTs τbound and τfree, respectively, and using the experimentally determined FLT τ2 can be expressed as the linear combination
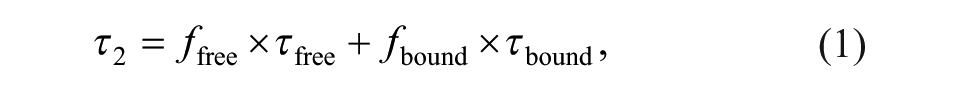
where ffree and fbound are the relative amounts (fractions) of the probe in the free and the unbound state, respectively.
The concentration of the receptor in the bound state, [CRbound], can be calculated as
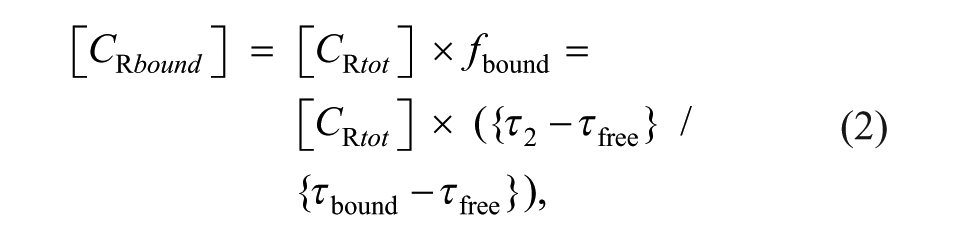
where [CRtot] is the total receptor concentration.
The concentration of the receptor in the bound state can be calculated for that setting by the law of mass action:
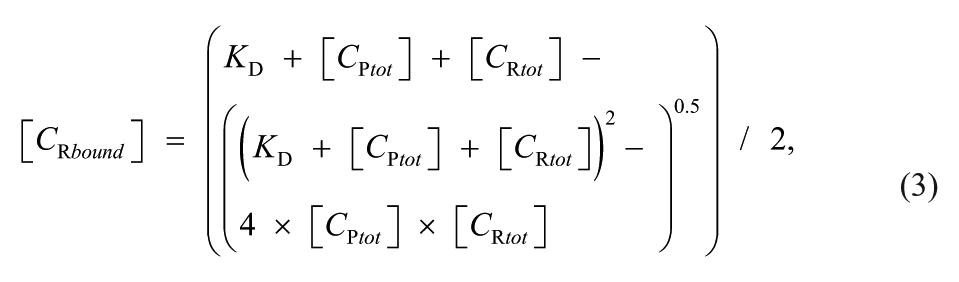
where KD is the equilibrium constant for the dissociation.
The combination of eqs 2 and 3 yields the correlation of the FLT τ2 determined in the experiment and the equilibrium constant KD, characterizing the affinity of the probe to the receptor:
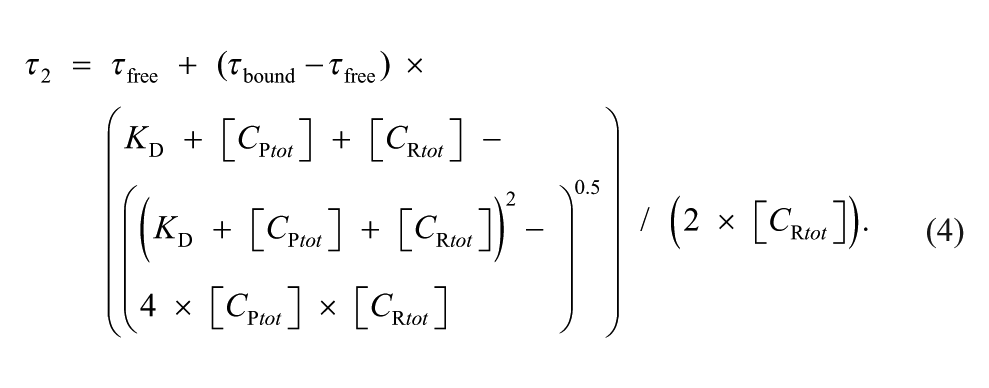
In eq 4, the assay window is described by the overall change in the FLT (τbound – τfree).
For the determination of the equilibrium constants for dissociation, KD, the experimental data were fitted using the nonlinear regression program Origin 8.5.0 SR1 (OriginLab Corporation, Northampton, MA).
Determination of Values for Half-Maximal Effective and Inhibitory Concentrations from Dose-Response Curves
For the determination of EC50 values, the assays were performed at room temperature in 384-well plates with a total assay volume of 25.25 µL per well. The test compound was dissolved in 90% (v/v) DMSO/water. For the assays, 250 nL of the 90% (v/v) DMSO/water solution or compound solution was added per well, followed by the addition of 12.5 µL protease solution (protease in 1× assay buffer).
The final assay concentration of the enzymes was nominally 20 nM. After 1 h of preincubation at room temperature, the competition for binding was started by the addition of 12.5 µL probe solution (probe dissolved in assay buffer). The final concentration of the probes in the assays was 25 nM. After the addition of the probe solution, the final DMSO concentration in the assay was 0.9% (v/v). The effect of the probe on the preestablished compound-enzyme equilibrium was determined after 1 h (t = 60 min). The EC50 value was calculated from the plot of percentage of enzyme saturation versus the inhibitor concentration by a logistics fit according to
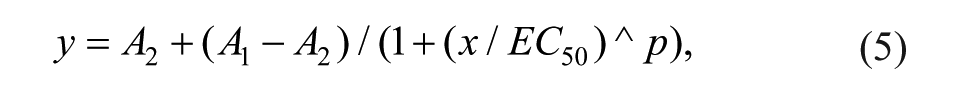
where y is the percentage saturation value at the inhibitor concentration x, A1 is the lowest saturation value (i.e., 0%), and A2 is the maximum saturation value (i.e., 100%). The exponent, p, is the Hill coefficient.
Values for the half-maximal inhibitory concentration, IC50 values, were calculated with an equation analogue to eq 5 as described previously. 3
The fitting of data with eq 5 was conducted with proprietary in-house software.
Tryptase Activity Assay
The enzyme activity assay-based monitoring of the cleavage of an artificial, short peptide substrate single labeled with PT14 as the FLT reporter dye was performed as described previously. 3
The fluorescent substrate Ac-Tyr-Ser-Ala-Lys↓C(PT14)-NH2 was purchased from Biosyntan (Cat. No. S-# 9048; Berlin, Germany). Within the substrate sequence given above, the arrow “↓” indicates the primary cleavage site for the protease confirmed by LC/MS. For the determination of IC50 values, the assay was performed at room temperature as described above with a final human β-tryptase concentration of 0.012 nM. After 1 h of preincubation of enzyme and inhibitors, the enzyme reaction was started by the addition of substrate (in assay buffer, final assay concentration was 1 µM). The effect of the compound on the enzymatic activity was obtained from the linear part of the progress curves and determined after 1 h (t = 60 min).
TR-FRET–Based Competitive Binding Assay for Second Serine Protease
The TR-FRET–based competitive binding assay for the second serine protease employed the protease site specifically labeled with biotin, enabling the binding of a Eu-streptavidin conjugate and a similar probe. The TR-FRET probe consisted of the same active site–directed binding moiety as the FLT probe 2 conjugated to the dye Cy5 instead of PT14.
For the determination of EC50 values, the assay was performed as described for the FLT measurements using biotinylated-protease (10 nM final concentration) as binding protein. After 1 h preincubation of enzyme and inhibitors at room temperature, a detection mix consisting of Eu-streptavidin conjugate (2 nM, final concentration) and a Cy5-labeled ligand (10 nM, final concentration, equivalent to its KD value) was added to each well. Competition for binding to the protease between the Cy5-labeled ligand and test compounds was allowed to proceed for 1 h to reach equilibrium before TR-FRET measurements as described above. Data were expressed as percentage inhibition of the maximal TR-FRET signal measured in the absence of competitor.
Results
Probe Design
For the design of an FLT-reporter probe, the fluorescent dye PT14 is ideally attached to the moiety of the inhibitor that is exposed to the solvent when the inhibitor is bound to the enzyme. The fluorescence emission of PT14 is sensitive to adjacent quenchers, such as nitrogen containing heterocyclic moieties, which should be located in the enzyme-buried part of the inhibitor. 2 The tryptase inhibitor 1 ( Fig. 1 ) characterized with an IC50 value of 36 nM in the previously established enzymatic activity assay, provided a good starting point for the design of an FLT probe. From in-house crystal structures of complexes with related compounds, it is known that the indole heterocycle binds into the S1′ pocket of tryptase ( Fig. 2 ). For the attachment of the PT14 dye, the long triazole-based side chain of tryptase inhibitor 2 (IC50 value of 40 nM in the activity assay) should be well suited. This residue is solvent exposed, and the incorporation of larger substituents is expected to leave the tryptase activity unchanged. Indeed, the combination of the PT14 dye on the solvent-exposed linker part of inhibitor 2 with the indole-based P1′ moiety of inhibitor 1 delivered the tryptase FLT probe 1, which retained strong inhibition of tryptase activity with an IC50 of 24 nM.
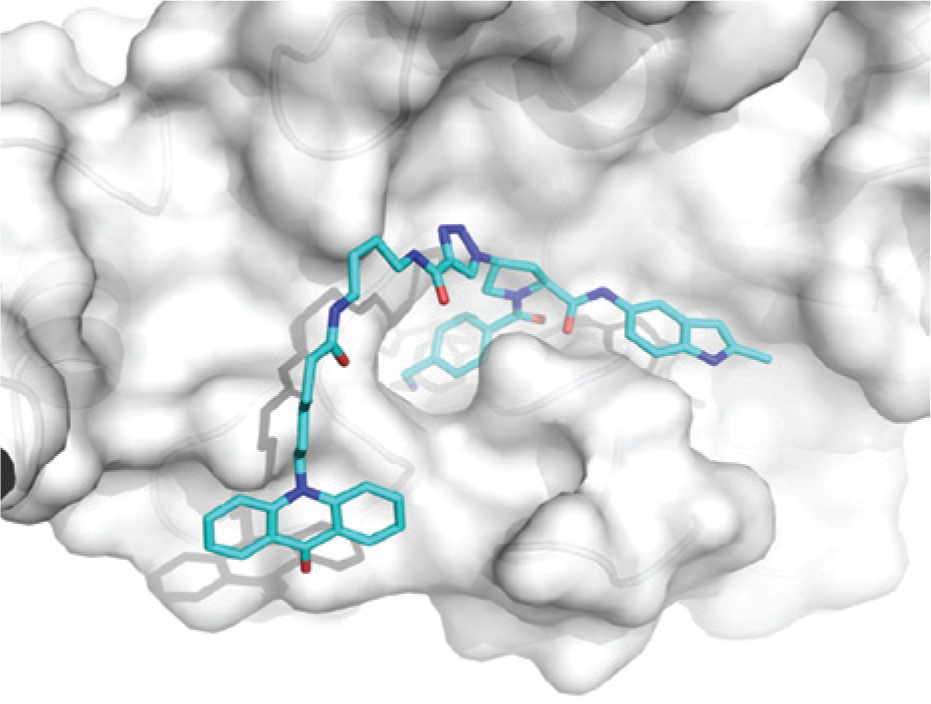
Structural model of the fluorescence lifetime (FLT) probe 1 bound to human β-tryptase. The model was based on an x-ray structure of the inhibitor 1 ( Fig. 1 ) bound to human β-tryptase.
The design of the FLT-reporter probe for the second serine protease was analogous to the probe for tryptase described above.
Probe Characterization
The binding of the novel FLT-based reporter probes to the enzymes was investigated by enzyme titrations at constant probe concentrations of 20 nM and 25 nM for β-tryptase and the second serine protease, respectively.
Figure 3
and
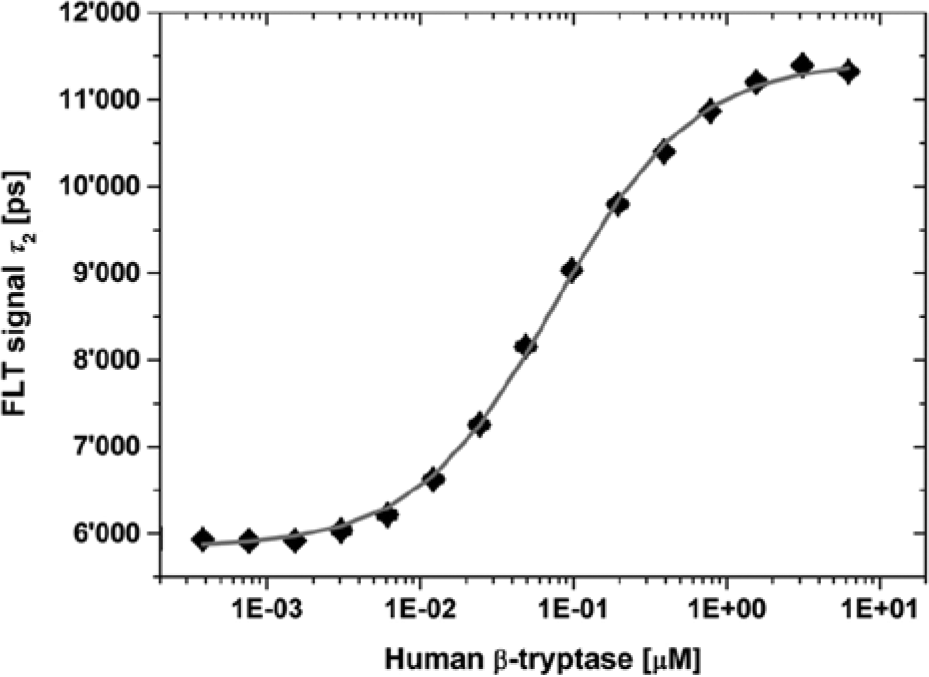
Binding of the fluorescence lifetime (FLT) probes to the human serine proteases, human β-tryptase. Result of an enzyme titration at constant probe 1 concentration of 20 nM in a semi-logarithmic plot. The tryptase was titrated in the concentration range of 0.4 nM to 6.25 µM. The fit of eq 5 to the data resulted in lifetime values, τ2, of 11,419 ± 77 ps and 5858 ± 28 ps for the probe in the unbound and the bound state, respectively; a value for the overall dynamic range, Δτ, of 5561 ps; an EC50 value of 79 ± 5 nM; and a Hill coefficient of 1.0. The nonlinear curve fit of eq 4 to the same data with the constraint of keeping the probe concentration constant at 20 nM results in a lifetime value, τ2, of 5867 ± 31 ps for the unbound probe; an overall dynamic range, Δτ, of 5543 ± 47 ps; and a KD value of 75 ± 3 nM.
The nonlinear fit of the data obtained from the titration experiment to eq 5 resulted in EC50 values of 79 ± 5 and 42 ± 2 nM for the binding of probe 1 to tryptase and probe 2 to the second protease, respectively. In both cases, the curve fit delivered Hill slopes of 1, indicating equimolar binding of the probes to the proteases.
The analysis of the data obtained for the titrations of probe 1 with tryptase and of probe 2 with the second protease with eq 4 (i.e., with a mathematical model describing an equimolar binding of the probe to the protein) yields KD values of 75 ± 3 and 29 ± 3 nM, respectively.
For tryptase, the KD value obtained for the PT14-labeled reporter probe is in good agreement with the IC50 value determined for the unlabeled, parent inhibitor with a previously developed, enzyme activity assay conducted with a substrate concentration below the Michaelis constant, KM, of 63 µM (KD = 62 nM vs IC50 = 82 nM). This finding suggests that the PT14 dye does not interfere with probe binding to the enzyme. In addition, the PT14 dye (6-(9-oxoacridin-10(9H)-yl)hexanoic acid) was tested for its inhibitory potency on both proteases by conducting dye titrations ranging from 0.4 nM to 40 uM in a β-tryptase activity assay employing the short peptide substrate Ac-Tyr-Ser-Ala-Lys-Rh110-DPro with rhodamine 110 (Rh110-DPro) as the fluorogenic leaving group and in the TR-FRET–based competitive binding assay for the second serine protease, respectively. In these assays, binding of the PT14 dye to the active enzymes was not detected (data not shown).
Assay Validation
To further validate the FLT-based competitive binding assay format, proprietary, reversibly binding, active site–directed inhibitors of both proteases were selected and tested in the novel assays. The results were compared with those obtained with alternative assay formats established previously.
In
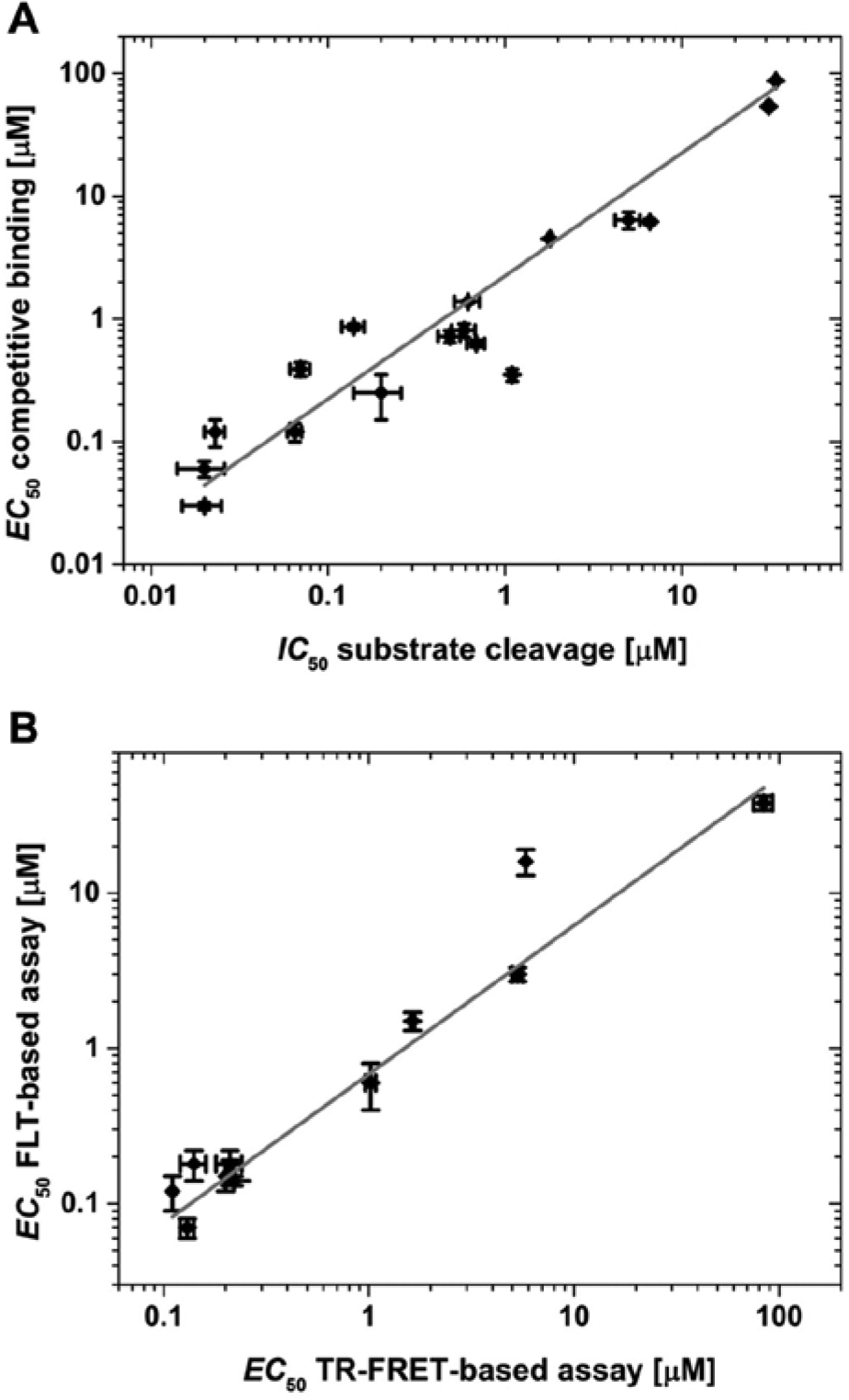
(
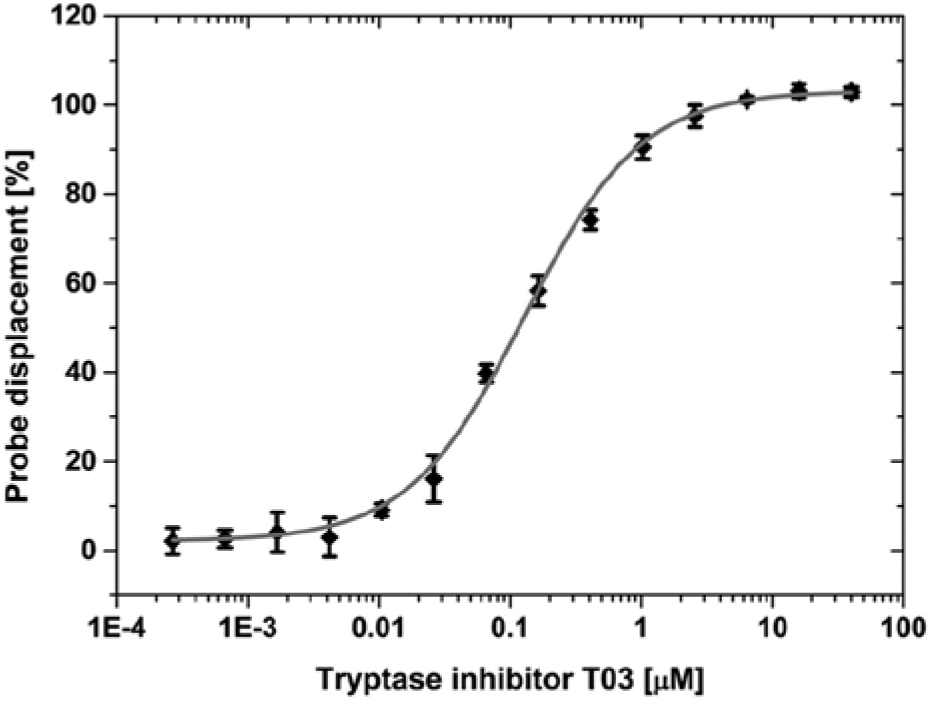
Representative example for the determination of EC50 values: tryptase inhibitor T03 (
In
For the second serine protease, data obtained with the novel FLT-based displacement assay are listed in
Discussion
In this report, we present a novel readout format for competitive binding assays based on changes in the FLT of the acridone dye PT14 as an integral part of the displacement probe.
The synthesis of the probe was driven by rational design. From our previous work on FLT-based enzyme activity assays, we learned that nitrogen-containing aromatic heterocycles are potential dynamic quenchers of PT14’s fluorescence. 2 We selected active site–directed, LMW inhibitors of S1 serine proteases comprising such heterocylic, putatively quenching molecular moieties as, for example, S1 or S1′ pocket binders, respectively. The PT14 dye was conjugated to a site of the inhibitor in a way that the dye is most likely solvent exposed when the probe is bound to the proteases. We hypothesized that in the bound state, the inhibitor’s quenching moieties will be completely or partially buried in the S1 or S1′ pockets, respectively; thus, the spatial separation of dye and quencher will result in a long observable FLT. In contrast, the probe’s unbound states will be characterized by a short FLT, because the probe’s structural flexibility will allow for interaction of the PT14 and the quenching heterocycle.
These hypotheses were shown to be correct. For two different human S1 serine proteases, competitive displacement assays were developed and used for characterizing selected inhibitors by measuring the binding potency of LMW compounds to the active site of the enzyme through competition with the probe.
Changes in the FLTs of greater than 5 ns for both probes were observed upon binding to the respective target enzyme. These values for the overall assay signal windows are very high compared with those reported previously. In 2007, Lebakken et al. 11 investigated FLT as a potential signal for assaying kinases. A change in the lifetime of a dye covalently bound to staurosporine was used to monitor the displacement of staurosporine from a kinase’s ATP binding pocket by LMW compounds. The choice of a red-fluorescent probe with a short lifetime less than 2 ns resulted in a very small assay signal window of only 0.6 ns, significantly limiting the practical usefulness of the proposed assay format.
For selected in-house inhibitors, EC50 data were compared with IC50 data from an FLT-based enzyme activity assay for β-tryptase and EC50 data from a TR-FRET–based competitive binding assay for the second protease, respectively. In both cases, the data from the novel FLT-based reporter displacement assays compare well with those from the assays established previously, thus proving the validity of the presented approach and its suitability for drug discovery.
Currently, HTS campaigns for proteases are usually based primarily on enzymatic activity and competitive binding assays employed in an orthogonal screening step to validate hits. The most common displacement assay format in this regard is TR-FRET. Such assays are relatively cost intensive because they require labor-intensive preparation of tools such as antibodies or proteins site-specifically labeled with Eu/Tb derivatives. 12 Moreover, they are following multiple-step protocols. We see the FLT-based displacement assays as an attractive alternative to TR-FRET assays. FLT-based reporter displacement assays are carried out on native protein instead of a tagged version. This noninvasive approach is less sensitive to assay artifacts and significantly simplifies the assay development.
In addition, FLT-based displacement assays follow a simple mix-and-measure protocol. The labeling of the displacement reporter compound has already proven to be straightforward. Thus, this new assay format can increase the efficiency of the lead-finding process. Decisively, the data quality is at least comparable, if not better than, that obtained with TR-FRET–based assay methods. On the other hand, the highest hurdle for the development of the FLT-based competitive displacement assays is the availability of a protein-binding reporter probe, having both a molecular moiety capable of quenching the fluorescence of the FLT dye as part of its structure and having that moiety tightly bound to the protein of interest. The probe design requires prior knowledge of the protein structure and the binding mode of compounds. Thus, the new FLT-based reporter displacement assay format will preferably be of use for a second wave of lead finding in an ongoing drug discovery program.
In addition to the identification of enzyme inhibitors by screening efforts, we see a high potential for the assay principle as presented here in the hit validation phase following the primary HTS campaign. Here, the competitive binding assay can be applied as a secondary, orthogonal assay principle for hit validation, following the primary screening run based on, for example, an enzyme activity assay. The probe design can be based on chemotypes of inhibitors either known before starting hit finding or on those identified during the hit-finding campaign.
We have demonstrated the development of FLT-based reporter displacement assays with high data quality for two human serine proteases. As a next step, we will transfer this new assay principle to other protease classes. We expect that the FLT-based displacement assay principle can be expanded to other enzyme classes and nonenzymatic targets. Several molecular entities capable of quenching the fluorescence of PT14 are already known. For many targets for which potent inhibitors are known, this approach will enable the design of reporter probes for FLT-based displacement assays.
In conclusion, FLT-based competitive binding assays are very attractive tools for the hit-to-lead and the lead optimization phases of drug discovery projects.
Footnotes
Acknowledgements
The authors thank Laurence Kieffer and Samuel Hassig for their excellent technical support.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.