
Abstract
Liver disease is a leading cause of morbidity worldwide and treatment options are limited, with organ transplantation being the only form of definitive management. Cell-based therapies have long held promise as alternatives to whole-organ transplantation but have been hindered by the rapid loss of liver-specific functions over a period of days in cultured hepatocytes. Hypothesis-driven studies have identified a handful of factors that modulate hepatocyte functions in vitro, but our understanding of the mechanisms involved remains incomplete. We thus report here the development of a high-throughput platform to enable systematic interrogation of liver biology in vitro. The platform is currently configured to enable genetic knockdown screens and includes an enzyme-linked immunosorbent assay–based functional assay to quantify albumin output as a surrogate marker for hepatocyte synthetic functions as well as an image-based viability assay that counts hepatocyte nuclei. Using this platform, we identified 12 gene products that may be important for hepatocyte viability and/or liver identity in vitro. These results represent important first steps in the elucidation of mechanisms instrumental to the phenotypic maintenance of hepatocytes in vitro, and we hope that the tools reported here will empower additional studies in various fields of liver research.
Introduction
Liver disease is a major health care burden with limited treatment options short of organ transplantation. Cell-based therapies represent promising alternatives, particularly for organs with complex repertoires of biochemical functions such as the liver. Progress in the field, however, has been hindered by the propensity of hepatocytes to lose phenotypic functions in vitro 1 and, consequently, the lack of a reliable in vitro model of human hepatocyte biology capable of predicting clinical outcome. The underlying mechanisms of this cellular decline are not well understood, but it is likely that the transformation is related to the loss of the native microenvironment.
In vivo, hepatocytes exist within the complex architecture of the liver and interact with diverse extracellular matrix (ECM) molecules, nonparenchymal cells, and soluble factors. Heterotypic interactions between parenchymal cells (hepatocytes) and their stromal neighbors are known to be important during development 2 and in the adult liver, under both physiologic and pathologic conditions.3,4 Conventional tissue cultures of primary cells lack such multifaceted cellular stimuli. Liver microsomes are currently used in high-throughput identification of detoxification enzymes, but their lack of cytoarchitecture and functional cellular machinery, including dynamic gene expression systems, limits their use in the study of many aspects of liver biology. Liver slices do contain intact cells but have extremely limited viability (~1 day) and are not readily adapted to high-throughput screening (HTS). Similarly, hepatic spheroids, and models that manipulate the ECM microenvironment of hepatocytes with Matrigel and/or collagen require a high degree of hepatocyte confluency for long-term survival and are difficult to miniaturize into a standard and high-throughput format.
In vitro, the viability and liver-specific functions of hepatocytes from multiple species can be maintained for several weeks upon cocultivation with stromal cell types. This coculture effect can be observed using a wide variety of nonparenchymal cells, both primary and immortalized, from intrahepatic and extrahepatic sources, and can be observed even across species barriers.5–7 Hepatocytes in cocultures, particularly with murine embryonic 3T3-J2 fibroblasts, maintain for weeks the distinct nuclei, polygonal morphology, well-demarcated cell-cell borders, and visible bile canaliculi network displayed by cells in vivo. Cocultures have been used to investigate various physiologic and pathologic processes and, more recently, to develop in vitro liver models for pharmaceutical drug screening, disease modeling (e.g., hepatitis C virus, hepatitis B virus, malaria), and engineered hepatic tissues.8–12
To better understand the molecular mechanisms driving the phenotypic maintenance of hepatocytes by cocultures, previous work characterized the type and duration of heterotypic cell-cell interactions required to mediate the coculture effect. Studies suggest that cell-cell contact between primary hepatocytes and nonparenchymal cells (such as murine embryonic 3T3 fibroblasts) is required for ~18 to 24 h, after which continuous stimulation with stromal-derived soluble signals alone over a distance of <400 µm is sufficient. 13 These critical soluble factors appear to be constitutively expressed by 3T3 fibroblasts, independent of hepatocyte interactions, and are not involved in any reciprocal signaling loops between hepatocytes and the supporting nonparenchymal cells. A handful of stromal-derived molecules have been implicated in this process. These include liver regulating protein (LRP), E-cadherin, transforming growth factor (TGF)–β1, and decorin.14–17 While these molecules have been shown to modulate hepatocyte functions in vitro, none of them are sufficient in replacing the stromal cells, nor are they all expressed by all cell types known to maintain the hepatocyte phenotype. Using a gene expression profiling approach, we had previously identified additional candidate fibroblast genes that may play a role in the stabilization of liver-specific functions. 17 Although these findings are promising, a complete picture of the mechanisms underlying the stabilizing effect of fibroblasts remains elusive. This includes the unknown identity of a single factor or cocktail of factors that can adequately support hepatocytes in culture, emphasizing the need to apply objective, genome-wide approaches to these studies.
We thus report here the development of a high-throughput genetic screening platform ( Fig. 1A ) to identify the most critical stromal cell gene products involved in the stabilization of phenotypic functions of primary human hepatocytes ( Fig. 1B ). We hope our findings will provide a foundation for a more complete understanding of liver phenotype maintenance, with implications for basic research, drug development, molecular therapeutics, and cell-based therapies.
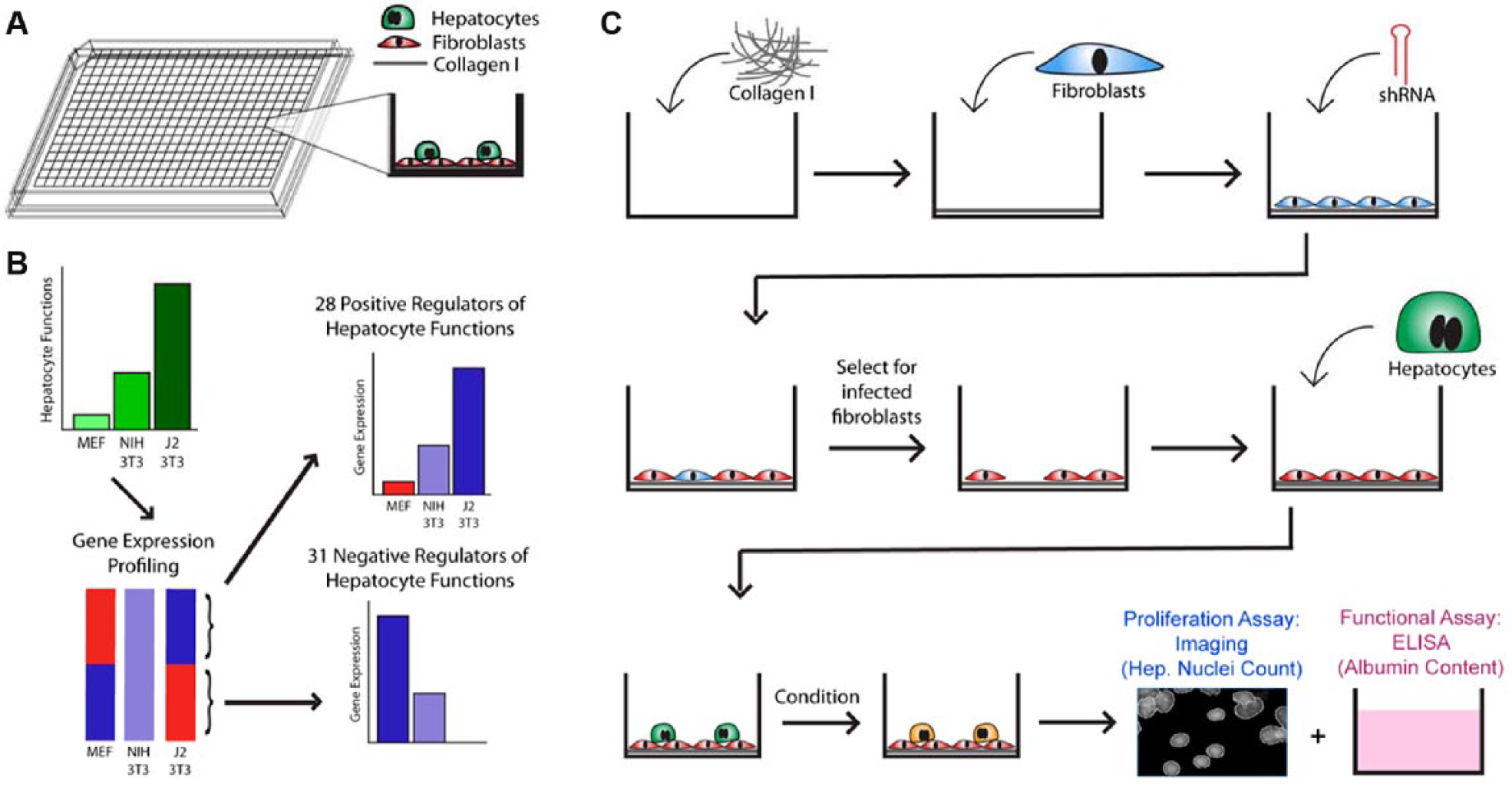
Platform and screen design. (
Materials and Methods
Cell Culture
3T3-J2 culture
Passage 2 3T3-J2 murine fibroblasts were obtained from Howard Green (Harvard) and kept in liquid nitrogen until use. Cells were maintained under standard tissue culture conditions, in 1× Dulbecco’s modified Eagle’s medium (DMEM) containing 10% Bovine Serum (BS) and 1% penicillin-streptomycin. Fibroblasts were grown in T-150 tissue culture flasks and passaged 1:10 using 0.25% trypsin-EDTA when cells reached confluency. Experiments used 3T3-J2s ranging in passage numbers from P7 to P9.
Hepatocyte culture
Primary human hepatocytes were purchased in cryopreserved suspension from Celsis In Vitro Technologies (Halethorpe, MD) and kept in liquid nitrogen until use. To thaw, cells were pelleted by centrifugation at 50 × g for 10 min. The supernatant was discarded before resuspension of cells in hepatocyte culture medium, which consisted of 1× DMEM supplemented with 10% fetal bovine serum (FBS), 15.6 µg/mL insulin, 7.5 µg/mL hydrocortisone, 16 ng/mL glucagon, and 1% penicillin-streptomycin.
Automated cell seeding
Cell suspensions were diluted to the desired densities and kept in suspension using a magnetic stir bar. A Thermo Combi robot (Thermo, Waltham, MA) was used to dispense cells into 384-well formats using speed setting low and standard cassette, 30 µL/well.
Functional Assays
Albumin competitive enzyme-linked immunosorbent assay
A saturating amount of human albumin (40 µL/well of 50 µg/mL albumin) was coated onto the walls of adsorptive 384-well plates (cat. NUNC 460372, NUNC MaxiSorp plates; NUNC, Roskilde, Denmark) at room temperature overnight under agitation. Then, 20 µL of sample supernatant was introduced and competed with coated albumin for binding to horseradish peroxidase (HRP)–conjugated antibodies (cat. 55235; MPBio, Santa Ana, CA). The amount of bound antibody was then quantified via an ultrasensitive chemiluminescent substrate (cat. 37070, Thermo SuperSignal ELISA Pico Chemiluminescent Substrate; Thermo). The competitive enzyme-linked immunosorbent assay (ELISA) signal was normalized to the standard deviation of all 44 control wells found on the same plate to yield a z score. Due to the competitive nature of the ELISA assay, higher ELISA z scores represent lower albumin output.
Biochemical assays
Urea concentration was quantified using a colorimetric assay that reacted diacetylmonoxime with acid and heat, following product instructions (ref. 0580-250, Stanbio Labs Urea Nitrogen Test; Stanbio Labs, Boerne, TX). In total, 3 µL of sample supernatant and 45 µL of reagent were used.
Cytochrome-P450 induction
25 µL of 7-benzyloxy-4-trifluoromethylcoumarin (BFC; BDGentest, Franklin Lakes, NJ) was added to cultures at 50 µM and incubated for 75 min at 37 °C in phenol red–free media. Many different CYP450 isoforms processed BFC into its fluorescent product of 7-hydroxy-4-trifluoromethylcoumarin (7-HFC), which was then quantified fluorometrically. Then, 15 µL of sample supernatant was analyzed following de-conjugation (to generate fluorescent products) by 7 µL of the enzyme β-glucuronidase/aryl-sulfatase.
Automated plate washing
Plate washing for ELISA was performed on the BioTek (Winooski, VT) ELx-405 HT, using the following optimized settings:
Prime: Prime_200 using DI water
Wash: Named program HEPELISA
Method
Number of cycles = 02 Wash format = Plate Soak/shake = Yes Soak duration = 010 s Shake before soak = Yes Shake duration = 005 s Shake intensity = 4 (18 cycles/s) Prime after soak = No Disp
Dispense volume = 100 µL/well Dispense flow rate = 05 Dispense height = 120 (15.240 mm) Horizontal X disp. pos. = 25 (1.143 mm) Horizontal Y disp. pos. = 20 (0.914 mm) Bottom wash first = No Prime before start = No Aspir
Aspir. height = 020 (2.540 mm) Horiz. X asp. pos. = 00 Horiz. Y asp. pos. = 00 Asp. rate = 05 (6.4 mm/s) Asp. delay = 0000 ms Crosswise aspir. = Yes Crosswise on = All Crosswise height = 020 (2.540 mm) Crosswise X horiz. pos. = 00 Crosswise Y horiz. pos. = 00 Final asp. = Yes Final asp. delay = 0000 ms
Automated plate reading
A PerkinElmer (Waltham, MA) Envision 2102 Multilabel Reader was used to quantify ELISA signal, integrated over 0.1 s using a luminescence 700 emission filter and measurement height of 6.5 mm.
Fibroblast Viability Assay (AlamarBlue)
Plated 3T3-J2 fibroblasts were incubated with the AlamarBlue (Thermo) reagent following manufacturer protocols. Stock solution was diluted 10× in culture medium and incubated with seeded 3T3-J2 fibroblasts for 1 h at 37 °C. The level of fluorescence was then read using an excitation wavelength of 540 to 570 nm (peak excitation is 570 nm) and emission wavelength of 580 to 610 nm (peak emission is 585 nm). It is important that all reagents and media are prewarmed to 37 °C prior to addition to cells; failure to do so leads to significant edge effects.
Hepatocyte Viability Assay (Imaging)
Image acquisition
Cultures of hepatocytes and 3T3-J2 fibroblasts were fixed using 4% paraformaldehyde (PFA) in black-walled, clear- and flat-bottomed 384-well plates (Corning, Tewksbury, MA). Fixed samples were then stained with Hoechst 33342. It is important to note that the cell membrane is much more permeable to Hoechst 33342 than Hoechst 33258; thus, an additional permeabilization step using 0.1% Triton-X for 30 min is necessary if visualizing nuclei with Hoechst 33258. Images of fluorescently labeled nuclei were acquired and digitized using an HTS microscope (IKM; Molecular Devices, Sunnyvale, CA) coupled to a barcode reader and robotic arm (Thermo) for automated plate loading. The microscope was configured to self-focus, first using lasers to identify the bottom of wells via differences in the refractive index of plastic and fluids, then using image-based focusing algorithms that scan through a z-stack of ~200 µm in ~50-µm steps in search of the plane with the sharpest images. Accurate examination of nuclear morphology required image acquisition with a 20× objective. Fifty percent of the well area was sampled in a checkerboard fashion, imaging a total of 21 sites per well to balance burden of analysis with potential sampling errors.
Image processing and nuclei identification
We used the freely available open-source software CellProfiler (Broad Institute, Cambridge, MA) for all image analysis, 18 and the configured image analysis pipelines are provided online (http://cellprofiler.org/published_pipelines.html). All images ( Fig. 2A ) underwent illumination correction, as the background intensities varied by up to 1.5-fold across a field of view and often caused unacceptable intensity artifacts ( Fig. 2B ). The illumination correction algorithm averaged all acquired images per plate to identify and normalize consistent discrepancies in the staining intensities across the field of view. 19 All corrected images then underwent a custom image analysis pipeline for nuclei identification (segmentation), by smoothing and using relative peaks in intensity to separate overlapping nuclei. For each identified nucleus, we measured a large number of features to construct a nuclear profile, including measures of nuclear size, shape, and texture, and the number of punctate subnuclear spots ( Fig. 2B , bottom). This per-cell data profile was stored in a MySQL database (Oracle, Redwood, City, CA) and was subsequently used to train supervised machine learning algorithms to automatically classify nuclei as hepatocytes or fibroblasts ( Fig. 2C ).
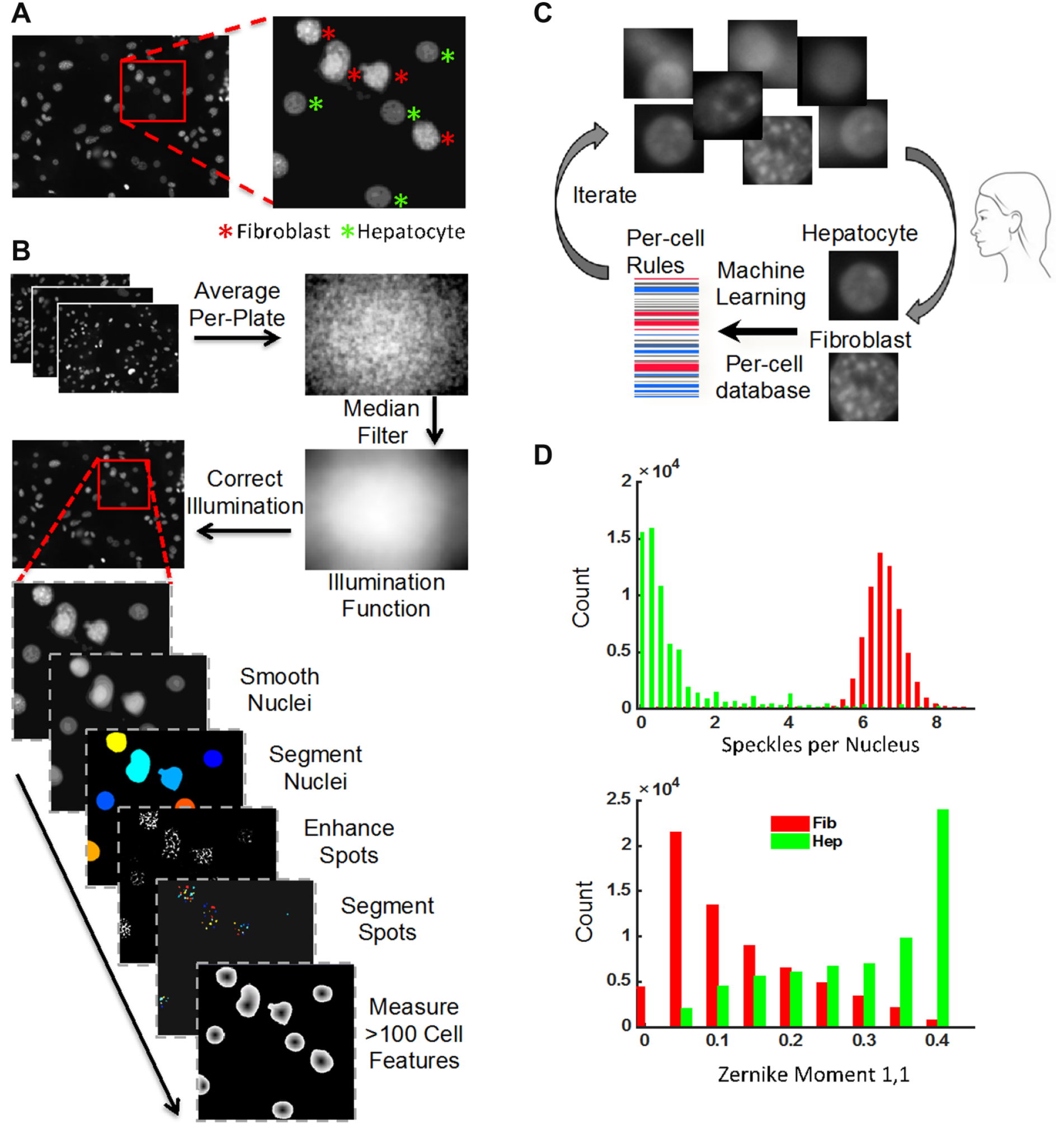
Image analysis and machine learning classification workflow. (
Nuclei classification and quantification
The nuclear profiles generated by CellProfiler were loaded into CellProfiler Analyst 20 for training of supervised machine learning algorithms to distinguish and count hepatocyte nuclei ( Fig. 2C ). Manually created training sets of representative hepatocytes and fibroblasts (50 example objects each) generate a preliminary set of rules for nuclei classification using the GentleBoosting algorithm applied to regression stumps. 20 This rule set was used by CellProfiler Analyst to classify a new batch of nuclei, outputting the results for manual error correction. Iteration of this process refined the rule set until an acceptable accuracy plateau was reached. The final rule set was then applied to the profiles of every nucleus in every image acquired to classify each object as a hepatocyte or fibroblast before outputting a count of each nucleus type per well. The final training set contained 577 objects with an accuracy plateau using a total of 100 rules. We observed that the single feature of nuclear morphology that most effectively distinguished fibroblast nuclei from hepatocyte nuclei was the punctate subnuclear structures present in fibroblasts but absent in hepatocytes, due to the murine origin of the former, which is known to have more textured chromatin. 21 The main pipeline, illumination correction pipeline, and the classifier rules are all available at http://cellprofiler.org/published_pipelines.html.
Short Hairpin RNA Library
The custom short hairpin (shRNA) library was assembled from The RNAi Consortium (TRC) library, designed and synthesized at the Broad Institute Genetic Perturbation Platform.22,23 The pLKO lentiviral delivery vector contains a U6 promoter for constitutive expression of the shRNA sequence and a PGK promoter to drive PAC conveying puromycin resistance. A total of 44 wells were devoted to control viruses that included 22 distinct pLKO-shRNAs targeting green fluorescent protein (GFP), LacZ, luciferase, and red fluorescent protein (RFP) (32 wells total), a GFP/puro fusion-expressing virus (8 wells), and the pLKO-EmptyT virus that expresses a nonhairpin sequence that therefore does not induce seed-based off-target effects (4 wells).
Functional Screen
The workflow of the high-throughput screen to assess the functional effects of implicated factors is summarized in
Figure 1C
. In total, 384-well screening plates (Corning) were incubated with a solution of type I collagen in water (100 mg/mL; BD Biosciences, San Jose, CA) for 1 h at 37 °C. A feeder layer of 3T3-J2 fibroblasts was robotically plated onto the collagen at a density of 4000 cells/well and allowed to acclimate over 24 h. Polybrene at 8 µg/mL and a library of 362 lentiviruses, of which 318 carried 318 different shRNAs representing the 59 genes of interest as well as 44 wells containing 24 different control vectors, were added to two duplicate plates, centrifuged for 30 min at 37 °C, and allowed to incubate for 24 h prior to selection with 5 µg/mL puromycin over 2 days. Infection efficiency was calculated as
Alamar Blue fluorescence prepuromycin selection / Alamar Blue fluorescence postselection
To avoid donor-to-donor variability, human primary hepatocytes from a single donor were plated onto successfully transduced fibroblasts at a density of 4000 cells/well and maintained under standard culture conditions with daily replacement of hepatocyte medium for 7 days, during which time the sample plates were kept in metal stacks with uniform air buffers between each plate to provide uniform gas and heat exchange. In addition, breathable membranes and extra water reservoirs were employed to minimize edge effects arising from fluid evaporation. On day 7 of coculture, culture supernatants were collected for automated ELISA analysis, and cells were fixed in 4% PFA for imaging and analysis, as described under “Functional Assays” and “Hepatocyte Viability Assay.”
Western Analysis
Samples from transduced 3T3-J2 fibroblasts were lysed in RIPA buffer (Upstate Biotechnology, Waltham, MA) with protease inhibitors cocktail (Roche, Indianapolis, IN) and analyzed by Western blot as previously described. 24 The following primary antibodies were used: decorin and GAPDH (Cell Signaling, San Jose, CA).
Statistical Analyses
Assay readiness was assessed via Z′ factor. The platform and assays were developed to detect 2-fold changes in hepatocyte populations; therefore, the positive control was set as “2× hepatocytes” or 4000 hepatocytes per well while the negative control was set as “1× hepatocytes” or 2000 hepatocytes per well.
The z score of each shRNA was calculated as its deviation from the mean of all 44 control wells found on the same plate, normalized to the standard deviation of all control wells on the plate. The control vectors comprised 23 distinct shRNAs, which target various reporter genes not found in wild-type 3T3-J2s to represent the distribution of off-target seed-based effects inherent to shRNAs, plus one GFP-PAC fusion-expressing vector and one vector, “EmptyT,” that expresses a short nonhairpin (nonpalidromic) sequence to avoid both on- and off-target effects. Hit candidates from the screen were selected according to their z score, as described under “Hit Selection.” Due to the competitive nature of the ELISA assay, higher ELISA z scores represent lower albumin output.
Results
Liver Platform Development
To best represent normal human physiology in our liver model and to maximize the model’s ability to predict clinical outcome, we opted to employ primary human hepatocytes in our in vitro platform. All cells were sourced from a single donor to eliminate innate genetic variations that exist within the human population. Eight different donors of cryopreserved human hepatocytes were tested in total. Three were nonplateable and thus incompatible with phenotypic screening. While the remaining five donors all yielded hepatocytes that adhered to rigid collagen in culture, one donor was too young (0.1 years) to exhibit a full repertoire of mature hepatocyte functions, while another two donors had poor functions at baseline. Ultimately, we chose donor GHA, a 1-year-old Caucasian female who died from dry drowning, whose hepatocytes attached well to rigid collagen and demonstrated good synthetic, detoxification, and metabolic functions.
To maintain GHA hepatocytes in culture, we cocultivated them with murine embryonic 3T3-J2 fibroblasts, which we had previously found to be the most effective nonparenchymal cell type at transiently stabilizing hepatocyte phenotype in vitro. 17 The platform contained a subconfluent population of hepatocytes on top of a confluent layer of 3T3-J2s within 384-well plates ( Fig. 1A ). This design enabled fibroblast-mediated hepatocyte stabilization for at least 9 days and is amenable to genetic manipulation of fibroblast populations in 384-well formats. The number of fibroblasts seeded per well was empirically optimized to 4000 cells/well to establish a confluent feeder layer while minimizing the risk of phenotypic transformations due to overcrowding at the end of a 4-day transduction process. The number of hepatocytes seeded per well was empirically optimized to 4000 cells/well, placing the functional protein output of both control and experimental groups within the linear detection range ( Fig. 3A ). Similarly, the amount of media used per well was empirically optimized to balance opposing needs of oxygen transport and nutrient supply—too much media presented a transport barrier for gas diffusion, causing observable steatosis in cultured hepatocytes; too little media caused nutrient deprivation. In this case, there was an additional instrument-based practical restriction that all fluids handled robotically could only be dispensed in volumes that are multiples of 10 µL. All cells were robotically seeded at the lowest possible speed setting to minimize shear stresses. During pilot testing, it was observed that fibroblasts had difficulty remaining attached to plain tissue culture plastic in 384-well formats, thus a matrix coating of collagen type I was added at a concentration of 100 µg/mL.
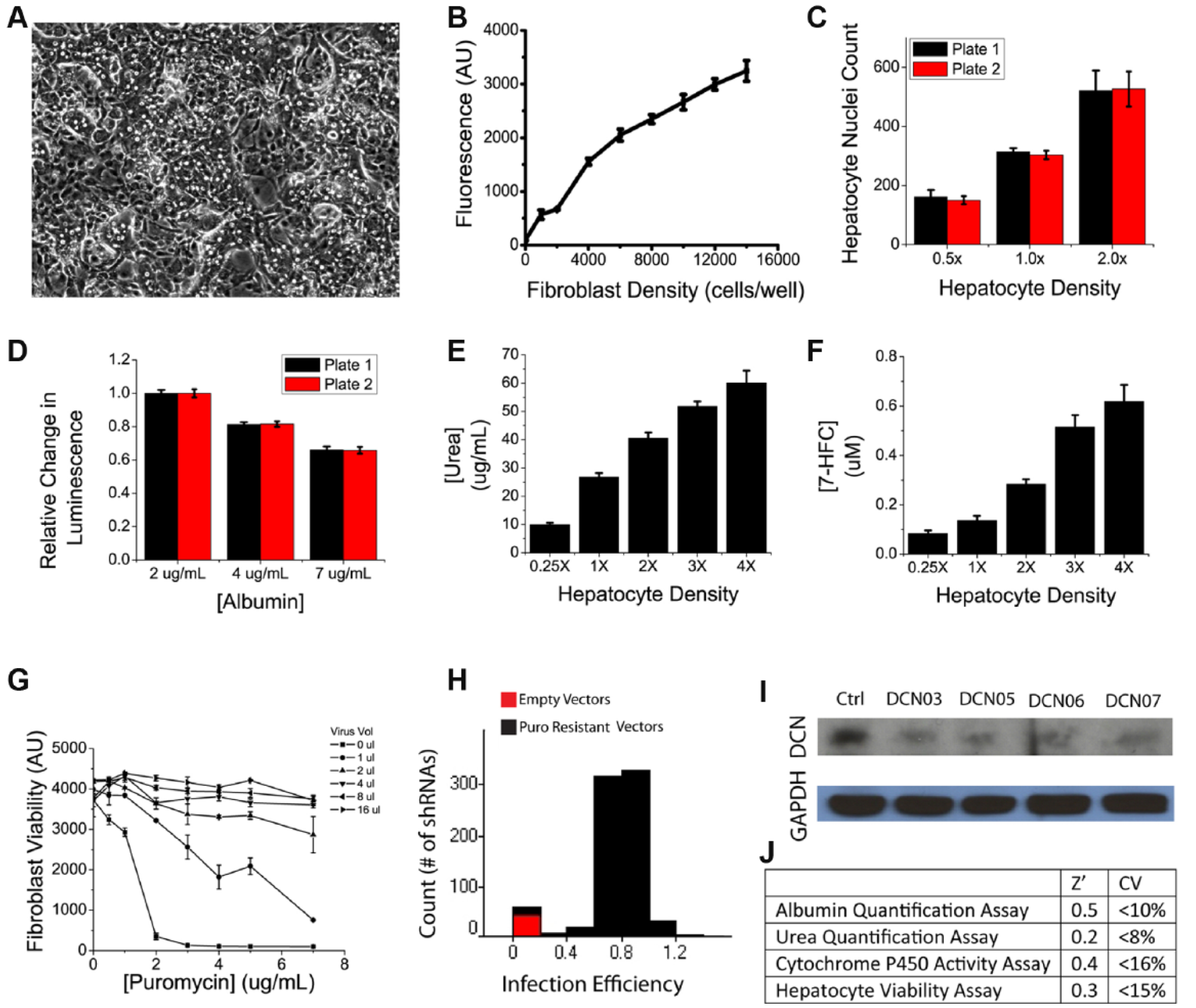
Platform and assay characterization. (
High-Throughput Assay Development
To assess cell fates in this platform, we developed three separate high-throughput readouts. The commercially available AlamarBlue assay was employed to quantify viability of the fibroblast feeder layer, prior to initiation of fibroblast-hepatocyte cocultures. This assay was also used to measure transduction efficiency and enabled optimization of transduction procedures by measuring viability in the absence and presence of puromycin for selection of cells carrying the puroR gene that was included in the shRNA viral vector. Two additional assays were developed to measure hepatocyte functions and cell numbers after 1 week of heterotypic fibroblast-hepatocyte interactions in the aforementioned coculture setting.
A number of commercial assays exist to measure cell viability in culture, including fluorescent-based live/dead cell stains as well as quantifications of cellular enzyme activity as surrogate markers of cell number. Many of these assays, however, are not amenable to automation due to cumbersome workflow and/or have cytotoxic properties that restrict them to end-point usage only. After testing a number of candidates, including CellTiter-Glo, Ki67 staining, and the MTT assay, we chose AlamarBlue to quantify fibroblast viability and transduction efficiency. AlamarBlue is based on the conversion of the molecule resazurin to resorufin through the reducing power of living cells. Resazurin is nontoxic, cell permeable, blue in color, and minimally fluorescent while Resorufin produces bright red fluorescence, thus providing both a colorimetric and fluorescent distinction between parent and daughter compounds. AlamarBlue yielded linear relationships between fluorescence and fibroblast cell numbers when tested on 3T3-J2 fibroblasts in 384-well formats, with an effective range of 2000 to 16,000 cells and a coefficient of determination of R2 = 0.98. The incubation period was empirically optimized to 1 h, although the data show an acceptable window between 1 and 5 h, during which the assay showed an excellent signal-to-noise ratio, with good reproducibility across wells, plates, and batches ( Fig. 3A ). Using the AlamarBlue assay, we automated and optimized lentiviral transduction conditions for delivering shRNAs to 3T3-J2 fibroblasts in 384-well formats to systematically knock down genetic factors of interest. Parameters examined include polybrene concentration for neutralization of cell surface charges, viral titers needed for effective transduction, and the concentration of puromycin required for selection of successfully transduced cells. Our results showed that a higher polybrene concentration of 8 µg/mL provided superior transduction than 4 µg/mL of polybrene. Similarly, puromycin was tested at a concentration range between 0 and 8 µg/mL and found to be optimal at 5 µg/mL ( Fig. 3B ). Using these transduction parameters, we were able to obtain a median infection efficiency of 79.3% ( Fig. 3B ). Knockdowns of target genes were also confirmed via Western analysis ( Fig. 3B ).
To assess hepatocyte phenotype in vitro, we equipped the high-throughput liver platform with several functional assays. Due to the diverse repertoire of the 500+ documented and yet unidentified biochemical functions of the liver, there does not exist a single all-inclusive, gold-standard assay for measuring hepatocyte functions. We thus opted to sample three major types of liver functions: (1) albumin output as a surrogate marker for protein synthesis functions of the liver through a competitive ELISA, (2) urea generation as a surrogate marker for amino acid metabolism functions of the liver through a colorimetric assay, and (3) cytochrome P450 activity as a surrogate marker for detoxification functions of the liver through an enzyme activity assay. For all three assays, we optimized parameters such as reagent type, concentration, and volume to develop them into biochemical assays compatible with HTS. Validation data demonstrated that these assays can confidently (Z′ > 0) detect 2-fold changes in hepatocyte populations with low variance (coefficient of variation [CV] <20%) and good reproducibility ( Fig. 3 ).
For our functional screen, we chose the ELISA-based albumin quantification as the functional readout because it exhibited the highest Z′ and was of the greatest clinical relevance. The most common form of the ELISA assay is a sandwich ELISA, which captures the antigen of interest in between two layers of antibodies. This assay is difficult to adapt to HTS due to a cumbersome protocol, which is difficult to program robotically, thus limiting throughput. Therefore, for our liver platform, we employed a competitive ELISA assay, which reduced the length of the workflow by approximately one-third. It is important to note here that the competitive nature of the assay inverts the readout such that a higher ELISA z score reflects a lower albumin content. When coupled with the optimized fibroblast and hepatocyte cell numbers as well as transduction procedure, the platform and assays are able to easily identify cultures where hepatocytes have lost their liver-specific synthetic functions ( Fig. 4A , “empty” red dots; the fibroblasts of these cultures were not treated with any lentivirus but still underwent puromycin selection; they thus effectively became hepatocyte-only cultures that lost viability and phenotype by day 7).
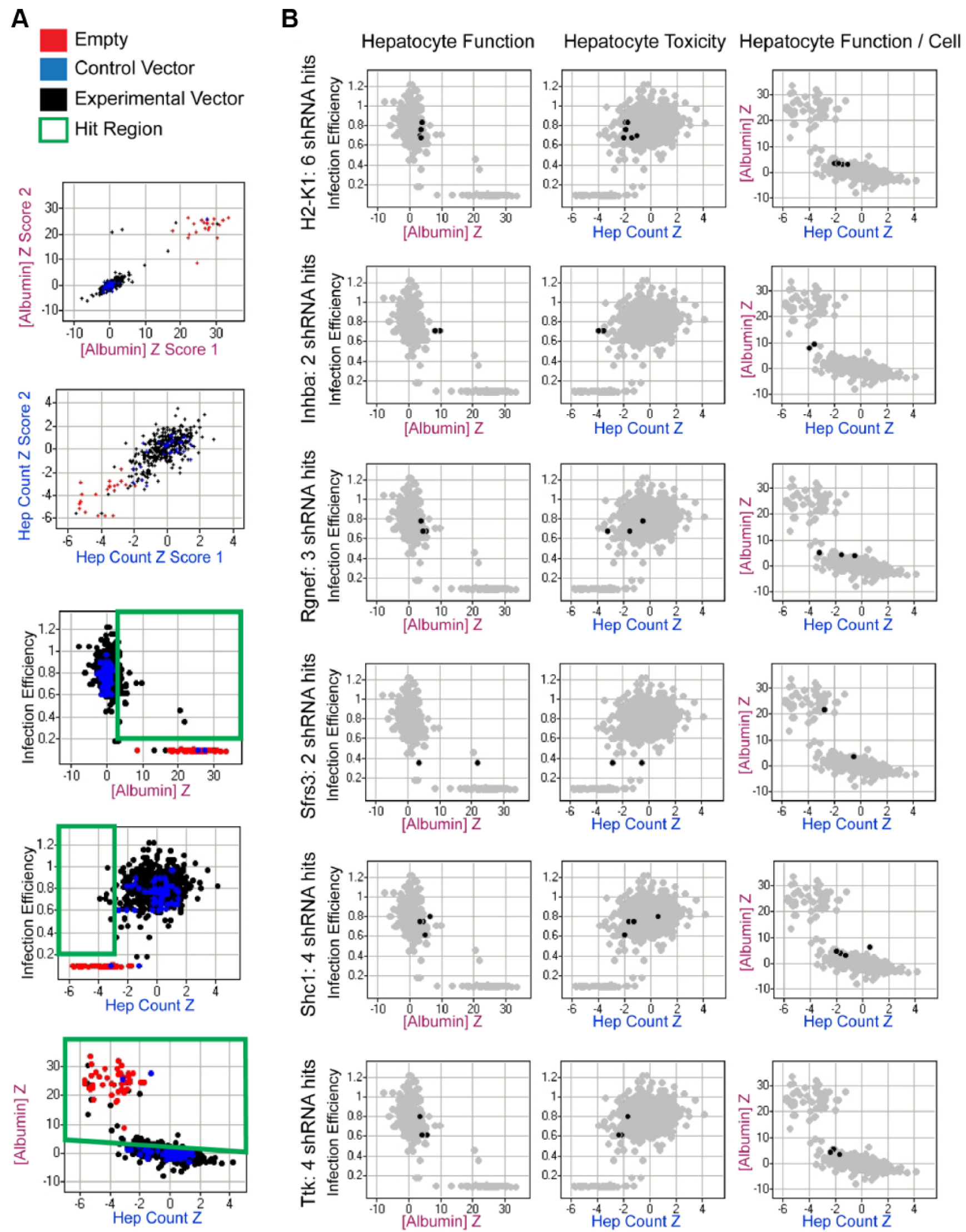
Functional screen. (
In addition to assessing the functional output of hepatocytes, we developed an image-based proliferation assay that uses nuclear morphology to quantify hepatocyte nuclei numbers in coculture. This allowed us to measure hepatocyte viability in addition to determining hepatocyte synthetic functions on a per-cell basis. When visualized with Hoechst stain, hepatocyte nuclei are more uniform in texture while fibroblast nuclei are punctate ( Fig. 2A ). The assay thus visualizes all cell nuclei in culture using a simple Hoechst stain, specifically identifies hepatocytes based on nuclear morphology, and provides a count of the number of hepatocyte nuclei in culture. Imaging of multiple 384-well plates containing untreated hepatocyte-fibroblast cocultures showed that the image-based readout can confidently (Z′ > 0) detect 2-fold changes in the number of hepatocyte nuclei, with low variance (CV <15%) and good reproducibility ( Fig. 3A , B ).
Functional Screening
Selection of genetic factors for systematic knockdown
A large number of nonparenchymal cell types are known to support the coculture phenomenon. They differ, however, in the degree of hepatic functions that they are able to induce when cocultivated with primary hepatocytes. 3T3-J2 fibroblasts, for example, are able to induce physiological or even supra-physiological levels of liver-specific functions in cocultivated hepatocytes, while the closely related NIH-3T3 fibroblasts and the primary mouse embryonic fibroblasts (MEFs) from whence these cell lines were originally derived are much lower inducers of the coculture effect. Previously, Khetani et al. 17 conducted gene expression profiling of these three fibroblast populations using Affymetrix (Santa Clara, CA) GeneChips. Here, we reanalyzed those data to identify 59 genes whose levels of expression correlated strongly with the induction of hepatocyte functions in vitro. Using our high-throughput liver platform together with virally delivered shRNAs, we tested the effect of loss-of-function perturbation of these genes in fibroblast feeder cells on phenotypes of cocultured hepatocytes to assess the potential role of these stromal factors in the coculture phenomenon.
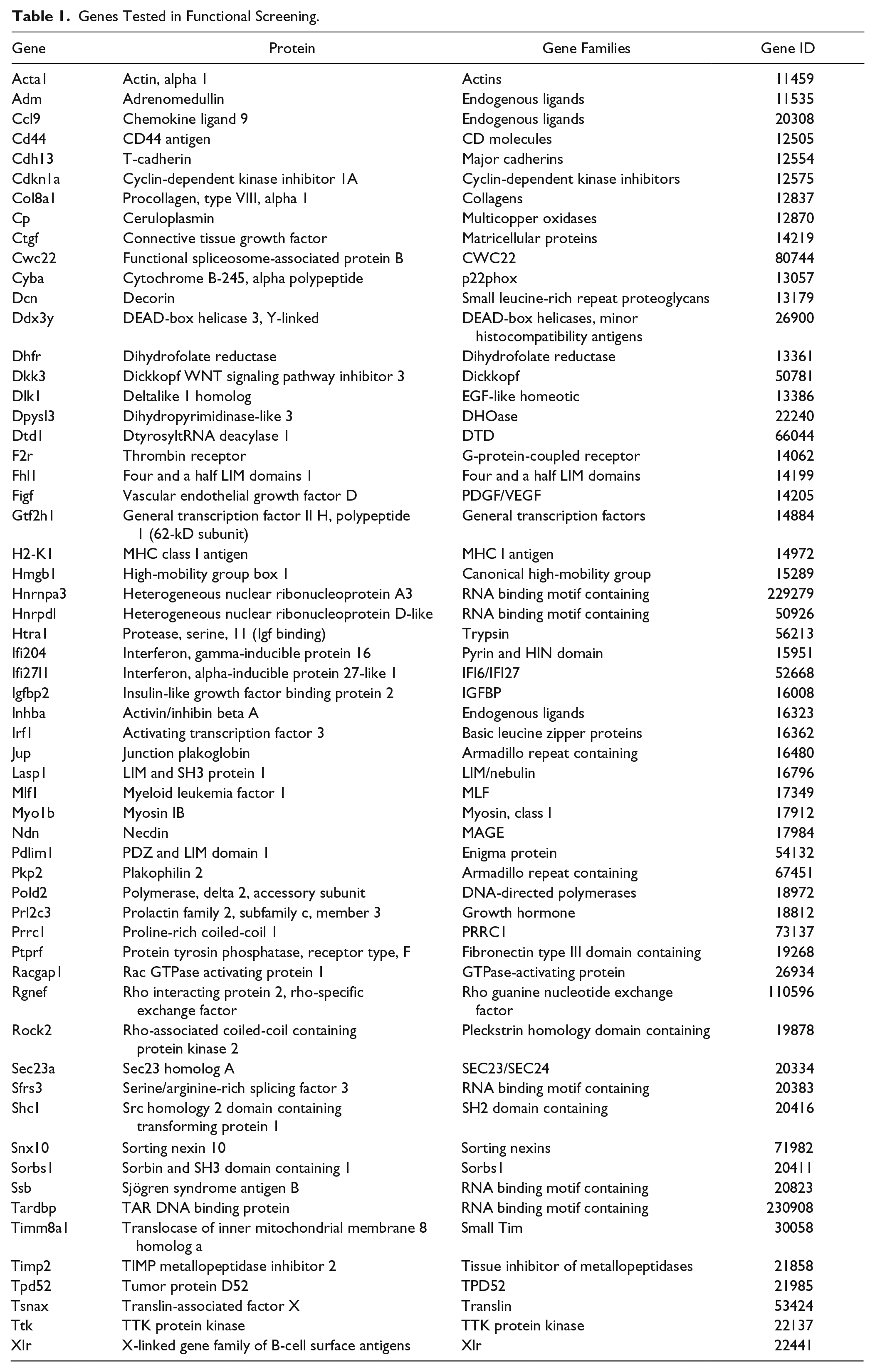
The 59 genes exhibited a minimum of a 5-fold difference in levels of expression between the high inducer 3T3-J2s and low inducer MEFs. We prioritized molecules with known functions in cell-cell communications, such as various cell surface (e.g., Delta-like homolog 1), ECM (e.g., decorin), and secreted factors (e.g., vascular endothelial growth factor D, ceruloplasmin) and eliminated genes that were flagged for having poor quality control data on the Affymetrix GeneChips. We also added factors that have been implicated in the coculture effect through previous investigations (e.g., T-cadherin). 25 Among these 59 genes were 31 genes whose differential levels of expression correlated positively with the pattern of hepatocyte induction observed (positive inducers); the remaining 33 factors showed expression patterns that correlated negatively with hepatocyte functions (negative inducers). All 59 genes are listed in Table 1 .
Genes Tested in Functional Screening.
Functional Screen and Hit Selection
To determine if any of these 59 differentially expressed fibroblast genes play a critical role in the maintenance of hepatocyte phenotype in vitro, we conducted knockdown studies in 3T3-J2 fibroblasts, targeting each gene through a custom shRNA library and monitoring effects on cocultured primary hepatocytes. The library contained 4 to 10 different shRNAs for each gene of interest, a redundancy designed to distinguish the real biological effects of gene knockdown from artifacts such as off-target effects and the disruption of potentially critical cellular functions through the random insertion of lentiviral genes into the host genome. Only genes represented by two or more different shRNAs were included in the screen hit list, although exceptions were made for those with particularly profound effects (z > 4). All shRNAs were tested in duplicate along with various controls: “empty” red dots represent cultures whose fibroblast feeder layer was not treated with lentivirus but still underwent puromycin selection; these effectively became hepatocyte-only cultures that lacked fibroblast coculture support; “control vector” blue dots represent treatments with viruses conferring expression of either shRNA targeting reporter genes not found in wild-type 3T3-J2s that are meant to assess the range of seed-based RNA interference (RNAi) off-target activities or of a nonhairpin (nonpalindromic) sequence that should not produce either on- or off-target RNAi-based effects. The resultant mutant 3T3-J2 fibroblasts were then cocultivated with primary human hepatocytes in the high-throughput liver platform, and their ability to induce liver-specific functions was examined via the competitive ELISA-based functional assay (where high z score indicates low albumin output) and the image-based hepatocyte viability assay.
Individual shRNAs were considered biologically active and selected for further analysis if their effect on the feeder cells was to significantly decrease either total albumin output from the hepatocytes or a hepatocyte-number adjusted albumin level. Population-level data were directly measured by the ELISA assay, and any shRNA with a z score greater than 3 was selected (p < 0.001). We would like to reemphasize here that a higher ELISA z score reflects lower albumin output due to the competitive nature of the ELISA assay. Albumin output was also assessed relative to the albumin versus HepCount trend observed for the control vectors. Regression analyses of cultures treated with control vectors (blue dots, which thus contained wild-type fibroblasts) revealed the following relationship between ELISA z scores (z_ELISA) and imaging z scores (z_HepCount):
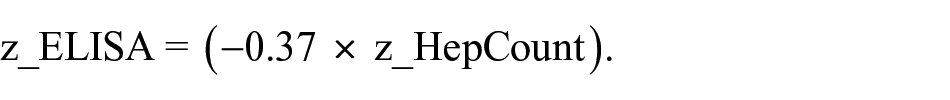
That is, as expected, albumin levels were higher (lower ELISA z score) when more hepatocyte numbers were higher (higher HepCount z score). Applying to this population-adjusted albumin level the same hit selection criterion of a z score greater than 3 (p < 0.001), hit shRNAs were considered to be those for which z_ELISA > (–0.37 × z_HepCount + 3). In addition, any shRNA with infection efficiency below 20% was eliminated from analyses. The final hit list was thus generated via the following algorithm:
[Infection efficiency >20%] AND
[z_ELISA > (–0.37 × z_HepCount
[number of active hairpins per gene > 2 OR z_ELISA > 4]
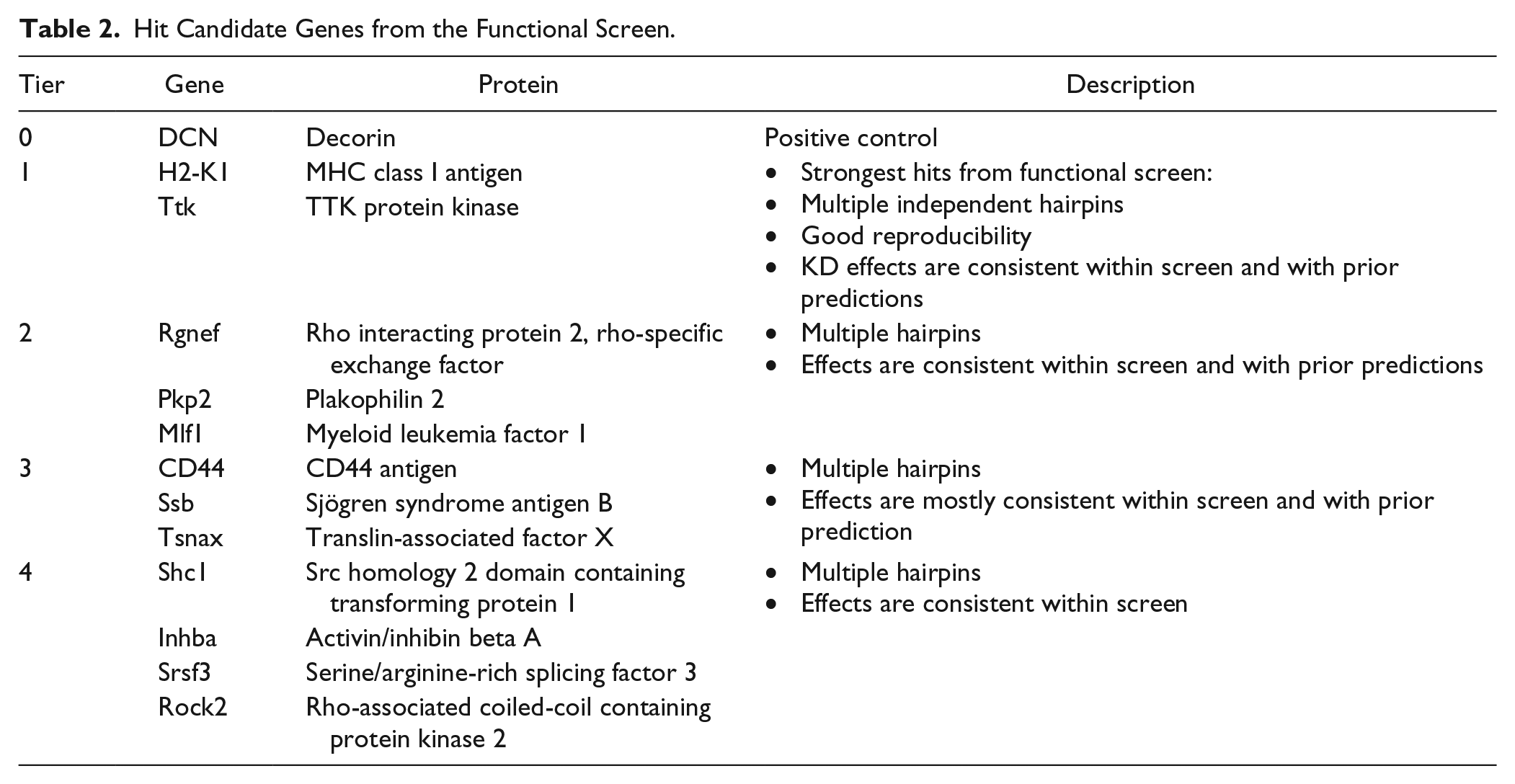
A total of 12 hit genes, listed in Table 2 , were nominated by this screen in agreement with previous hypotheses that these molecules play an important role in the maintenance of hepatocyte phenotype in vitro. The magnitude and consistency of effect of some representative active shRNAs in the hit list are plotted in Figure 4B .
Hit Candidate Genes from the Functional Screen.
Discussion
Sourcing of hepatic cells is a fundamental challenge for many fields of liver research. Many are forced to employ xenogeneic sources such as rodent and porcine hepatocytes or immortalized human hepatocyte cell lines. Animal hepatocytes are extensively studied and easily obtained but exhibit numerous species-specific differences in hepatocellular functions, including apolipoprotein expression, metabolic regulation of cholesterol, and phase I detoxification enzymes. Human hepatocyte cell lines, while expandable, contain mutations and exhibit an abnormal repertoire of liver functions, limiting their clinical relevance.
Primary human hepatocytes present the best functional output, and recent advances in cryopreservation technologies have also enabled the storage of entire livers of human hepatocytes, enough to power multiple batches of phenotypic screens. This enables hundreds of thousands of knockdown or overexpression studies on a constant genetic background over a period of several months. We recognize that it would be important to test additional donors and genetic backgrounds and recommend changing donors after the completion of primary screening, such as during various follow-up studies. While extensive previous characterizations have shown that high-quality cryopreserved primary human hepatocytes exhibit phenotypes that approach fresh hepatocytes and are thus very useful for in vitro liver studies, our findings here indicate that not all donors are suitable for use in phenotypic screens. Therefore, we propose that empirical characterization of each donor of cryopreserved primary human hepatocytes is an indispensable first step to their use in liver research.
Maintenance of cryopreserved primary human hepatocytes in vitro can be achieved transiently via cocultivation with a variety of nonparenchymal cells. There exist multiple configurations of cocultures, with varying degrees of architectural organization. The simplest implementation consists of a coplanar distribution of randomly mixed hepatocytes and 3T3-J2 fibroblasts on a matrix of rigid collagen type I. More sophisticated designs use semiconductor-driven microtechnology to organize primary hepatocytes into in vitro colonies of empirically optimized island sizes, subsequently surrounded by 3T3-J2 fibroblasts (MPCC). All configurations of hepatocyte-J2 cocultures were found to maintain primary human hepatocyte functions in vitro for at least 9 days.12,26 Generally, increased architectural organization of cells in culture leads to longer term stabilization of hepatocyte functions, with MPCC being the most optimal configuration, enabling maintenance of hepatocyte functions in vitro for 4 to 6 weeks. However, such segregation of hepatocyte and fibroblast populations limits the number of hepatocytes that engage in heterotypic cell-cell interactions. In addition, MPCCs are difficult to miniaturize beyond 96-well platforms, and in any case, such prolonged periods of hepatocyte functions are neither necessary nor practical for most whole-cell screens. We thus designed our high-throughput liver platform to assume a feeder layer coculture configuration, which ensures that every hepatocyte has access to a fibroblast and can participate in heterotypic cell-cell signaling.
Significant efforts were dedicated to the development of an image-based hepatocyte viability assay. The coexistence of two different cell types in each well renders their differentiation challenging. Existing measurements of cellular numbers, such as AlamarBlue, CellTiter-Glo and live/dead stains, all reflect the joint state of the whole well, which allows behavior of the more populous 3T3-J2 fibroblasts to mask the number of hepatocytes in culture. We also considered generating stable fluorescent clones of hepatocytes; however, the rapid decline in hepatocyte viability ex vivo as well as their inability to proliferate in vitro renders such lengthy manipulations technically unfeasible. Generating stable clones of fibroblasts is possible, but doing so alters their biology such that they lose their protective phenotype. Therefore, we needed to develop a custom readout to isolate and quantify the hepatocyte subpopulation. Human hepatocytes in culture can be distinguished from underlying 3T3-J2 fibroblasts via a variety of methods, including phase-contrast microscopy, staining for hepatocyte-specific markers, and striking species-specific differences in nuclear morphology. Bright-field images, while easy to acquire, are difficult to quantify, particularly in a high-throughput manner. Immunofluorescent staining of particular antigens, while easy to measure in an automated fashion, is difficult and expensive to execute in 384-well and smaller formats with often unacceptable signal-to-noise ratios. Therefore, we chose to leverage differences in nuclear morphology. It should be noted that the highly textured nature of fibroblast nuclei rendered their segmentation difficult, often leading to the breakup of a single nucleus into multiple nuclei. Therefore, while the assay does ostensibly report numbers of fibroblast nuclei as well as hepatocyte nuclei, it is optimized for accurate detection of hepatocyte nuclei only. We point to our later work, which yielded an image analysis pipeline for more accurate measurement of the number of fibroblasts in coculture. 21
In addition, we recognize that counting nuclei is not always equivalent to counting cells. Hepatocytes in particular are well known for their ability to assume polyploid states and can sometimes contain eight or more nuclei. While nuclear proliferation does not always translate into cellular proliferation, cell death is always accompanied by a decrease in nuclei numbers. Therefore, nuclei number is still a viable surrogate marker for cell number, albeit without a strict one-to-one correlation. We note that polyploidy can be detected visually in images in follow-up analysis of hits.
Analyses of the functional screen focused on finding only positive mediators of coculture and neglected to examine negative mediators, whose knockdown would enhance hepatocyte functions. This decision was made because 3T3-J2 fibroblasts are strong inducers of the coculture effect, often supporting physiological to supra-physiological levels of liver-specific synthetic functions in vitro. We thus believe that it would not be rewarding to look for negative mediators in a cell type that is inherently already a high inducer of hepatocyte functions. We also prioritized the search for positive mediators because their gene products have the potential to substitute for nonparenchymal cells and directly advance the development of fibroblast-free tissue-engineered hepatic systems. Although negative mediators are less useful in this regard, studies of such factors can provide insight into the signaling pathways involved in the phenotypic maintenance of hepatocytes ex vivo and should be conducted in the future. Such studies might be best suited for knockdown studies in nonparenchymal cells known to be low inducers of the coculture effect, such as primary MEFs.
As a first step to analysis of the functional screen results, we examined the effect of decorin, a putative positive control, in our screen. Decorin is a chondroitin sulfate–dermatan sulfate proteoglycan that binds to collagen, whose role as a positive mediator of the coculture effect was not only theoretically predicted by gene expression profiling but also confirmed via studies that showed upregulation of hepatic functions in vitro when cocultivated with stromal cells on adsorbed decorin in a dose-dependent manner. 17 In our screen, two active shRNAs were found to represent decorin, and both showed a decrease in hepatocyte functions upon knockdown, consistent with prior findings. Similar to other top hits of the screen (e.g., H2-K1, which encodes for the K region of a histocompatibility protein with a human ortholog of HLA-A, or Inhba, which encodes for a subunit of both activin A and inhibin A, both secreted growth factors involved in a myriad of biological processes, including cell cycle regulations), it is not immediately apparent how decorin might be maintaining liver functions or what signaling pathways may be involved. These questions are the subject of ongoing investigations.
We also noted that knockdown of decorin did not completely remove the rescue effects of coculture on hepatocyte phenotype, again consistent with prior studies that decorin alone cannot stabilize hepatocytes in vitro. In fact, while knockdown of the hit candidates each had significant effects on hepatocyte functions, the coculture effect was not consistently abolished in any of the cultures with mutant fibroblasts, as evidenced by significantly higher albumin output compared with hepatocytes cultured alone, without fibroblast support ( Fig. 4A ). Although it is possible this is related to incomplete knockdown of protein products under our experimental conditions, we suspect that multiple signaling molecules are involved in the maintenance of hepatocyte phenotype ex vivo and that a cocktail of stromal factors will ultimately be needed to replace nonparenchymal cells in hepatic tissue engineering applications.
In conclusion, we report here the development of a high-throughput human liver model and attendant automatable assays capable of reflecting human liver physiology in vitro. These tools were used to conduct a small genetic knockdown screen of fibroblast-derived factors to identify molecules important to the maintenance of hepatocyte functions in vitro. Overall, we identified 12 genes as priority candidates for further experimental validation and hope the tools reported here will empower additional studies in the various fields of liver research.
Footnotes
Acknowledgements
We thank the members of the Bhatia Lab, especially Dr. Heather Fleming, the Broad Institute Imaging Platform, and the Broad Institute Genetic Perturbation Platform, especially Dr. Serena Silver and Dr. Federica Piccioni, for helpful guidance throughout.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health (NIH UH3 EB017103, to SNB), Koch Institute Support Grant P30-CA14051 from the National Cancer Institute (Swanson Biotechnology Center), as well as a grant from the National Science Foundation (NSF CAREER DBI 1148823, to AEC). Dr. Bhatia is an HHMI Investigator and Merkin Institute Fellow at the Broad Institute of MIT and Harvard.